

Abstract
Background: Diabetes is proved to be one of the independent risk factors for cognitive dysfunction. The pathophysiologic changes caused by diabetes including hyperglycemia and tissue hypoxia may contribute greatly to cognitive decline. In the present study, we demonstrate E3 Ubiquitin Ligase Siah-1 downregulates the key synaptic protein Synaptophysin expression under high glucose and hypoxia condition which may be the underlying factor leading to cognitive dysfunction in diabetic patients. Methods: In this study, hypoxia (2% oxygen) and high glucose (50 mM) were used to treat primary neuronal culture. By using quantitative PCR and western blotting we determined the influence of hypoxia and high glucose on the expression of synaptophysin and Siah-1 and the phosphorylated forms of extracellular signal-regulated kinase (ERK). Knockdown of Siah-1, inhibitors for proteasome, lysosome and ERK kinase was employed to evaluate the role of Siah-1 and ERK activity on the expression of synaptophysin. By immunoprecipitation we also examined the role of Siah-1 in the ubiquitination of synaptophysin under hypoxic and hyperglycemic condition. Results: We demonstrated that hypoxia and high glucose together but not hypoxia or high glucose along mediated posttranscriptional reduction of synaptophysin with increased ERK phosphorylation and Siah-1 expression. The downregulation of synaptophysin was reversed by inhibition of ERK and Siah-1 knockdown. Overexpression of Siah-1 accelerated the degradation of synaptophysin under hypoxia and high glucose conditions and promoted the ubiquitination of synaptophysin. Conclusions: The present results demonstrate that Siah-1 is the key factor that contributes to hypoxia and high glucose mediated synaptophysin degradation.
Keywords: Siah-1, diabetes, synaptophysin, ERK, cognitive dysfunction
Introduction
Vascular risk factors such as hypertension, stroke, diabetes, have been demonstrated in close relationship with cognitive decline in the elderly. Among them, diabetes has been affirmed to cause cognitive dysfunction in clinical research [1-5].
A multi-center European prospective study (LADIS) found that diabetes is one of the independent risk factors for cognitive dysfunction [5]. There is also evidence showing that blood glucose control could significantly improve cognitive function in elderly patients [6-8]. Animal experiments showed that diabetes can cause significantly reduced hippocampal mossy fiber endings and the number of synaptic vesicles, leading to cognitive decline in animals [9,10].
Studies have indicated that people with diabetes may also have obviously reduced oxygen supply to the tissue [11-14]. Considering hypoxia as another important risk factor for cognitive decline, diabetes related cognitive dysfunction may be caused by chronic hyperglycemia and tissue hypoxia.
Synaptophysin (Syp) is a key synaptic vesicle protein which is involved in synaptic vesicle exo-endocytosis and synaptic vesicle biogenesis. Syp was demonstrated to relate closely with cognitive function [15-17]. In neurons, Syp is mainly degraded through the ubiquitin-proteasome system. Studies have shown that E3 ubiquitin ligase Siah (Mammalian Homologues of Seven in Absentia) is the main protease that degrades Syp [18]. The expression of Siah was elevated significantly by both hypoxia and hyperglycemia. We postulated that in diabetes, both hyperglycemia and hypoxia might cause great synaptic changes, resulting in cognitive dysfunction. Thus, in this study, we examined the influence of hypoxia together with higher glucose level on the expression of Syp in cultured neurons, and evaluated the role of Siah in this pathological change.
Methods
Primary hippocampus neuron culture
Sprague-Dawley rats were purchased from the Shanghai Institute of the Chinese Academy of Science. All experimental procedures were carried out in accordance with the experimental standards of Tongji University, as well as international guidelines on the ethical treatment of experimental animals. Neurons were isolated from embryonic day (E) 14.5 embryos [19]. Briefly, the embryos were removed and placed into ice-cold Dulbecco’s minimal essential medium. Using a dissecting microscope, the hippocampus was dissected away from the brainstem, the meninges were removed and the telencephalon was placed into ice-cold HBSS. The tissue was digested in trypsin-EDTA (0.1%) for 20 min, washed three times in Dulbecco’s minimal essential medium, and dispersed in neurobasal medium containing B27, L-glutamine and glutamic acid. The cells were plated on poly-L-lysine coated-glass coverslips, 96-well plates or 100 mm dishes at a density of 3-10×105 cells/ml and maintained at 37°C in a humidified 5% CO2 incubator. Under these conditions, neuronal purity was approximately 90%, as estimated by positive immunocytochemical staining for neurofilament proteins and the negative immunocytochemical staining for glial fibrillary acidic protein (data not shown). The cultured neurons were used for studies on in vitro days 8-10 (DIV 8-10).
Cell treatment
Optimal survival rate and neurite growth of hippocampal neurons require 25 mM basal glucose [20], reflecting the fact that neurons have high metabolic rates. Neurobasal medium containing 25 mM glucose (normal cultured, NC) meets these metabolic requirements. After 7 days in culture, cells were incubated with 50 mM of glucose (high glucose, HG) or with 25 mM glucose + 25 mM mannitol (osmotic control, OC), Then maintained for further 3 days under hypoxia (2% oxygen) or Normoxia (21% oxygen) condition. The proteasome inhibitor MG132 or lactacystin (20 μM for both; Calbiochem, San Diego, CA), the lysosome inhibitor E64 (50 μM; Sigma, St.Louis, MO, USA), the extracellular signal-regulated kinase (ERK) kinase inhibitor U0126 (10 μM; Cell Signaling Technology, Beverly, MA) or PD98059 (10 μM; Calbiochem) or vehicle were added before the hypoxia procedure.
Cell injury assays-LDH measurement
Cellular injury was determined by measuring concentration of lactate dehydrogenase (LDH) released into the medium [21]. Briefly, the media was removed and LDH in the medium and total cellular LDH were determined using the CytoTox 96® Non-Radioactive Cytotoxicity Assay (Promega, Madison, WI, USA). The maximal LDH release was obtained in each well following repeated cycles of freezing and thawing. Each experimental condition was repeated in triplicate. Results were expressed as a percentage of maximal LDH release, after the subtraction of background levels was determined from the medium alone.
Cell viability assay
The viability of cells was examined by 3-(4,5-dimethylthiazole-2-yl)-2,5-dipenyltetrazolium bromide (MTT) assay. After hypoxia and high glucose exposure, MTT was added to a final concentration of 0.5 mg/mL for 4 h. The supernatant was removed and 150 uL dimethylsulfoxide was added for 20 min. The MTT optical density values were measured on a microplate reader at 570 nm and 630 nm wavelengths. Each experimental condition was repeated in triplicate.
Quantitative PCR
The knockdown efficiency of siah-1 siRNA was confirmed by measuring siah-1 expression using quantitative real-time PCR. And the expression level of synaptophysin was also been detected. Briefly, total RNA was purified using RNA easy columns (Qiagen, Valencia, CA, USA) and cDNA synthesis was performed using SuperScript III (Invitrogen, Carlsbad, CA, USA). Primers were designed to amplify a segment to rat siah-1 and synaptophysin. Rat β-actin primers were used in the same reactions to control for the amount of starting template. All samples were measured in duplicate and were compared to standard curves of known concentrations of different genes; all mRNA expression data is expressed relative to β-actin.
Immuno blotting
The cultured primary neurons cells were placed into RIPA buffer. The whole cell lysate (40 μg) were separated using SDS-PAGE and transferred to nitrocellulose membranes (Millipore, Bedford, MA, USA). After nonspecific binding was blocked with 4% skim milk, the membranes were incubated at 4°C overnight with a rabbit polyclonal antibody against phosphorylated forms of ERK or total ERK (1:1,000, 1:1,000, respectively; Cell Signaling Technology), a mouse monoclonal antibody against synaptophysin (1:500; Sigma), a goat polyclonal antibody against Siah-1 (1:2000; Abcam, Cambridge, MA, USA) or mouse monoclonal antibody against tubulin (1:2,000; Sigma). The membranes were then incubated with a horseradish peroxidase-conjugated goat antibody against rabbit or mouse immunoglobulins or with a biotinylated secondary antibody followed by avidin-biotin horseradish peroxidase complexes (Vectastain Elite ABC Kit; Vector, Burlingame, CA). The signals were visualized with chemiluminescence (ECL Blotting Analysis System; Amersham, Arlington Heights, IL), measured by ImageJ software (National Institutes of Health, Bethesda, MD) and normalized to -tubulin.
siRNA mediated siah-1 knockdown
For siah-1 knockdown, hippocampus neurons were transfected with siRNA against rat siah-1 (5’-GAAAUCCGACAACAUCCUUUU-3’; Dharmacon, Lafayette, CO, USA) or a scrambled control siRNA with no significant homology to any known gene sequences (ID#4611; Ambion, Carlsbad, CA, USA) using Lipofectamine™ 2000 (Invitrogen), according to the manufacturer’s instructions. After 5 h incubation, the medium was replaced with regular culture medium, and the cells were cultured for an additional 48 h. The efficiency of siah-1 knockdown was confirmed by real time RT-PCR and Immuno blot analysis.
Transient transfection of constructs
Cultured HEK 293 cells were transfected with Myc-synaptophysin, Flag-Siah-1 or HA-ubiquitin plasmid (1 ug per 1×105 cells) using LipofectamineTM LTX (Invitrogen) according to company instructions. Six hours later, medium was replaced with DMEM containing 10% fetal bovine containing either 50 mM glucose or 25 mM glucose + 25 mM mannitol. Transfected cells were detected by immunoblotting with antibodies against Myc (1:1000, Millipore), Flag (1:1000, Sigma) or HA (1:1000, Roche).
Ubiquitination assays
For immunoprecipitation (IP), HEK293 cells were grown in 100 mm dishes and transiently transfected with 1 μg of each plasmid containing HA-ubiquitin, Myc-synaptophysin in the presence or absence of Flag-Siah-1. Cells were then collected and lysed as described [27] in IP buffer [1X PBS, 0.5% Triton-X-100, Complete Mini protease inhibitor (Roche)] and rotated for 1 h at 4°C. Supernatant was collected for IP after centrifugation at 17,500 g for 15 min at 4°C . Primary monoclonal anti-Myc antibody (2 μg, Millipore) or 2 μg mouse IgG (Santa Cruz) was bound to Dynabeads Protein G (Invitrogen) at room temperature for 30 min by rotation. The beads-Ab complexes were then washed in PBS containing 0.01% Tween 20 prior to being incubated with the soluble cell lysates (500 μg) overnight at 4°C with rotation. The beads-Ab-Ag complexes were then washed with citrate-phosphate buffer (pH 5.0). After washes of nonspecific binding to beads, the bound protein was solubilized in sample buffer, loaded on an SDS-PAGE, and processed for immunoblotting.
Statistical analysis
All values are expressed as mean ± S.E. Differences were analyzed using either one-way or two-way ANOVA followed by Newman-Keuls post hoc testing for pairwise comparisons using SigmaStat Version 3.5. The null hypothesis was rejected when the p value < 0.05.
Results
The cell viability was decreased when the primary cultured neurons were exposed to both hypoxia and high glucose
To evaluate the effect of both hypoxia and high glucose on the neuronal cell viability, We cultured primary hippocampus neurons and treated the cells with either 50 mM glucose or 25 mM glucose + 25 mM mannitol as osmotic control for 3 days under hypoxia (2% oxygen) or Normoxia (21% oxygen) condition. Our data demonstrated that neither hypoxia nor high glucose exposure alone could elicit decrease in cell viability and increase in LDH release (Figure 1). However, when the cultured neurons were exposed to hypoxia and high glucose together, the cell viability were dramatically decreased. This evidence demonstrates the neuronal damage caused by diabetes may relate to both tissue chronic hypoxia and high glucose environment in pathological conditions.
Figure 1.
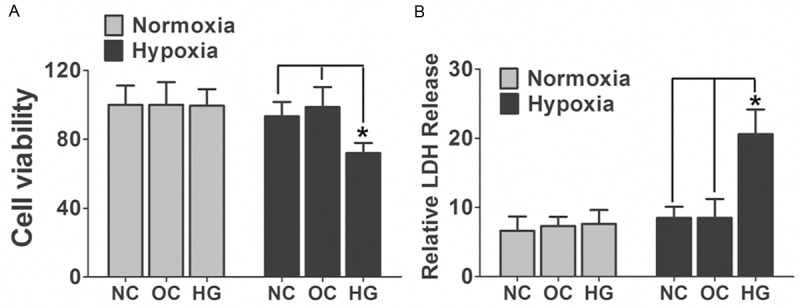
Neuronal cell death was induced by both hypoxia and high glucose exposure. A. Primary cultured hippocampus neurons were treated by 50 mM glucose (HG) under hypoxia (2% oxygen) or Normoxia (21% oxygen) condition for 3 days. Cells treated by 25 mM glucose + 25 mM mannitol were used as osmotic control (OC). LDH released was measured. Data are mean ± SD from six independent experiments. B. Primary cultured hippocampus neurons treated either by 50mM glucose or 25 mM glucose + 25 mM mannitol were subjected to 3 days hypoxia or normoxia exposure and cell viability was assessed with MTT assay. Cells under normal cultured medium were used as normal control (NC). Data are mean ± SD from six independent experiments. *, P<0.05 vs. NC or OC group. NC, normal control; OC, osmotic control; HG, high glucose.
Hypoxia and high glucose mediated posttranscriptional reduction of synaptophysin was regulated by the ubiquitin-proteasome system in neuronal cells
To elucidate the molecular mechanisms involved in the diabetes related cognitive decline. We used the in vitro culture system with primary hippocampus neurons stimulated with high glucose or isomotic medium and exposure to hypoxia for 3 days. As we showed in Figure 2A and 2B, the exposure of hippocampus neurons to elevated glucose or hypoxia did not affect the mRNA or protein content of synaptophysin, but in vitro application with high glucose and hypoxia together significantly reduced synaptophysin protein, but not mRNA. Because synaptic vesicle proteins including synaptophysin have recently proven to be physiologically degraded by the ubiquitin-proteasome system (UPS) for the maintenance of synaptic plasticity [18,22,23], we examined the involvement of UPS with hypoxia and high glucose posttranscriptional decrease in synaptophysin protein. As we showed in Figure 3, application with the proteasome inhibitor MG132 or lactacystin, but not the lysosome inhibitor E64, led to significant suppression of hypoxia and high glucose induced degradation of synaptophysin. In contrast, the baseline levels of synaptophysin in neurons were unaffected with these inhibitors.
Figure 2.
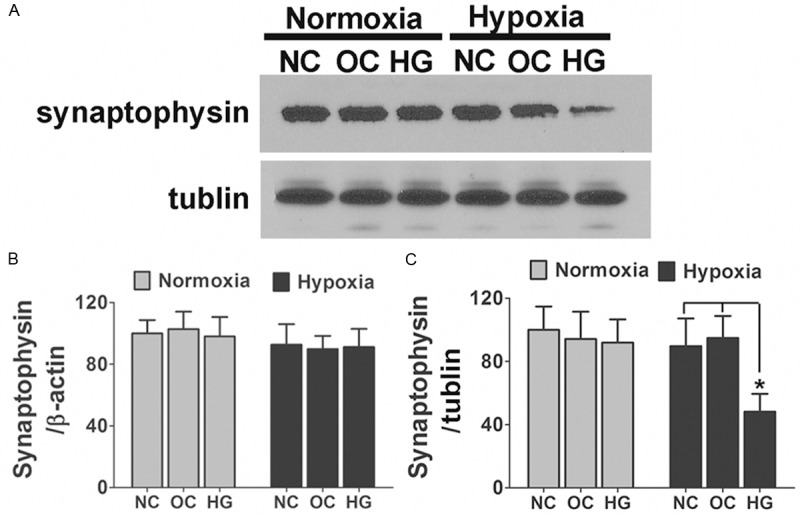
Posttranscriptional reduction of synaptophysin was detected in primary cultured hippocampus neurons after hypoxia and high glucose exposure. A. Primary cultured hippocampus neurons treated either by 50 mM glucose or 25 mM glucose + 25 mM mannitol were subjected to 3 days hypoxia or normoxia exposure. The levels of synaptophysin were analyzed by immunoblot. Shown are representative blots from six independent experiments with similar results. B. Neurons were treated as described above; the mRNA expression levels of synaptophysin are shown. There was no significant difference between different groups. Data are mean ± SD from six independent experiments. C. Densitometric quantification of synaptophysin in hippocampus neuronal cells treated with different culture medium in response to hypoxia or normoxia exposure. Data are mean ± SD from six independent experiments. *, P<0.05 vs. NC or OC group. NC, normal control; OC, osmotic control; HG, high glucose.
Figure 3.
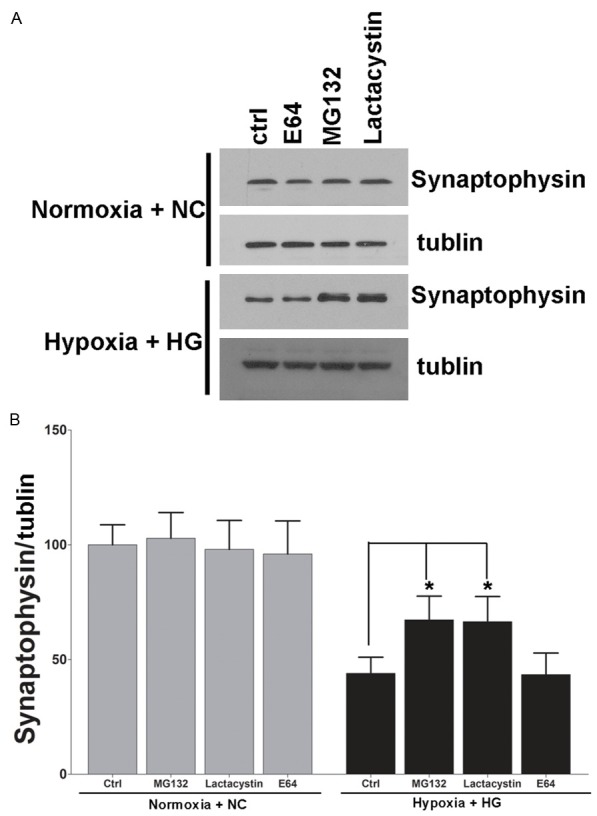
Hypoxia and high glucose induced synaptophysin degradation by the ubiquitin-proteasome pathway. A. Representative blots showed that the effect of hypoxia and high glucose on synaptophysin degradation is blocked by proteasome inhibitors. Primary cultured hippocampus neurons were treated by 50 mM glucose under hypoxia condition for 3 days. Cells under normal cultured condition were used as control. Proteasome inhibitor MG132 or Lactacysin but not the lysosome inhibitor E64 significantly blocked the hypoxia and high glucose induced degradation of synaptophysin. The expression level of synaptophysin under normal condition was not affected by these inhibitors. B. Densitometric quantification of synaptophysin in hippocampus neuronal cells exposed to hypoxia and high glucose or normal conditions. Data are mean ± SD from six independent experiments. *, P<0.05 vs. Normoxia + NC group.
Hypoxia and high glucose mediated ERK activation was required for synaptophysin degradation in neuronal cells
Synaptophysin protein may be degraded by the mammalian homolog of Drosophila seven in absentia (sina), an E3-ligase selective for synaptophysin named seven in absentia homologue (Siah). Since Drosophila sina is regulated by ERK signaling [24,25], the Siah may also be regulated by ERK activation. We further examined the involvement of ERK activation with hypoxia and high glucose mediated degradation of synaptophysin in the neuronal cells. Consistent with synaptophysin degradation, ERK phosphorylation was significantly enhanced after hypoxia and high glucose exposure. Hypoxia or high glucose exposure alone also could induce the phosphorylation of ERK, but not as dramatic as in Hypoxia + HG group (Figure 4A and 4B). Inhibition of ERK activation with U0126 or PD98059 led to a significant suppression of hypoxia and high glucose induced degradation of synaptophysin (Figure 5A and 5D). In addition to ERK signaling, Siah-1 expression levels were also been detected. In the neuronal cells, consistent with phosphorylation of ERK and synaptophysin degradation, Siah-1 was also dramatically upregulated and this elevation was totally reversed by ERK phosphorylation inhibitor (Figures 4A, 4C, 5A and 5C). These results suggested that ERK activation is responsible for hypoxia and high glucose induced UPS degradation of synaptophysin through the regulation of E3-ligase Siah-1.
Figure 4.
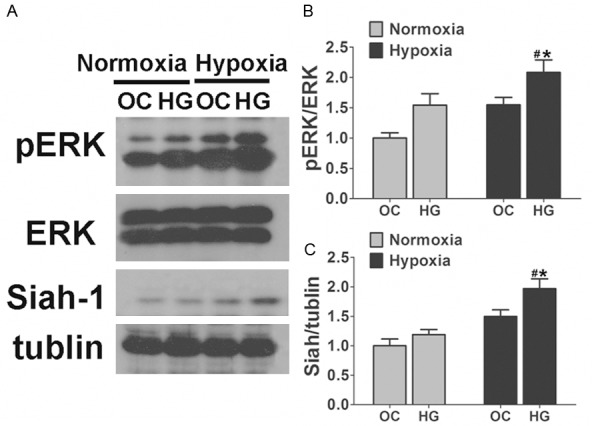
Hypoxia and high glucose induced ERK activation and production of Siah-1. A. Immunoblot analyses showing the ERK activation and the production of Siah-1 in hippocampus neurons under hypoxia and high glucose condition. ERK phosphorylation, elevated in either hypoxia or high glucose exposure alone, was significantly upregulated when the cells were exposed to hypoxia and high glucose together. Consistent with the phosphorylation of ERK, Siah-1 was also elevated after hypoxia and high glucose exposure. B, C. Densitometric quantification of ERK and Siah-1 in hippocampus neuronal cells exposed to 50 mM glucose (HG) under hypoxia or Normoxia conditions. Cells treated by 25 mM glucose + 25 mM mannitol were used as osmotic control (OC). Data are mean ± SD from six independent experiments. *, P<0.05 vs. Normoxia + OC group; #, P<0.05 vs. Hypoxia + OC group.
Figure 5.
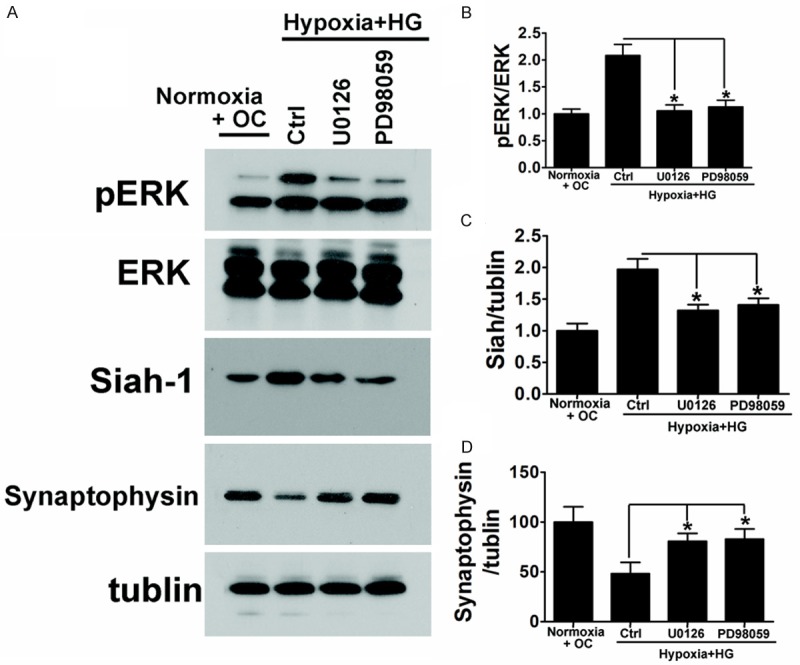
Hypoxia and high glucose mediated ERK activation and Siah-1 production was required for synaptophysin degradation in neuronal cells. A. Inhibition of ERK activation with U0126 or PD98059 led to a significant suppression of Hypoxia and high glucose induced upregulation of Siah-1. Consistent with this result, inhibition of ERK activation also blocked the decline of synaptophysin under hypoxia and high glucose condition. B-D. Densitometric quantification of ERK Siah-1 and Synaptophysin in hippocampus neuronal cells exposed to high glucose under hypoxia condition. Data are mean ± SD from six independent experiments. *, P<0.05 vs. control group.
Posttranscriptional reduction of synaptophysin in hypoxia and high glucose exposed neuronal cells were reversed by Siah-1 gene knockdown
To further determine whether Siah-1 protein regulates the degradation of synaptophysin, we examined the effect of knockdown Siah-1 on synaptophysin protein expression levels in hypoxia and high glucose exposed primary cultured hippocampus neurons. Here, we generated a hypoxia and high glucose model, and manipulated Siah-1 by knocking down endogenous Siah-1 expression by siRNA. Siah-siRNA significantly reduced siah-1 mRNA and protein levels over 80% in the primary neurons (Figure 6A-C). Silencing of Siah-1 significantly reversed hypoxia and high glucose induced reduction of synaptophysin (Figure 7A-C). Taken together, these results indicate that Siah-1 proteins have the ability to regulate synaptophysin expression under hypoxia and high glucose conditions.
Figure 6.
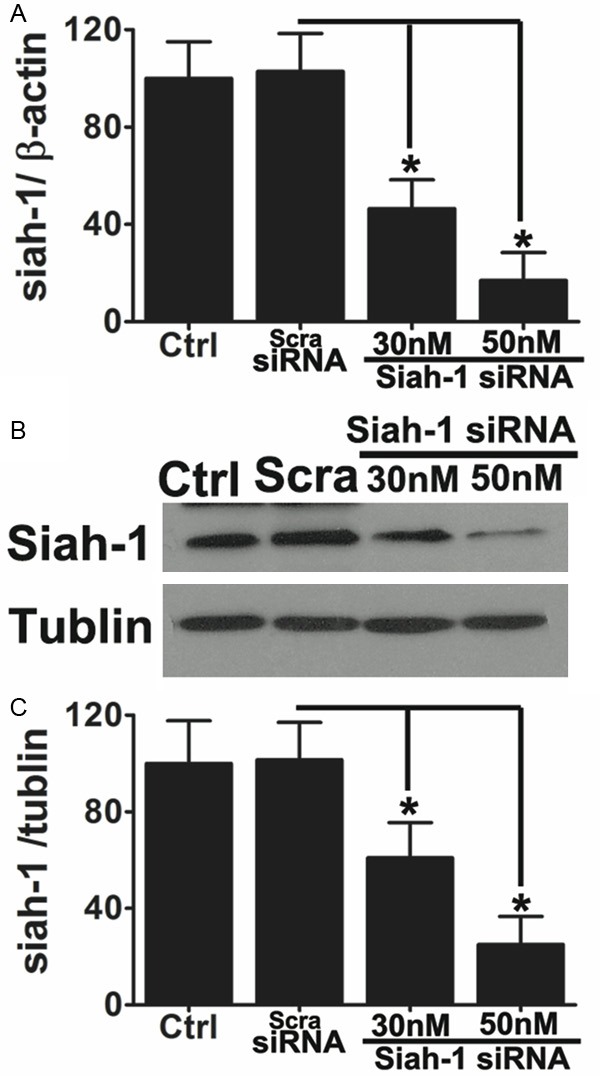
siRNA-mediated knockdown of Siah-1 in primary cultured hippocampus neurons. Primary culture hippocampus neurons were transfected with different concentrations of Siah-1-siRNA indicated for 48 hours. Cells without transfection or transfected with scramble siRNA (Scra-siRNA) were used as controls. A. Quantitative real-time RT-PCR was performed to quantify the extent to which endogenous Siah-1 was knocked down. Data are mean ± SD from three independent experiments. *P<0.05 compared to scramble siRNA. B. Representative immunoblots of Siah-1 showing knockdown of Siah-1 by the specific Siah-1 siRNA in a dose-dependent manner but not by the scramble siRNA. C. Densitometric quantification of Siah-1 immunoblots in hippocampus neurons transfected with scramble siRNA or Siah-1 specific siRNA. Data are mean ± SD from three independent experiments. *, P<0.05 compared with scramble siRNA.
Figure 7.
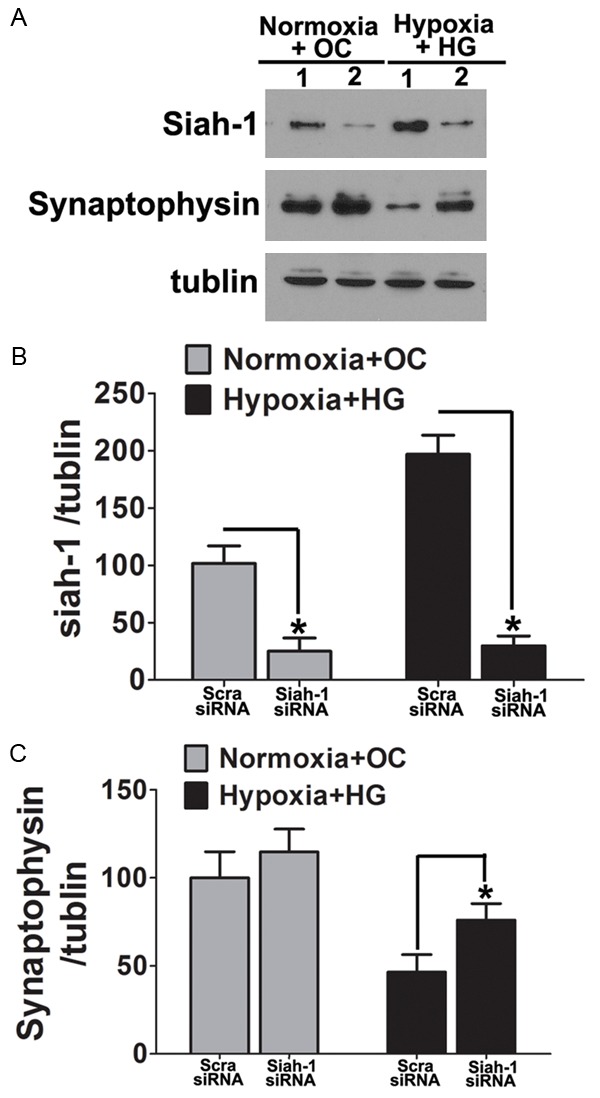
Siah-1 mediates the hypoxia and high glucose induced-synaptophysin degradation in neuronal cells. A. Primary cultured hippocampus neuron were transfected with either siRNA targeting Siah-1 or non-targeting scrambled control. The cells were then treated by hypoxia and high glucose for 3 days. Cells incubated in isotonic medium under normoxia condition were used as control. Protein were isolated and immunoblotted for Siah-1 and synaptophysin. Representative blots showed silencing of siah-1 significantly reversed hypoxia and high glucose induced reduction of synaptophysin; B, C. Densitometric quantification of Siah-1 and Synaptophysin in neuronal cells. Data represent mean ± SD from 4 independent experiments and are expressed as either the ratio of Siah-1 or Synaptophysin to tubulin and normalized as percentage control to the scramble-siRNA cells under Normoxia + OC condition. *, P<0.05 compared with scramble siRNA.
Siah-1 regulates synaptophysin degradation via the ubiquitin-proteasome pathway during hypoxia and high glucose conditions
Next, we sought to determine whether the enhanced degradation of synaptophysin by Siah-1 is mediated by the proteasome pathway. HEK293 cells exposed to hypoxia and high glucose that have been co-transfected with myc-synaptophysin and Flag-Siah-1 were treated by various inhibitors of proteolytic pathways, and the synaptophysin levels were then analyzed by Western blotting. As shown in Figure 8, the enhanced degradation of synaptophysin by Siah-1 overexpression is blocked by MG132, a potent inhibitor of proteasome function [26]. A similar effect was also observed when cells were treated with lactacystin, an irreversible inhibitor of the proteasome pathway (data not shown). In contrast, E64, an inhibitor of lysosomal cysteine proteases, had no effect.
Figure 8.
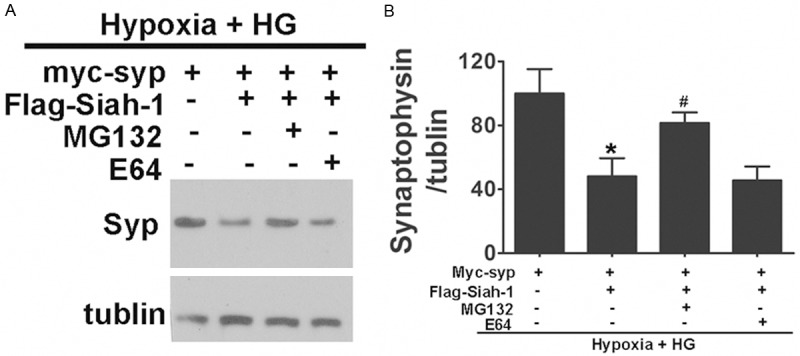
Siah-1 targets synaptophysin for degradation by the ubiquitin-proteasome pathway under hypoxia and high glucose condition. A. The effect of Siah-1 on synaptophysin degradation is blocked by proteasome inhibitors. HEK293 cells were co-transfected with Myc-synaptophysin and Flag-Siah-1. HEK293 cells were then treated by 50 mM glucose under hypoxia condition for 3 day and incubated with proteasome inhibitor MG132 or cysteine protease inhibitor E64. Cells were then lysed, and an equal amount of protein from each lysate was analyzed by immunoblotting for synaptophysin and tubulin. B. Densitometric quantification of Synaptophysin in HEK293 cells. Data represent mean ± SD from 4 independent experiments and are expressed as the ratio of Synaptophysin to tubulin. *, P<0.05 compared with Myc-syp transfection alone. #, P<0.05 compared with Myc-syp and Flag-Siah-1 co-transfected cells. Syp, synaptophysin.
Because proteasome-dependent proteolysis involves the ubiquitination of target proteins, we investigated whether Siah-1 accelerates the degradation of synaptophysin under hypoxia and high glucose conditions by promoting the ubiquitination of synaptophysin. After hypoxia and high glucose exposure, HEK293 cells were co-expressed with Myc-tagged synaptophysin along with HA tagged ubiquitin in the absence or presence of exogenous Siah-1. Cell lysates were subjected to immunoprecipitation with an anti-myc antibody followed by immunoblotting with an anti-HA antibody to detect ubiquitin-conjugated synaptophysin (Figure 9). Compared to normoxia + OC group of cells, hypoxia and high glucose exposure dramatically increased the ubiquitination of synaptophysin. Under hypoxia and high glucose conditions, in the absence of exogenous Siah proteins, synaptophysin was ubiquitinated, and the ubiquitinated synaptophysin could be detected by the anti-HA antibody immunoblotting. Overexpression of Siah-1 enhanced the ubiquitination of synaptophysin, because increased levels of HA-tagged ubiquitin were detected on synaptophysin. In the control IP using mouse IgG pulled down, no synaptophysin or HA-ubiquitin was detected from triple transfected cell lysates (Figure 9). In combination, these data indicate that Siah-1 target synaptophysin for ubiquitin-mediated degradation under hypoxia and high glucose conditions.
Figure 9.
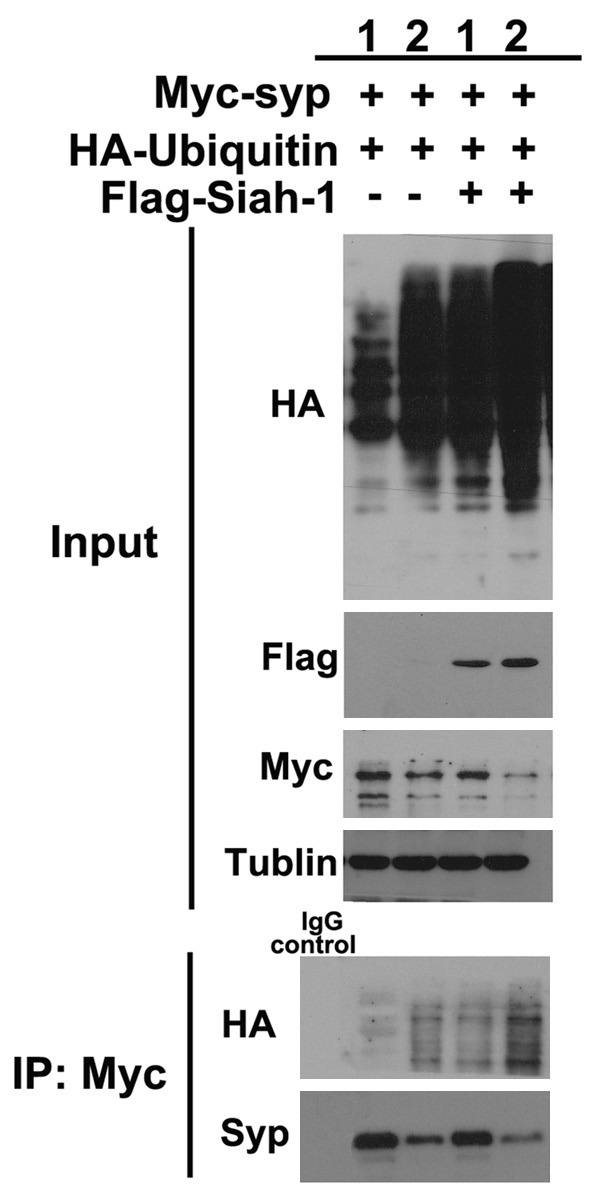
Synaptophysin is ubiquitinated by Siah-1 under hypoxia and high glucose condition. Total cell lysates from HEK 293 cells transfected with indicated plasmids, in the presence or absence of hypoxia and high glucose treatment for 3 days, were immunoblotted with anti-HA, anti-myc, and anti-Flag antibodies (Input panel). Tubulin was used as loading control. Cells incubated in isotonic medium under normoxia condition were used as control. Myc-tagged synaptophysin proteins were then immunoprecipitated from cell lysates with an anti-myc antibody. The immunoprecipitates were analyzed by immunoblotting with an anti-HA antibody to identify synaptophysin proteins to which the HA-ubiquitin polypeptide had been attached. The blot was then stripped and reprobed with an anti-synaptophysin antibody. Shown are representative blots from 4 independent experiments with similar results. Syp, synaptophysin; 1, normoxia + OC, 2, Hypoxia + HG.
Discussion
Studies have shown that diabetes is an independent factor for cognitive dysfunction [27-29]. The positive control of blood glucose is thought to improve cognitive function in the elderly [30-32]. Yet more than high glucose level maybe involved in diabetes related cognitive dysfunction. For example, there was evidence showing that more than 75% of diabetic patients suffered from obstructive sleep apnea (OSA) [11,33,34]. This indicated that chronic hypoxia may also participate in the pathophysiologic changes caused by diabetes. Hypoxia has been proved to be one of the most important risk factor for cognitive impairment [35,36]. Hypoxia could cause animal short-term memory decline in animal models [37,38]. In this context, we hypothesized that both high glucose level and hypoxia may contribute to cognitive dysfunction cause by diabetes. Synaptic transmission is important to cognitive function. And synaptophysin as the specific marker of synapses, has been proved to be involved in neurotransmitter release, synaptic plasticity, synaptic vesicle formation and reuse [17,39,40]. It is involved in learning, memory, etc. and abnormal synaptophysin expression has been observed in a variety of neurodegenerative disease [41-43].
In this study we employed high glucose and hypoxia in cultured hippocampal neurons to mimic the pathological changes in diabetes, and then detected synaptophysin expression. It is quite interesting that in the different conditions we used, only the coexist of high glucose and hypoxia could cause significant down regulation of synaptophysin protein level while its mRNA levels were not significantly changed. This makes us speculate that post translational processes which regulate the synaptophysin degradation may be involved in the down regulation of synaptophysin. By using ubiquitin inhibitors MG-132 and lactacystin or lysosomal inhibitor E64 we tested synaptophysin expression in hippocampal neurons under different conditions. Although there was slight upregulation of synaptophysin expression in MG-132 treated neurons, under normal culture condition no significant changes were observed by these inhibitors. While in high glucose and hypoxia treated neurons both ubiquitin inhibitors can improve the expression of synaptophysin, yet the lysosomal inhibitor E64 could not change the expression of synaptophysin. These results suggest the ubiquitination process may be involved in the down regulation of synaptophysin by high glucose and hypoxia.
E3 ubiquitin ligase Siah (Mammalian Homologues of Seven in Absentia) is the key enzyme degradation of Synaptophysin [18]. Siah protein, the Drosophila SINA (seven in absentia) homologous, is an E3 ubiquitin ligase which ubiquitinates the substrate protein and induces its degradation by the proteasome pathway [44,45]. Mammalian animal SIAH proteins are highly conservative in evolution. SIAH-1a, SIAH-1b and SIAH-2 are the main forms in rodents while in human SIAH-1 and SIAH-2 dominant [46-48]. SIAH has been proved to be involved in the regulation of a variety of substrate protein, those participate in different signal transduction pathway, including cell cycle, cell differentiation, apoptosis, and neurodegenerative diseases [49-51].
We have identified the ubiquitination inhibitor MG132 can inhibit high glucose and hypoxia induced downregulation of synaptophysin, this suggest ubiquitination is involved in the degradation of synaptophysin under high glucose and hypoxia condition. To demonstrate whether Siah-1 is involved in the ubiquitination of synaptophysin under this condition. We further demonstrated that high glucose and hypoxia can promote synaptophysin ubiquitination, and increase the expression of SIAH-1 can promote an increase in synaptophysin ubiquitination, and reduced expression of synaptophysin. This suggests that Siah-1 is the key factor that result in the degradation of synaptophysin. The next question is what signaling pathway may control the expression of Siah-1 under this condition.
Because the ERK pathway is involved in regulating Drosophila Sina expression [52], we hypothesized that ERK pathway maybe activated under hypoxia and high glucose condition, thus influence Siah expression. In this research we observed high glucose or hypoxia along can cause increase in the phosphorylation of ERK, while the co-effect of high glucose and hypoxia induced ERK phosphorylation more significantly. To determine whether the change of ERK phosphorylation affect Siah-1 expression, we use ERK activation inhibitor U0126 or PD98059 to inhibit ERK phosphorylation. As expected, there was obvious decrease of SIAH-1 expression concomitantly with an upregulation of synaptophysin level. We hypothesized that ERK signaling pathway regulates E3 ligase Siah-1 under our high glucose and hypoxia condition.
In summary, the co-effect of high glucose and hypoxia can cause the degradation of synaptophysin this may explain partially the diabetes related cognitive dysfunction. In high glucose under hypoxia environment synaptophysin degradation is mainly regulated by the E3 ligase Siah-1.
Acknowledgements
This work was supported by National Natural Science Foundation of China 81471173 (to M.C) 81171023 (to Y.Z) Shanghai Rising-Star Program (11QA1400900) and Shanghai Pujiang Program (12PJ1407200) and Shanghai Science and Technology Committee (No. 11DZ1920902).
Disclosure of conflict of interest
None to disclose.
References
- 1.Yaffe K, Blackwell T, Kanaya AM, Davidowitz N, Barrett-Connor E, Krueger K. Diabetes, impaired fasting glucose, and development of cognitive impairment in older women. Neurology. 2004;63:658–663. doi: 10.1212/01.wnl.0000134666.64593.ba. [DOI] [PubMed] [Google Scholar]
- 2.Biessels GJ, Strachan MW, Visseren FL, Kappelle LJ, Whitmer RA. Dementia and cognitive decline in type 2 diabetes and prediabetic stages: towards targeted interventions. Lancet Diabetes Endocrinol. 2014;2:246–255. doi: 10.1016/S2213-8587(13)70088-3. [DOI] [PubMed] [Google Scholar]
- 3.Cukierman T, Gerstein HC, Williamson JD. Cognitive decline and dementia in diabetes--systematic overview of prospective observational studies. Diabetologia. 2005;48:2460–2469. doi: 10.1007/s00125-005-0023-4. [DOI] [PubMed] [Google Scholar]
- 4.Geroldi C, Frisoni GB, Paolisso G, Bandinelli S, Lamponi M, Abbatecola AM, Zanetti O, Guralnik JM, Ferrucci L. Insulin resistance in cognitive impairment: the InCHIANTI study. Arch Neurol. 2005;62:1067–1072. doi: 10.1001/archneur.62.7.1067. [DOI] [PubMed] [Google Scholar]
- 5.Verdelho A, Madureira S, Moleiro C, Ferro JM, Santos CO, Erkinjuntti T, Pantoni L, Fazekas F, Visser M, Waldemar G, Wallin A, Hennerici M, Inzitari D. White matter changes and diabetes predict cognitive decline in the elderly: the LADIS study. Neurology. 2010;75:160–167. doi: 10.1212/WNL.0b013e3181e7ca05. [DOI] [PubMed] [Google Scholar]
- 6.Cognitive function is unaffected by tight glucose control in paediatric intensive care. BMJ. 2012;345:e7065. doi: 10.1136/bmj.e7065. [DOI] [PubMed] [Google Scholar]
- 7.Umegaki H, Kawamura T, Mogi N, Umemura T, Kanai A, Sano T. Glucose control levels, ischaemic brain lesions, and hyperinsulinaemia were associated with cognitive dysfunction in diabetic elderly. Age Ageing. 2008;37:458–461. doi: 10.1093/ageing/afn051. [DOI] [PubMed] [Google Scholar]
- 8.Yaffe K, Falvey C, Hamilton N, Schwartz AV, Simonsick EM, Satterfield S, Cauley JA, Rosano C, Launer LJ, Strotmeyer ES, Harris TB. Diabetes, glucose control, and 9-year cognitive decline among older adults without dementia. Arch Neurol. 2012;69:1170–1175. doi: 10.1001/archneurol.2012.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sasaki-Hamada S, Sacai H, Oka JI. Diabetes onset influences hippocampal synaptic plasticity in streptozotocin-treated rats. Neuroscience. 2012;227:293–304. doi: 10.1016/j.neuroscience.2012.09.081. [DOI] [PubMed] [Google Scholar]
- 10.Ryan CM, Geckle M. Why is learning and memory dysfunction in Type 2 diabetes limited to older adults? Diabetes Metab Res Rev. 2000;16:308–315. doi: 10.1002/1520-7560(2000)9999:9999<::aid-dmrr141>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 11.Hermans MP, Ahn SA, Mahadeb YP, Rousseau MF. Sleep apnoea syndrome and 10-year cardiovascular risk in females with type 2 diabetes: relationship with insulin secretion and insulin resistance. Diabetes Metab Res Rev. 2013;29:227–234. doi: 10.1002/dmrr.2387. [DOI] [PubMed] [Google Scholar]
- 12.Zubkova ST, Derevianko LP, Evtushenko AM. [Oxygen supply and physical working capacity in patients with diabetes mellitus] . Ter Arkh. 1988;60:24–27. [PubMed] [Google Scholar]
- 13.Pillai A, Warren G, Gunathilake W, Idris I. Effects of sleep apnea severity on glycemic control in patients with type 2 diabetes prior to continuous positive airway pressure treatment. Diabetes Technol Ther. 2011;13:945–949. doi: 10.1089/dia.2011.0005. [DOI] [PubMed] [Google Scholar]
- 14.Braslavskaia GM, Shteingardt Iu N. [Tissue hypoxia in diabetes mellitus, its pathogenesis and treatment] . Ter Arkh. 1980;52:83–88. [PubMed] [Google Scholar]
- 15.Sarnat HB, Born DE. Synaptophysin immunocytochemistry with thermal intensification: a marker of terminal axonal maturation in the human fetal nervous system. Brain Dev. 1999;21:41–50. doi: 10.1016/s0387-7604(98)00068-0. [DOI] [PubMed] [Google Scholar]
- 16.Wiedenmann B, Franke WW. Identification and localization of synaptophysin, an integral membrane glycoprotein of Mr 38,000 characteristic of presynaptic vesicles. Cell. 1985;41:1017–1028. doi: 10.1016/s0092-8674(85)80082-9. [DOI] [PubMed] [Google Scholar]
- 17.Tarsa L, Goda Y. Synaptophysin regulates activity-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 2002;99:1012–1016. doi: 10.1073/pnas.022575999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wheeler TC, Chin LS, Li Y, Roudabush FL, Li L. Regulation of synaptophysin degradation by mammalian homologues of seven in absentia. J Biol Chem. 2002;277:10273–10282. doi: 10.1074/jbc.M107857200. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Lim S, Hoffman D, Aspenstrom P, Federoff HJ, Rempe DA. HUMMR, a hypoxia- and HIF-1alpha-inducible protein, alters mitochondrial distribution and transport. J Cell Biol. 2009;185:1065–1081. doi: 10.1083/jcb.200811033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- 21.Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- 22.Bao H, Reist NE, Zhang B. The Drosophila epsin 1 is required for ubiquitin-dependent synaptic growth and function but not for synaptic vesicle recycling. Traffic. 2008;9:2190–2205. doi: 10.1111/j.1600-0854.2008.00832.x. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM. Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci U S A. 2000;97:13354–13359. doi: 10.1073/pnas.240347797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carthew RW, Neufeld TP, Rubin GM. Identification of genes that interact with the sina gene in Drosophila eye development. Proc Natl Acad Sci U S A. 1994;91:11689–11693. doi: 10.1073/pnas.91.24.11689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ozawa Y, Kurihara T, Tsubota K, Okano H. Regulation of posttranscriptional modification as a possible therapeutic approach for retinal neuroprotection. J Ophthalmol. 2011;2011:506137. doi: 10.1155/2011/506137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alexandrova A, Petrov L, Georgieva A, Kirkova M, Kukan M. Effects of proteasome inhibitor, MG132, on proteasome activity and oxidative status of rat liver. Cell Biochem Funct. 2008;26:392–398. doi: 10.1002/cbf.1459. [DOI] [PubMed] [Google Scholar]
- 27.Strachan MW, Reynolds RM, Marioni RE, Price JF. Cognitive function, dementia and type 2 diabetes mellitus in the elderly. Nat Rev Endocrinol. 2011;7:108–114. doi: 10.1038/nrendo.2010.228. [DOI] [PubMed] [Google Scholar]
- 28.Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol. 2004;61:661–666. doi: 10.1001/archneur.61.5.661. [DOI] [PubMed] [Google Scholar]
- 29.Wang KC, Woung LC, Tsai MT, Liu CC, Su YH, Li CY. Risk of Alzheimer’s disease in relation to diabetes: a population-based cohort study. Neuroepidemiology. 2012;38:237–244. doi: 10.1159/000337428. [DOI] [PubMed] [Google Scholar]
- 30.Launer LJ, Miller ME, Williamson JD, Lazar RM, Gerstein HC, Murray AM, Sullivan M, Horowitz KR, Ding J, Marcovina S, Lovato LC, Lovato J, Margolis KL, O’Connor P, Lipkin EW, Hirsch J, Coker L, Maldjian J, Sunshine JL, Truwit C, Davatzikos C, Bryan RN. Effects of intensive glucose lowering on brain structure and function in people with type 2 diabetes (ACCORD MIND): a randomised open-label substudy. Lancet Neurol. 2011;10:969–977. doi: 10.1016/S1474-4422(11)70188-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hewer W, Mussell M, Rist F, Kulzer B, Bergis K. Short-term effects of improved glycemic control on cognitive function in patients with type 2 diabetes. Gerontology. 2003;49:86–92. doi: 10.1159/000067947. [DOI] [PubMed] [Google Scholar]
- 32.Mussell M, Hewer W, Kulzer B, Bergis K, Rist F. Effects of improved glycaemic control maintained for 3 months on cognitive function in patients with Type 2 diabetes. Diabet Med. 2004;21:1253–1256. doi: 10.1111/j.1464-5491.2004.01322.x. [DOI] [PubMed] [Google Scholar]
- 33.Nannapaneni S, Ramar K, Surani S. Effect of obstructive sleep apnea on type 2 diabetes mellitus: A comprehensive literature review. World J Diabetes. 2013;4:238–244. doi: 10.4239/wjd.v4.i6.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Foster GD, Sanders MH, Millman R, Zammit G, Borradaile KE, Newman AB, Wadden TA, Kelley D, Wing RR, Sunyer FX, Darcey V, Kuna ST. Obstructive sleep apnea among obese patients with type 2 diabetes. Diabetes Care. 2009;32:1017–1019. doi: 10.2337/dc08-1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peers C, Dallas ML, Boycott HE, Scragg JL, Pearson HA, Boyle JP. Hypoxia and neurodegeneration. Ann N Y Acad Sci. 2009;1177:169–177. doi: 10.1111/j.1749-6632.2009.05026.x. [DOI] [PubMed] [Google Scholar]
- 36.Yaffe K, Laffan AM, Harrison SL, Redline S, Spira AP, Ensrud KE, Ancoli-Israel S, Stone KL. Sleep-disordered breathing, hypoxia, and risk of mild cognitive impairment and dementia in older women. JAMA. 2011;306:613–619. doi: 10.1001/jama.2011.1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Row BW. Intermittent hypoxia and cognitive function: implications from chronic animal models. Adv Exp Med Biol. 2007;618:51–67. doi: 10.1007/978-0-387-75434-5_5. [DOI] [PubMed] [Google Scholar]
- 38.Salmaso N, Silbereis J, Komitova M, Mitchell P, Chapman K, Ment LR, Schwartz ML, Vaccarino FM. Environmental enrichment increases the GFAP+ stem cell pool and reverses hypoxia-induced cognitive deficits in juvenile mice. J Neurosci. 2012;32:8930–8939. doi: 10.1523/JNEUROSCI.1398-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kwon SE, Chapman ER. Synaptophysin regulates the kinetics of synaptic vesicle endocytosis in central neurons. Neuron. 2011;70:847–854. doi: 10.1016/j.neuron.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gordon SL, Leube RE, Cousin MA. Synaptophysin is required for synaptobrevin retrieval during synaptic vesicle endocytosis. J Neurosci. 2011;31:14032–14036. doi: 10.1523/JNEUROSCI.3162-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmitt U, Tanimoto N, Seeliger M, Schaeffel F, Leube RE. Detection of behavioral alterations and learning deficits in mice lacking synaptophysin. Neuroscience. 2009;162:234–243. doi: 10.1016/j.neuroscience.2009.04.046. [DOI] [PubMed] [Google Scholar]
- 42.Tampellini D, Capetillo-Zarate E, Dumont M, Huang Z, Yu F, Lin MT, Gouras GK. Effects of synaptic modulation on beta-amyloid, synaptophysin, and memory performance in Alzheimer’s disease transgenic mice. J Neurosci. 2010;30:14299–14304. doi: 10.1523/JNEUROSCI.3383-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Callahan LM, Vaules WA, Coleman PD. Progressive reduction of synaptophysin message in single neurons in Alzheimer disease. J Neuropathol Exp Neurol. 2002;61:384–395. doi: 10.1093/jnen/61.5.384. [DOI] [PubMed] [Google Scholar]
- 44.Zhao J, Wang C, Wang J, Yang X, Diao N, Li Q, Wang W, Xian L, Fang Z, Yu L. E3 ubiquitin ligase Siah-1 facilitates poly-ubiquitylation and proteasomal degradation of the hepatitis B viral X protein. FEBS Lett. 2011;585:2943–2950. doi: 10.1016/j.febslet.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 45.Jumpertz S, Hennes T, Asare Y, Vervoorts J, Bernhagen J, Schutz AK. The beta-catenin E3 ubiquitin ligase SIAH-1 is regulated by CSN5/JAB1 in CRC cells. Cell Signal. 2014;26:2051–9. doi: 10.1016/j.cellsig.2014.05.013. [DOI] [PubMed] [Google Scholar]
- 46.Carthew RW, Rubin GM. seven in absentia, a gene required for specification of R7 cell fate in the Drosophila eye. Cell. 1990;63:561–577. doi: 10.1016/0092-8674(90)90452-k. [DOI] [PubMed] [Google Scholar]
- 47.Della NG, Senior PV, Bowtell DD. Isolation and characterisation of murine homologues of the Drosophila seven in absentia gene (sina) Development. 1993;117:1333–1343. doi: 10.1242/dev.117.4.1333. [DOI] [PubMed] [Google Scholar]
- 48.Reed JC, Ely KR. Degrading liaisons: Siah structure revealed. Nat Struct Biol. 2002;9:8–10. doi: 10.1038/nsb0102-8. [DOI] [PubMed] [Google Scholar]
- 49.Bruzzoni-Giovanelli H, Faille A, Linares-Cruz G, Nemani M, Le Deist F, Germani A, Chassoux D, Millot G, Roperch JP, Amson R, Telerman A, Calvo F. SIAH-1 inhibits cell growth by altering the mitotic process. Oncogene. 1999;18:7101–7109. doi: 10.1038/sj.onc.1203187. [DOI] [PubMed] [Google Scholar]
- 50.Hara MR, Snyder SH. Nitric oxide-GAPDH-Siah: a novel cell death cascade. Cell Mol Neurobiol. 2006;26:527–538. doi: 10.1007/s10571-006-9011-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee JT, Wheeler TC, Li L, Chin LS. Ubiquitination of alpha-synuclein by Siah-1 promotes alpha-synuclein aggregation and apoptotic cell death. Hum Mol Genet. 2008;17:906–917. doi: 10.1093/hmg/ddm363. [DOI] [PubMed] [Google Scholar]
- 52.Wen YY, Yang ZQ, Song M, Li BL, Zhu JJ, Wang EH. SIAH1 induced apoptosis by activation of the JNK pathway and inhibited invasion by inactivation of the ERK pathway in breast cancer cells. Cancer Sci. 2010;101:73–79. doi: 10.1111/j.1349-7006.2009.01339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]