

Abstract
Background: The efficacy of chemotherapy in patients with hepatocellular carcinomas still poor due to multidrug resistance. This study aimed to investigate the impact of the over-expressed mitochondrial human telomerase reverse transcriptase on multidrug resistance of hepatocellular carcinomas. Methods: HepG2 and SK-Hep1 cell lines were used. And sensitivity to chemotherapeutic drugs was detected. Results: Mitochondrial human telomerase reverse transcriptase over-expression in hepatocellular carcinomas cells could significantly reduce its sensitivity to multiple chemotherapeutic drugs in vitro and in vivo. Hepatocellular carcinomas cells over-expressing mitochondrial human telomerase reverse transcriptase showed a significantly higher mitochondrial membrane potential, a markedly lower activated caspase-3 after drug treatment, and an increased mtDNA copy number, which explained the drastically decreased drug-induced apoptosis of hepatocellular carcinomas cells with mitochondrial human telomerase reverse transcriptase over-expression. Conclusion: Over-expressed mitochondrial human telomerase reverse transcriptase may increase the mtDNA copy number and inhibit the activation of mitochondrial apoptotic pathway to contribute to the multidrug resistance of hepatocellular carcinomas cells.
Keywords: Telomerase, mitochondria, multidrug resistance, hepatocellular carcinoma
Introduction
Multidrug resistance (MDR) usually accounts for the poor clinical response to chemotherapy in hepatocellular carcinoma (HCC) patients. The mechanisms underlying MDR have been widely studied, but the specific cause is still poorly understood. According to the available studies, MDR is closely associated with some genetic and/or epigenetic factors that can increase the export and decrease the import of chemotherapeutics; activate the DNA repair pathway and promote the drug metabolism; suppress the apoptosis pathway, etc [1-4]. Most of the chemotherapeutics, such as cisplatin, 5-fluorouracil and doxorubicin, may induce cell apoptosis [5-7]. The abnormal expression and dysfunction of apoptosis-associated molecules, including p53, Bax, and Bcl-2, may contribute to the resistance to drug-induced apoptosis in HCC cells [8], which otherwise requires the participation of mitochondria [9]. In addition, studies have confirmed that the telomerase is associated with the drug resistance of tumor cells [10]. Therefore, telomerase may serve as an MDR-associated molecule in HCC cells, but the relationship between telomerase and MDR remains elusive.
Telomerase, a reverse transcriptase, consists of human telomerase reverse transcriptase (hTERT) and telomerase RNA component (TERC). Studies have shown that telomerase is able to not only maintain the length of telomeres, but also play important roles in the cell proliferation [11,12], the DNA damage response of cells [13,14] and the cell apoptosis [15,16]. Telomerase is mainly localized in the nucleus. It has been demonstrated that active telomerase TERT can translocate from nuclei to mitochondria under certain stress conditions [17,18]. The mitochondrial translocation of TERT is associated with the tyrosine phosphorylation, which involves the tyrosine kinase Src kinase family [19] and tyrosine phosphatase Shp-2 [20].
Mitochondrial TERT and nuclear TERT have different roles. Mitochondrial TERT can use mitochondrial tRNA instead of TERC as a template, functioning as a reverse transcriptase (RdDP) [21]. Mitochondrial TERT binds to the RNA component of mitochondrial RNA ribonuclease (RMRP) and plays a role in the gene silencing at the transcriptional level by influencing a component similar to RNA dependent RNA polymerase (RdRP) [22,23]. Moreover, mitochondrial TERT can reduce the mitochondrial reactive oxygen species (ROS) [24,25] and attenuate DNA damage [18] to inhibit the apoptosis pathway [26].
In addition, our previous studies showed that hTERT gradually translocates from the nuclei to the mitochondria accompanied by an increase in the resistance index of drug-resistant HCC cells [27]. Therefore, the mitochondrial translocation of hTERT may be involved in the acquired MDR of HCC cells. How the mitochondrial hTERT regulates the drug resistance of HCC cells is still unclear. In this study, we investigated the effect of mitochondrial hTERT over-expression on the resistance of HCC cells to multiple chemotherapeutic drugs in vitro and in vivo, and explored the relationship between mitochondrial translocation of hTERT and MDR in human HCC cells.
Materials and methods
Cell lines and cell culture
HepG2 and SK-Hep1 cell lines were provided by the Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. HepG2 cells and SK-Hep1 cells were maintained in 1640 medium (Gibco, Life Technologies, Grand Island, NY) containing 10% fetal bovine serum (FBS) (Gibco, Life Technologies, Grand Island, NY), 100 U/mL penicillin and 100 μg/mL streptomycin (Amresco LLC., Solon, OH) in an atmosphere with 5% CO2 at 37°C.
Lentivirus (PLeno-mito-hTERT-GTP) construction and cell transfection
Lentivirus (PLeno-mito-hTERT-GTP) was constructed by Invitrogen Biotechnologies Co., Ltd through molecular cloning technology as described previously [28,29]. The mitochondrial targeted sequences (MTS), approximately 87 bp of combined mito-hTERT, were as follows: 5’-ATGTCCGTCCTGACGCCGCTGCTGCTGCGGGGCTTGACAGGCTCGGCCCGGCGGCTCCCAGTGCCGCGCGCCAAGATCCATTCGTTG-3’, and the combined sequences were cloned into the PLeno-GTP vector (Invitrogen, Life Technologies). The production of lentivirus containing mito-hTERT or vector plasmid was performed according to conventional protocols.
HepG2 cells and SK-Hep1 cells were seeded in 6-well plates (3×104 cells/well) and used for lentivirus transfection when 70% confluence was observed. The multiplicity of infection (MOI) was calculated with the following formula: MOI = (lentivirus concentration × lentivirus amount)/cell number. At 24 h after transfection, the medium was refreshed with normal medium. The transfection efficiency at 48 h was approximately 90-95%.
Immunofluorescence staining
Approximately 0.5×103 cells were seeded on coverslips and then processed 24 h late. Briefly, cells were washed thrice with PBS and incubated with 300-500 µl of 400 nM MitoTracker Deep Red (Molecular probes, Invitrogen, Life Technologies) at 37°C for 30 min. Then, cells were fixed in 4% paraformaldehyde for 15 min at room temperature, permeabilized with 0.3% Triton X-100 (Amresco LLC., Solon, OH) for 10 min at 4°C and blocked with 2 mg/ml of BSA (Amresco LLC., Solon, OH) for 1 h at 37°C, followed by incubation with an rabbit anti-hTERT monoclonal antibody (1:50, Abcam, Cambridge, MA) and rabbit anti-activated caspase-3 (1:10, Epitomics, Burlingame, CA) for 12 h at 4°C. Thereafter, cells were incubated with Alexa Fluor 488 or FITC goat anti-rabbit IgG (1:1000) (Abcam, Cambridge, MA) for 1 h at 37°C. DAPI (Roche Diagnostics Corp., Indianapolis, IN) was used for nuclear counterstaining. Immunofluorescence figures were captured with a Leica TCS SP2 Confocal Microscope (Leica Microsystems Inc., Buffalo Grove, IL) and were subsequently analyzed with Image J.
Western blot assay
Cells and tissue homogenate were lysed in lysis buffer. The mitochondrial proteins were extracted with a mitochondrial protein extraction kit (Nanjing Keygen Biotech. Co. Ltd., Nanjing, China) according to the manufacturer’s instructions. The extraction of nucleoproteins was performed with a nucleoprotein extraction kit (Nanjing Keygen Biotech Co. Ltd., Nanjing, China) according to the manufacturer’s instructions. The nuclear and mitochondrial protein concentrations of tissues and cells were determined with a BCA protein quantification kit (Beyotime, Shanghai, China). Proteins of equal amount from tissues and cells (100 µg) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and electrophoretically transferred onto polyvinylidene fluoride (PVDF) membranes. After incubation in 5% skim milk in TBST buffer (10 mM Tris HCl, pH 8.0, 150 mM NaCl, and 1% Tween-20) for 1 h at room temperature, the membranes were incubated overnight with hTERT (Abcam, 1:500) or GAPDH (Epitomics, 1:3000) at 4°C and then with a horseradish peroxidase conjugated secondary antibody (Abcam, Cambridge, MA, 1:2000) for 2 h at room temperature. The film was exposed and developed in the presence of ECL substrate (Thermo Scientific, Tewksbury, MA) for 1 min, and the densitometry of protein bands was quantified and analyzed with Gel-Pro Analyzer Software (Media Cybernetics, Inc., Rockville, MD).
Detection of sensitivity to chemotherapeutic drugs by CCK-8 assay
SK-Hep1 cells and HepG2 (1×104 each) were seeded into 96-well plates. After incubation for 24 hg, CDDP (1, 2, 4, 8, and 16 g/ml), DOX (0.15, 0.3, 0.6, 1.25, and 2.5 g/ml) and 5-FU (1, 2, 4, 8, 16, and 32 g/ml) (Sigma) at different concentrations were added. After incubation at 37°C for 24 h, 10 µL of CCK-8 (Do jindo Laboratories, Rockville, MD) was added to each well, followed by incubation at 37°C for 2 h. The absorbance was measured at 450 nm with a microplate reader (Thermo Scientific, Tewksbury, MA). The relative inhibitory rate of cell growth was determined with the following formula: inhibitory rate = (1-OD450 [treatment]/OD450 [vehicle control] × 100%.
Detection of cell apoptosis by FCM after Annexin V-PE/7-AAD staining
Non-transfected and lentivirus-transfected HepG2 cells were plated into a 6-well plate at a density of 1×105 cells/ml and treated with 0.5 mg/ml DOX, 5 g/ml CDDP, or 10 g/ml 5-FU for 24 h. Cells (>1×107) were then centrifuged at 1000 rpm for 5 min re-suspended with PBS, stained with the Annexin V-PE/7-AAD kit (Roche Diagnostics Corp., Indianapolis, IN) and incubated for 15 min. A total of 5×105 cells were detected for FCM of apoptotic cells (BD FACSVantage SE; Becton Dicknson Company, Franklin Lakes, NJ, USA) at 488 nm. Data were analyzed using CellQuest software.
DNA extraction and real-time Q-PCR
Non-transfected and lentivirus-transfected HepG2 cells were grown in a 6-well plate and treated with 0.5 g/ml DOX, 5 g/ml CDDP, or 10 g/ml 5-FU for 24 h. Total DNA was extracted with DNA extraction kits (Qiagen China, Shanghai, China). Q-PCR was carried out according to the manufacturer’s instructions (Takara, Dalian, China). The copy number of mtDNA was calculated as 2ΔCt, where ΔCt differs in the nuclear and mitochondrial genomes. MYH10 was designed to expand a 116-bp fragment from the nuclear genome with a forward primer: 5’-ttgccagaccatgggattgtctca-3’ and a reverse primer: 5’-ttcctaccgaacgaggactccaaa-3’. The mtDNA D-loop was designed to expand a 199-bp fragment from the mitochondrial genome using 5’-gatttgggtaccacccaagtattg-3’ and 5’-aatattcatggtggctggcatgta-3’. PCR was carried out at 94°C for 4 min, followed by 35 cycles of 94°C for 20 s, 60°C for 30 s and 72°C for 30 s, and the Ct value was calculated [30,31].
Detection of mitochondrial membrane potential (MMP)
Non-transfected and lentivirus-transfected HepG2 cells were grown in a 6-well plate and treated with 0.5 g/ml DOX, 5 g/ml CDDP, or 10 g/ml 5-FU for 12 h. Cells were incubated with 40 nM TMRE (Molecular probes, Invitrogen) for 30 min, and then the MMP was detected as the percentage of cytoplasmic signal in the total cytoplasmic area based on images obtained by confocal laser scanning microscopy. Data were analyzed with Image J.
In vivo drug sensitivity assay
The in vivo drug sensitivity assay was performed according to a previously described method [32]. Approximately 1.0×107 HepG2 cells transfected with lenti- mito-hTERT vector or control vector were subcutaneously injected into nude mice at both armpits. After two weeks, these mice were intraperitoneally injected with 1.5 mg/kg DOX, 25 mg/kg 5-FU or 2 mg/kg CDDP (Sigma) once daily for one week and twice weekly in the following week. Control mice were injected with PBS of equal volume. The tumor volume was measured with a Vernier caliper on days 7, 14, 21, and 28 and calculated as follow: volume = length × width2/2. All the mice were sacrificed on day 28, and the xenograft tumors were photographed.
Immunohistochemistry
The xenograft tumors were collected from nude mice, fixed in 10% neutral buffered formalin (NBF) and embedded in paraffin. Sections (5 m) were obtained, deparaffinized in xylene and rehydrated in a series of ethanol. The classical SP staining method was performed in the sections. The expression of PCNA (Epitomics) and hTERT was determined by calculating the proportion of positive cells in each section with Image J software.
Statistical analysis
Data are presented as mean ± standard deviation (SD). Statistical analysis was done with the STATISTICA program (StatSoft, Tulsa, OK). A value of P<0.05 was considered statistically significant.
This study has been approved by the ethical committee of Xinqiao Hospital affliated to Third Military Medical University.
Results
Increased expression of mito-hTERT after transfection with PLeno-mito-hTERT-GTP lentivirus
It has been reported that MTS can promote the import of nuclear genome coding proteins into the mitochondria [28,29]. To over-express the mitochondrial hTERT in HCC cells, MTS (87 bp) was connected to hTERT sequences and the MTS-hTERT sequences were subsequently cloned into the PLeno- GTP vector (Figure 1A). DNA sequencing showed that the PLeno-mito-hTERT-GTP vector was constructed successfully (Figure 1B). Three types of hTERT can be found in cells: nuclear TERT (N), mitochondrial TERT (C) and intermediary TERT (I). The co-localization of hTERT and mitochondria in HepG2 cells and SK-Hep1 cells was examined by immunofluorescence staining (Figure 1C). The nuclei and mitochondria of these cells were visualized by DAPI and Mitotracker staining, respectively. When compared with negative control and parental cells, co-localization signals significantly increased in HepG2 cells and SK-Hep1 cells transfected with PLeno-mito-hTERT-GTP lentivirus (P<0.01) (Figure 1D). The expression of mitochondrial hTERT and nuclear hTERT in HepG2 cells and SK-Hep1 cells was detected by western blot assay (Figure 1E). The mitochondrial hTERT was normalized to GAPDH, and nuclear hTERT was normalized to PCNA. In contrast to negative control and parental cells, mitochondrial hTERT expression significantly increased in HepG2 cells and SK-Hep1 cells transfected with PLeno-mito-hTERT-GTP lentivirus (P<0.05), while nuclear hTERT expression remained unchanged (P>0.05) (Figure 1F).
Figure 1.
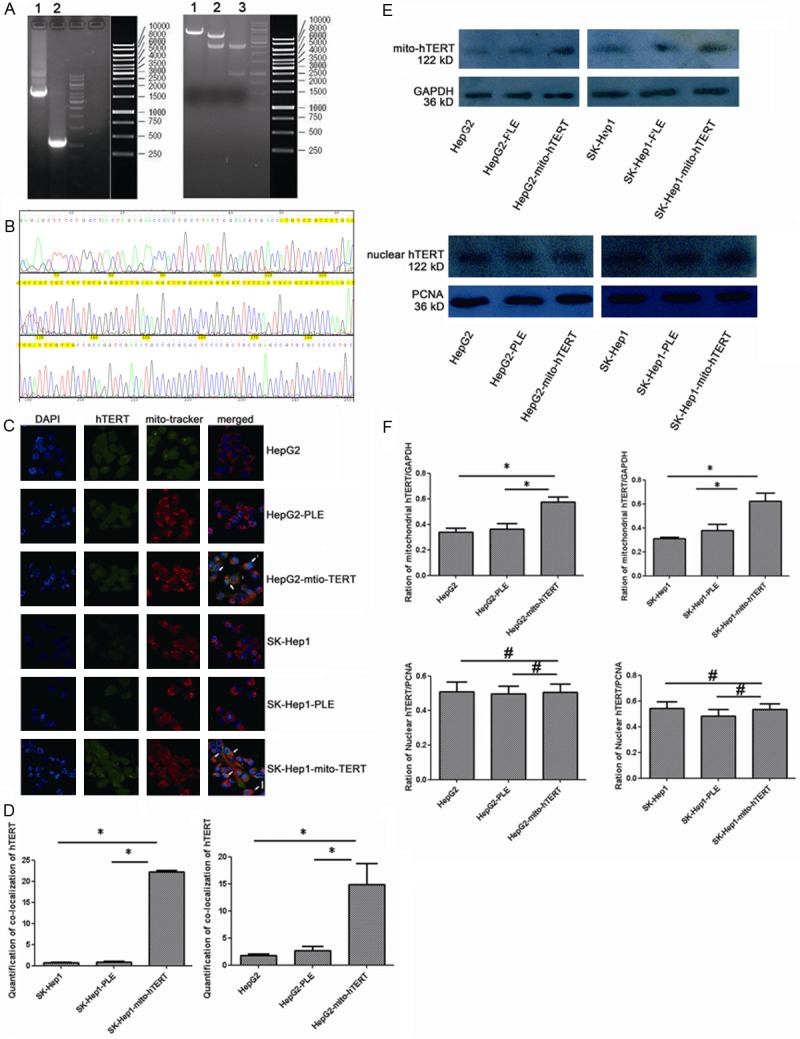
Increased expression of mito-hTERT in cells after transfection with PLeno-mito-hTERT-GTP lentivirus (A)Agarose Gel Electrophoresis of Plasmid DNA. Left Lane 1: MTS-hTERT (4000 bp). Left Lane 2: PDOWN-vehicle (1000 bp). Right Lane 1: PLe-plasmid after EcoRI/XhoI cutting (5000 bp). Right Lane 2: PLe-MTS-hTERT-plasmid after EcoRI/XhoI cutting (5000 bp+4000 bp). Lane 3: PDOWN-MTS-hTERT-vehicle after EcoRI/XhoI cutting (4000 bp+1000 bp). (B) DNA sequencing of MTS-hTERT fusion gene. MTS (87 bp) was located in 50-136 bp (yellow). (C) Immunofluorescence staining shows that the co-localization (yellow) of mitochondrial hTERT (green) and mitochondria (red) in non-transfected and lentivirus-transduced HepG2 and SK-Hep1 cells. Nuclei were counterstained blue with DAPI. Magnification: 1200×. N: nuclear hTERT, C: cytoplasmic hTERT, I: intermediate hTERT. (D) Histogram represents the quantitative analysis of co-localization signals, comparing the average fluorescence intensity per unit area. *P<0.01. (E) Western blot shows expression of mitochondrial and nuclear hTERT: mitochondrial hTERT was normalized to GAPDH and nuclear hTERT was normalized to PCNA. (F) Histogram Histogram represents the quantitative analysis of gray ratio, *P<0.05. #P>0.05.
Effect of mito-hTERT over-expression on drug resistance
Our previous findings showed that hTERT gradually translocates from the nuclei to the mitochondria accompanied by an increases in the resistance index of drug-resistant HCC cells [27]. In this study, when compared with negative control and parental cells, HepG2 cells over-expressing mito-hTERT showed a significantly higher growth inhibition ratio after treatment with CDDP (1-16 g/ml) (Table 1), 5-FU (4-32 g/ml) (Table 2) and DOX (0.15-2.5 g/ml) (Table 3) (P<0.05); this resistance gradually reduced with the increase in drug concentrations.
Table 1.
The relative inhibition rate of CDDP
| Drug (µg/ml) | HepG2 | HepG2-PLE | HepG2-mito-hTERT |
|---|---|---|---|
| 1 | 0.372±0.024 | 0.318±0.013 | 0.120±0.018*,** |
| 2 | 0.479±0.010 | 0.463±0.001 | 0.246±0.014*,** |
| 4 | 0.589±0.005 | 0.572±0.005 | 0.344±0.003*,** |
| 8 | 0.604±0.017 | 0.609±0.006 | 0.478±0.007*,** |
| 16 | 0.680±0.005 | 0.694±0.004 | 0.623±0.006*,** |
Footnotes: HepG2-mito-hTERT vs. HepG2 cells;
P<0.05.
HepG2-mito-hTER vs. HepG2-PLE;
P<0.05.
mean ± SD, n=3.
Table 2.
The relative inhibition rate of 5-FU
| Drug (µg/ml) | HepG2 | HepG2-PLE | HepG2-mito-hTER |
|---|---|---|---|
| 1 | 0.111±0.020 | 0.120±0.004 | 0.053±0.005 |
| 2 | 0.220±0.013 | 0.228±0.013 | 0.181±0.001 |
| 4 | 0.350±0.006 | 0.310±0.015 | 0.207±0.016*,** |
| 8 | 0.442±0.023 | 0.430±0.019 | 0.320±0.006*,** |
| 16 | 0.606±0.018 | 0.594±0.004 | 0.499±0.018*,** |
| 32 | 0.678±0.011 | 0.717±0.024 | 0.555±0.019*,** |
Footnotes: HepG2-mito-hTERT vs. HepG2 cells;
P<0.05.
HepG2-mito-hTER vs. HepG2-PLE;
P<0.05.
mean ± SD, n=3.
Table 3.
The relative inhibition rate of DOX
| Drug (µg/ml) | HepG2 | HepG2-PLE | HepG2-mito-hTER |
|---|---|---|---|
| 0.15 | 0.383±0.020 | 0.385±0.006 | 0.165±0.023*,** |
| 0.3 | 0.420±0.006 | 0.400±0.008 | 0.229±0.007*,** |
| 0.6 | 0.547±0.005 | 0.522±0.005 | 0.410±0.011*,** |
| 1.25 | 0.634±0.015 | 0.628±0.020 | 0.509±0.066*,** |
| 2.5 | 0.750±0.008 | 0.741±0.008 | 0.562±0.009*,** |
Footnotes: HepG2-mito-hTERT vs. HepG2 cells;
P<0.05.
HepG2-mito-hTER vs. HepG2-PLE;
P<0.05.
mean ± SD, n=3.
Similarly, SK-Hep1 cells over-expressing mito-hTERT showed a significantly higher growth inhibition ratio after treatment with CDDP (1-16 g/ml) (Table 4), 5-FU (2-8 g/ml) (Table 5) and DOX (0.15-1.25 g/ml) (Table 6) (P<0.05), when compared with negative control and parental cells.
Table 4.
The relative inhibition rate of CDDP
| Drug (µg/ml) | SK-Hep1 | SK-Hep1-PLE | SK-Hep1-mito-hTER |
|---|---|---|---|
| 1 | 0.189±0.029 | 0.190±0.026 | 0.083±0.017*,** |
| 2 | 0.284±0.042 | 0.277±0.042 | 0.151±0.063*,** |
| 4 | 0.368±0.019 | 0.376±0.021 | 0.234±0.021*,** |
| 8 | 0.479±0.048 | 0.497±0.065 | 0.387±0.022*,** |
| 16 | 0.699±0.016 | 0.709±0.020 | 0.624±0.013*,** |
Footnotes: SK-Hep1-mito-hTERT vs. SK-Hep1 cells;
P<0.05.
SK-Hep1-mito-hTER vs. SK-Hep1-PLE;
P<0.05.
mean ± SD, n=3.
Table 5.
The relative inhibition rate of CDDP
| Drug (µg/ml) | SK-Hep1 | SK-Hep1-PLE | SK-Hep1-mito-hTERT |
|---|---|---|---|
| 1 | 0.182±0.031 | 0.173±0.055 | 0.145±0.022 |
| 2 | 0.575±0.025 | 0.570±0.035 | 0.275±0.058*,** |
| 4 | 0.639±0.040 | 0.643±0.040 | 0.458±0.042*,** |
| 8 | 0.801±0.005 | 0.779±0.011 | 0.670±0.064*,** |
Footnotes: SK-Hep1-mito-hTERT vs. SK-Hep1 cells;
P<0.05.
SK-Hep1-mito-hTER vs. SK-Hep1-PLE;
P<0.05.
mean ± SD, n=3.
Table 6.
The relative inhibition rate of DOX
| Drug (µg/ml) | SK-Hep1 | SK-Hep1-PLE | SK-Hep1-mito-hTERT |
|---|---|---|---|
| 0.15 | 0.548±0.014 | 0.512±0.006 | 0.112±0.008*,** |
| 0.3 | 0.665±0.022 | 0.628±0.008 | 0.294±0.007*,** |
| 0.6 | 0.715±0.006 | 0.764±0.020 | 0.475±0.009*,** |
| 1.25 | 0.823±0.013 | 0.811±0.006 | 0.740±0.018*,** |
Footnotes: SK-Hep1-mito-hTERT vs. SK-Hep1 cells;
P<0.05.
SK-Hep1-mito-hTER vs. SK-Hep1-PLE;
P<0.05.
mean ± SD, n=3.
Mito-hTERT over-expression reduces cell apoptosis induced by chemotherapeutic drugs
Most chemotherapeutics (such as cisplatin, 5-fluorouracil and doxorubicin) exert the anti-tumor effects through inducting apoptosis [5-7]. Thus, the effect of mito-hTERT over-expression on the drug-induced apoptosis was examined by immunofluorescence staining and flow cytometry. First, cell apoptosis was verified by immunostaining of activated caspase-3 (Figure 2A). When compared with negative control HepG2 cells and SK-Hep1 cells, the activated caspase-3 significantly decreased in HepG2 cells and SK-Hep1 cells over-expressing mito-hTERT after CDDP, 5-FU and DOX treatment (Figure 2C) (P<0.05). Furthermore, flow cytometry revealed that apoptotic cells after treatment with different drugs decreased markedly in HepG2 cells over-expressing mito-hTERT when compared with negative control and parental cells (Figure 2B) (P<0.05).
Figure 2.

Mito-hTERT reduces cell apoptosis induced by chemotherapeutic drugs (A) Activited caspase-3 in lentivirus-transduced SK-Hep1 and HepG2 cells was detected by immunofluorescence assay after treated with 0.5 mg/ml DOX, 5 mg/ml CDDP or 10 mg/ml 5-FU for 12 h. Magnification: 800×. (B) Apoptosis induced by different drugs in non-transfected and lentivirus-transduced HepG2 cells was determined by flow cytometry after treated with 0.5 mg/ml DOX, 5 mg/ml CDDP or 10 mg/ml 5-FU for 24 h. (C) Histogram represents the quantitative analysis of activited caspase-3, comparing the average fluorescence intensity per unit area. *P<0.05.
Effect of mito-hTERT over-expression on MMP
MPT is closely related to the apoptosis [9]. Our results showed the MMP increased dramatically in HepG2 cells over-expressing mito-hTERT when compared with negative controls and non-transfected cells after CDDP, 5-FU and DOX treatment (Figure 3A, 3B) (P<0.05).
Figure 3.

Effect of mito-hTERT on mtDNA and MMP. A. MMP expression in non-transduced and lentivirus-transduced HepG2 cells was detected by immunofluorescence staining after treatment with 0.5 mg/ml DOX, 5 mg/ml CDDP or 10 mg/ml 5-FU for 12 h. Magnification: 800× and 1200×. B. Histogram represents the quantitative analysis of MMP, comparing the average fluorescence intensity per unit area. *P<0.05. #P>0.05. C. MtDNA copy number in non-transduced and lentivirus-transduced HepG2 cells was detected by qRT-PCR before and after treated with 0.5 mg/ml DOX, 5 mg/ml CDDP or 10 mg/ml 5-FU for 2 h. *P<0.05. #P>0.05.
Effects of mito-hTERT over-expression on mtDNA
mtDNA is an important indicator of mitochondrial function. In the present study, the effect of mito-hTERT over-expression on mtDNA copy number was investigated with a previously described method [30,31]. Q-PCR revealed a significant increase was observed in the mtDNA copy number of mito-hTERT over-expressing HepG2 cells when compared with negative controls and non-transfected cells before and after CDDP, 5-FU and DOX treatment (Figure 3C) (P<0.05).
mito-hTERT over-expression increases drug resistance in vivo
The effects of mito-hTERT over-expression on the drug resistance were further examined in vivo with previously described methods [32] (Figure 4A). The tumor volume in negative control group was significantly lower than that in nude mice inoculated with mito-hTERT-over-expressing HepG2 cells after CDDP, 5-FU or DOX treatment (Figure 4B) (P<0.01). Moreover, the tumor volume in negative control group was not significantly different from that in PBS-treated nude mice inoculated mito-hTERT-over-expressing HepG2 cells (Figure 4B) (P>0.05).
Figure 4.

Over-expression of mito-hTERT increases drug resistance in vivo (A) HepG2-PLE (left) cells and HepG2-mito-hTERT cells (right) were subcutaneously injected into the armpit of nude mice. After two weeks, mice were intraperitoneally injected with 1.5 mg/kg DOX, 25 mg/kg 5-FU or 2 mg/kg CDDP in PBS once daily for one week and twice weekly for 1 week. Control mice were injected with PBS. All the mice were sacrificed and xenograft tumors were collected on day 30 and photographed. (B) Tumor volume was measured with a Vernier calliper on days 7, 14, 21, and 28, and calculated as follow: volume = length × width2/2. *P<0.01, #P>0.05. (C) PCNA expression and hTERT distribution in xenografted tumors were examined by immunohistochemistry. Magnification: 400×. (D) Quantitative analysis of PCNA expression normalized to the average fluorescence intensity per unit area, while mitochondrial hTERT and nuclear hTERT expression was expressed as the percentage of positive cells. *P<0.05, #P>0.05. (E) Western blot assay shows expression of mitochondrial and nuclear hTERT in xenograft tumors: mitochondrial hTERT was normalized to GAPDH and nuclear hTERT to PCNA. (F) Histogram represents the quantitative analysis of gray ratio, *P<0.05. #P>0.05.
The expression of mitochondrial TERT in xenograft tumors was examined by immunohistochemistry (Figure 4C) and western blot assay (Figure 4E). Immunohistochemistry showed the number of mito-TERT positive cells was significantly higher in mito-hTERT-over-expressing HepG2 cells group than that in negative control group (P<0.05) (Figure 4D). Moreover, the amount of nuclear-TERT-positive cells in mito-hTERT-over-expressing HepG2 cells group was comparable to that in control HepG2 cells (P>0.05) (Figure 4D). Similarly, western blot assay indicated that the expression of mitochondrial hTERT significantly increased in mito-hTERT-over-expressing HepG2 cells group when compared with negative control group (P<0.05), while the expression of nuclear hTERT was remained unchanged (P>0.05) (Figure 4F).
Discussion
The mechanisms underlying the MDR of HCC cells, including congenital and acquired MDR, are complex. Our previous results showed that hTERT gradually translocates from the nuclei to the mitochondria accompanied by an increase in the resistance index of drug-resistant HCC cells [27]. Therefore, mitochondrial translocation of hTERT may be involved in the acquired MDR of HCC cells.
The mitochondrion is a special organelle in the eukaryotic cells and has a special transport system. However, cytoplasmic proteins with a MTS on the N terminal can translocate into the mitochondria. MTS and mitochondrial outer membrane protein receptor are involved in almost all the processes of mitochondrial translocation [23,24]. Therefore, to over-express mitochondrial hTERT in HCC cells, an MTS (87 bp) was cloned into the hTERT sequence to construct a PLeno-mito-hTERT-GTP lentivirus. DNA sequencing and mitochondrial hTERT detection showed the PLeno-mito-hTERT-GTP lentivirus was constructed successfully.
Since 1997, many studies have confirmed that the hTERT expression is associated with the drug resistance of tumor cells [10]. Our previous studies also suggested that the increased expression of mitochondrial hTERT in SK-Hep1/CDDP cells is positively related to their drug-resistance to multiple drugs [27]. Consistent with these findings, the present study showed that mitochondrial hTERT over-expression in HCC cells could reduce their sensitivity to multiple chemotherapeutics.
When compared with parental cells, drug-resistant cancer cells show enhanced mitochondrial function, lower ROS and mtDNA damage after injury [33]. In the present study, mitochondrial hTERT over-expressing HCC cells also showed a higher mtDNA copy number before and after drug treatment, suggesting that mitochondrial hTERT might increase mtDNA replication and be involved in drug resistance of HCC cells, which might be ascribed to following mechanisms: mitochondrial TERT can function as reverse transcriptase (RdDP) [21]; mitochondrial TERT can play a role in gene silencing at the transcriptional level [22,23]; mitochondrial TERT can reduce DNA damage [18] to inhibit the apoptosis pathway [26].
Mitochondria play a key role in the cell apoptosis. In the early stage of apoptosis, mitochondrial changes (swelling or dilution) are reversible. However, irreversible mitochondrial permeability transition (MPT) occurs once mitochondrial injury continues, resulting in release of a large number of pro-apoptotic molecules into cytoplasm and activation of caspase family [9]. Our study revealed that mitochondrial hTERT over-expressing HCC cells had a higher MMP and a lower caspase-3 activity after drug treatment, which explains the decreased drug-induced apoptosis in HCC cells over-expressing mitochondrial hTERT.
These findings indicate that mito-hTERT may serve as an anti-apoptotic regulator through protecting the mitochondria. In addition, we further validated the function of mitochondrial hTERT in vivo that mitochondrial hTERT also regulates the drug resistance in nude mice. In summary, mitochondrial hTERT over-expression can increase the sensitivity of HCC cells to chemotherapeutics by increasing the mtDNA damage and elevating cells apoptosis.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 81101892) and Chongqing Provincial Natural Science Foundation of China (No. 2013C290).
References
- 1.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 2.Hussain SP, Schwank J, Staib F, Wang XW, Harris CC. TP53 mutations and hepatocellular carcinoma: insights into the etiology and pathogenesis of liver cancer. Oncogene. 2007;26:2166–2176. doi: 10.1038/sj.onc.1210279. [DOI] [PubMed] [Google Scholar]
- 3.Townsend DM, Tew KD. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene. 2003;22:7369–7375. doi: 10.1038/sj.onc.1206940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fabregat I, Roncero C, Fernandez M. Survival and apoptosis: a dysregulated balance in liver cancer. Liver Int. 2007;27:155–162. doi: 10.1111/j.1478-3231.2006.01409.x. [DOI] [PubMed] [Google Scholar]
- 5.Evans DL, Dive C. Effects of cisplatin on the induction of apoptosis in proliferating hepatoma cells and nonproliferating immature thymocytes. Cancer Res. 1993;53:2133–2139. [PubMed] [Google Scholar]
- 6.Kinsella AR, Smith D, Pickard M. Resistance to chemotherapeutic antimetabolites: a function of salvage pathway involvement and cellular response to DNA damage. Br J Cancer. 1997;75:935–945. doi: 10.1038/bjc.1997.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asakura T, Sawai T, Hashidume Y, Ohkawa Y, Yokoyama S, Ohkawa K. Caspase-3 activation during apoptosis caused by glutathione-doxorubicin conjugate. Br J Cancer. 1999;80:711–715. doi: 10.1038/sj.bjc.6690414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Z, Cheng M, Cao M. Potential targets for molecular imaging of apoptosis resistance in hepatocellular carcinoma. Biomed Imaging Interv J. 2011;7:e5. doi: 10.2349/biij.7.1.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tait SW, Green DR. Mitochondrial regulation of cell death. Cold Spring Harb Perspect Biol. 2013:5. doi: 10.1101/cshperspect.a008706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang L, Li PF, Geng M, Cao YC, Yin YC. Correlation between chemosensitivity to anticancer drugs and telomerase reverse transcriptase mRNA expression in gastric cancer. Diagn Pathol. 2013;8:33. doi: 10.1186/1746-1596-8-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith LL, Coller HA, Roberts JM. Telomerase modulates expression of growth-controlling genes and enhances cell proliferation. Nat Cell Biol. 2003;5:474–479. doi: 10.1038/ncb985. [DOI] [PubMed] [Google Scholar]
- 12.Geserick C, Tejera A, Gonzalez-Suarez E, Klatt P, Blasco MA. Expression of mTert in primary murine cells links the growth-promoting effects of telomerase to transforming growth factor-beta signaling. Oncogene. 2006;25:4310–4319. doi: 10.1038/sj.onc.1209465. [DOI] [PubMed] [Google Scholar]
- 13.Gu BW, Bessler M, Mason PJ. A pathogenic dyskerin mutation impairs proliferation and activates a DNA damage response independent of telomere length in mice. Proc Natl Acad Sci U S A. 2008;105:10173–10178. doi: 10.1073/pnas.0803559105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Masutomi K, Possemato R, Wong JM, Currier JL, Tothova Z, Manola JB, Ganesan S, Lansdorp PM, Collins K, Hahn WC. The telomerase reverse transcriptase regulates chromatin state and DNA damage responses. Proc Natl Acad Sci U S A. 2005;102:8222–8227. doi: 10.1073/pnas.0503095102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mondello C, Bottone MG, Noriki S, Soldani C, Pellicciari C, Scovassi AI. Oxidative stress response in telomerase-immortalized fibroblasts from a centenarian. Ann N Y Acad Sci. 2006;1091:94–101. doi: 10.1196/annals.1378.058. [DOI] [PubMed] [Google Scholar]
- 16.Lee J, Sung YH, Cheong C, Choi YS, Jeon HK, Sun W, Hahn WC, Ishikawa F, Lee HW. TERT promotes cellular and organismal survival independently of telomerase activity. Oncogene. 2008;27:3754–3760. doi: 10.1038/sj.onc.1211037. [DOI] [PubMed] [Google Scholar]
- 17.Ahmed S, Passos JF, Birket MJ, Beckmann T, Brings S, Peters H, Birch-Machin MA, von Zglinicki T, Saretzki G. Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J Cell Sci. 2008;121:1046–1053. doi: 10.1242/jcs.019372. [DOI] [PubMed] [Google Scholar]
- 18.Haendeler J, Drose S, Buchner N, Jakob S, Altschmied J, Goy C, Spyridopoulos I, Zeiher AM, Brandt U, Dimmeler S. Mitochondrial telomerase reverse transcriptase binds to and protects mitochondrial DNA and function from damage. Arterioscler Thromb Vasc Biol. 2009;29:929–935. doi: 10.1161/ATVBAHA.109.185546. [DOI] [PubMed] [Google Scholar]
- 19.Haendeler J, Hoffmann J, Brandes RP, Zeiher AM, Dimmeler S. Hydrogen peroxide triggers nuclear export of telomerase reverse transcriptase via Src kinase family-dependent phosphorylation of tyrosine 707. Mol Cell Biol. 2003;23:4598–4610. doi: 10.1128/MCB.23.13.4598-4610.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jakob S, Schroeder P, Lukosz M, Buchner N, Spyridopoulos I, Altschmied J, Haendeler J. Nuclear protein tyrosine phosphatase Shp-2 is one important negative regulator of nuclear export of telomerase reverse transcriptase. J Biol Chem. 2008;283:33155–33161. doi: 10.1074/jbc.M805138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharma NK, Reyes A, Green P, Caron MJ, Bonini MG, Gordon DM, Holt IJ, Santos JH. Human telomerase acts as a hTR-independent reverse transcriptase in mitochondria. Nucleic Acids Res. 2012;40:712–725. doi: 10.1093/nar/gkr758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maida Y, Yasukawa M, Furuuchi M, Lassmann T, Possemato R, Okamoto N, Kasim V, Hayashizaki Y, Hahn WC, Masutomi K. An RNA-dependent RNA polymerase formed by TERT and the RMRP RNA. Nature. 2009;461:230–235. doi: 10.1038/nature08283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maida Y, Masutomi K. RNA-dependent RNA polymerases in RNA silencing. Biol Chem. 2011;392:299–304. doi: 10.1515/BC.2011.035. [DOI] [PubMed] [Google Scholar]
- 24.Santos JH, Meyer JN, Van Houten B. Mitochondrial localization of telomerase as a determinant for hydrogen peroxide-induced mitochondrial DNA damage and apoptosis. Hum Mol Genet. 2006;15:1757–1768. doi: 10.1093/hmg/ddl098. [DOI] [PubMed] [Google Scholar]
- 25.Indran IR, Hande MP, Pervaiz S. hTERT overexpression alleviates intracellular ROS production, improves mitochondrial function, and inhibits ROS-mediated apoptosis in cancer cells. Cancer Res. 2011;71:266–276. doi: 10.1158/0008-5472.CAN-10-1588. [DOI] [PubMed] [Google Scholar]
- 26.Singhapol C, Pal D, Czapiewski R, Porika M, Nelson G, Saretzki GC. Mitochondrial telomerase protects cancer cells from nuclear DNA damage and apoptosis. PLoS One. 2013;8:e52989. doi: 10.1371/journal.pone.0052989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ling X, Wen L, Zhou Y. Role of mitochondrial translocation of telomerase in hepatocellular carcinoma cells with multidrug resistance. Int J Med Sci. 2012;9:545–554. doi: 10.7150/ijms.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pfanner N, Craig EA, Meijer M. The protein import machinery of the mitochondrial inner membrane. Trends Biochem Sci. 1994;19:368–372. doi: 10.1016/0968-0004(94)90113-9. [DOI] [PubMed] [Google Scholar]
- 29.Schatz G. The protein import system of mitochondria. J Biol Chem. 1996;271:31763–31766. doi: 10.1074/jbc.271.50.31763. [DOI] [PubMed] [Google Scholar]
- 30.Santos JH, Meyer JN, Mandavilli BS, Van Houten B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol Biol. 2006;314:183–199. doi: 10.1385/1-59259-973-7:183. [DOI] [PubMed] [Google Scholar]
- 31.Ussakli CH, Ebaee A, Binkley J, Brentnall TA, Emond MJ, Rabinovitch PS, Risques RA. Mitochondria and tumor progression in ulcerative colitis. J Natl Cancer Inst. 2013;105:1239–1248. doi: 10.1093/jnci/djt167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shang Y, Zhang Z, Liu Z, Feng B, Ren G, Li K, Zhou L, Sun Y, Li M, Zhou J, An Y, Wu K, Nie Y, Fan D. miR-508-5p regulates multidrug resistance of gastric cancer by targeting ABCB1 and ZNRD1. Oncogene. 2014;33:3267–3276. doi: 10.1038/onc.2013.297. [DOI] [PubMed] [Google Scholar]
- 33.Wardell TM, Ferguson E, Chinnery PF, Borthwick GM, Taylor RW, Jackson G, Craft A, Lightowlers RN, Howell N, Turnbull DM. Changes in the human mitochondrial genome after treatment of malignant disease. Mutat Res. 2003;525:19–27. doi: 10.1016/s0027-5107(02)00313-5. [DOI] [PubMed] [Google Scholar]