

Abstract
FHL2, a member of the four and one half LIM domain protein family, is a critical transcriptional modulator. Here, we identify FHL2 as a critical regulator of hematopoietic stem cells (HSCs) that is essential for maintaining HSC self-renewal under regenerative stress. We find that Fhl2 loss has limited effects on hematopoiesis under homeostatic conditions. In contrast, Fhl2-null chimeric mice reconstituted with Fhl2-null bone marrow cells developed abnormal hematopoiesis with significantly reduced numbers of HSCs, hematopoietic progenitor cells (HPCs), red blood cells and platelets as well as hemoglobin levels. In addition, HSCs displayed a significantly reduced self-renewal capacity and were skewed toward myeloid lineage differentiation. We find that Fhl2 loss reduces both HSC quiescence and survival in response to regenerative stress, probably as a consequence of Fhl2-loss-mediated down-regulation of cyclin dependent kinase (CDK)-inhibitors, including p21(Cip) and p27(Kip1). Interestingly, FHL2 is regulated under control of a tissue specific promoter in hematopoietic cells and it is down-regulated by DNA hypermethylation in the leukemia cell line and primary leukemia cells. Furthermore, we find that down-regulation of FHL2 frequently occurs in myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) patients, raising a possibility that FHL2 down-regulation plays a role in the pathogenesis of myeloid malignancies.
Introduction
Human FHL2, originally known as DRAL, was first identified as a four and a half LIM-domain protein that was down-regulated in rhabdomyosarcomas1. The LIM domain, an acronym derived from LIN-11, ISL-1 and MEC-3, is characterized by a double zinc finger motif which mediates protein-protein interaction 2. FHL2 belongs to the FHL protein family, a subfamily of LIM-only proteins, which includes the members FHL1, FHL3, FHL4 and ACT in humans 3-6. FHL2 is conserved between humans and mice with 91% amino acid identity. It is a potential adapter protein involved in multiple protein complexes6-8. FHL2 interacts with a variety of transcription factors, including the androgen receptor, β-catenin, AP1, Smad, SKI, TRAF2, TRAF4, TRAF6, CREB, PLZF, Runx2, Foxk1, SRF, TUCAN and WT1, and functions as either a transcriptional coactivator or a corepressor in a cellular context-dependent manner 9-23, indicating that FHL2 has important cellular functions.
FHL2 expression is often deregulated in cancer with down-regulation or overexpression in various types of tumors including rhabdomyosarcoma1, prostate cancer24, ovarian cancer25, human melanoma13, lung cancer6, breast cancer26 and liver cancer27, suggesting that FHL2 may act as an oncogene or as a tumor suppressor in a tissue-dependent manner. The dualistic nature of FHL2 is also reflected by the fact that FHL2 can act as a transcriptional repressor or activator depending on cell context2. We previously found that FHL2 is expressed in normal human CD34+ stem-enriched populations28. Its function in hematopoiesis was first documented in our previous study examining overexpression of FHL2 in bone marrow cells28. However, the function of endogenous FHL2 in vivo has not been reported yet.
To understand the biological role of Fhl2 in hematopoiesis and HSC function in vivo, we characterized a Fhl2 knockout mouse model. We found that Fhl2 is essential for maintaining the function of HSCs by regulating the cell survival and quiescence of HSCs under regenerative stress, but that it has limited effects on hematopoiesis under homeostatic conditions. In addition, Fhl2 loss leads to down-regulation of CDK inhibitors including p21(Cip1), p27(Kip1) and p57(Kip2) in HSC-enriched populations. However, we showed that forced expression of p21(Cip1) or p27(Kip1) but not p57 (Kip2) in HSC-enriched population partially rescued Fhl2 depletion-induced quiescence loss. We also found that FHL2 is down-regulated in both MDS and AML patients, and have identified a tissue specific promoter of FHL2 in hematopoietic cells. Of interest, our results revealed that down-regulation of FHL2 is associated with DNA hypermethylation of FHL2 hematopoietic specific promoter region, and it can be re-activated by hypomethylating agent in the KG1 myeloid leukemia cell line and primary AML cells. Together, these results suggest that FHL2 is an important regulator of HSC self-renewal in response to stress, and that FHL2 down-regulation is mediated by aberrant DNA methylation in a subset of AML patients, thereby contributing to leukemogenesis.
Materials and Methods
Mice and Blood Cell Counts
Fhl2-/- knockout mice were bread into C57Bl/6 background for more than eight generations. All animal experiments were approved by the University of Illinois at Chicago Institutional Animal Care and Use Committee. Peripheral blood (PB) samples were collected by tail bleeding into tubes containing EDTA. Complete blood counts and differentials were obtained using a Hemavet 950FS (Drew Scientific).
Flow cytometry
Bone marrow cells were obtained by flushing of femurs and tibiae in DPBS with 2% FBS. PB was obtained by tail bleeding. Phenotypic analyses of HPCs, LSK and HSCs have been described in our previous studies 29. The following mAbs were used: Streptavidin-PE-CY5, PE-Sca-1, APC-C-kit, PE-Cy7-CD48, APC-CD150, Streptavidin-APC-Cy7, APC-C-kit, PE-Cy7-CD16/32, e450-CD34, PE-CD45.1, FITC -CD45.2. A mixture of mAbs recognizing CD3e, B220, TER-119, CD19, Mac-1 and Gr-1 was used to identify Lin+ cells. All mAbs were obtained from eBioscience except CD150, which was from Biolegend. For lineage analysis, whole BM cells were stained with various combinations of antibodies for different cell populations: Percp-B220 and APC-IgM for B cells; PE-Gr-1 and APC-Mac-1 for myeloid cells; APC-Ter119 and PE-CD71 for erythroid cells; CD4 and CD8 for mature T cells; FACS analysis was performed using a CyAn ADP flow cytometer (Beckman Coulter). All data were analyzed by FlowJo software (TreeStar, Inc).
Cell cycle analysis and apoptosis
For Hoechst 33342/Pyronin Y staining, BM cells were stained with 1μg/mL Hoechst 33342 and 50μM Verapamil at 37°C for 45 mins, followed by staining with 1μg/mL Pyronin Y at 37°C for 45 mins. Subsequently, the cells were stained with mAbs against cell surface markers to identify the Lin-Sca-1+C-kit+ population. For DAPI staining, cells were stained with antibodies to identify HSCs, then fixed, stained with 5μg/mL DAPI. For the apoptosis analysis, freshly isolated BM cells were first stained with antibodies to identify Lin-Sca-1+C-kit+ cells, then washed in binding buffer and incubated with Annexin V APC and DAPI.
Homing assay for hematopoietic progenitor cells
In vivo homing assays were performed as described by Foudi et al. with some modifications30. Briefly, 20×106 BM cells from Fhl2-/- or WT mice were suspended in 1 ml PBS with added CSFE to a final concentration of 2.5μM, incubate cells at 37°C for 15 minutes, then an equal volume of pre-warmed FBS (100%, filtered) was added to stop labeling. Cells were washed twice with PBS, followed by injection into the retro-orbital sinus of recipient mice that had been irradiated 24 hours before injection. Bone marrow was harvested 6 hours after injection, stained with antibodies against lineage markers and Sca-1, the frequency of CFSE+ cells in Lin-Sca-1+ population was determined by FACS. The formula is based on the percentage of CFSE+ Lin-Sca-1+ donor cells that homed to the BM as determined by flow cytometry (A), multiplied by the cellularity of recovered BM from the lethally irradiated recipients at 6 hours after transplantation (B), then divided by the number of CFSE+ Lin-Sca-1+ cells injected per mice (N): % homing= (A×B)/N.
Transplantation assays
For competitive repopulation assays, 2.5×106 BM cells (CD45.2) from Fhl2-/- or Fhl2+/+ (control) mice were mixed 1:1 with the competitor BM cells (CD45.1/CD45.2) from B6.SJL/Ly5.1 mice and transplanted into lethally irradiated Ly5.1 mice by retroorbital injection. For generating BM chimeras, 106 BM cells from Fhl2-/- or Fhl2+/+ (control) were injected into lethally irradiated Ly5.1 mice.
Quantitative real-time PCR
Total RNA was isolated from LSK cells of the transplanted Fhl2+/+ and Fhl2-/- mice using an RNeasy Mini Kit (QIAGEN). And cDNAs were reverse-transcribed from total RNA prepared from LSK cells. Then cDNA was subjected to real-time PCR using SYBR Green Supermix (BIO-RAD) in realtime PCR System I-cycler (BIO-RAD). Amplification of β-actin was used for mice sample normalization. The housekeeping form of porphobilinogen dehydrogenase gene (HMBS)31 was used for human sample normalization. Primer sequences are listed in supplementary materials and methods.
5-Aza-2′-deoxycytidine treatment
KG-1 leukemia cells or primary leukemia cells were incubated for 72 hours in medium containing 2μM 5-aza-2′-deoxycytidine; the drug was replenished daily.
Bisulfite sequencing
Analysis of methylation of promoter region of bisulfite-treated KG-1 cells is described in supplementary materials and methods.
Retroviral production and infection
Retroviral constructs are described in supplementary material and methods. Retroviruses were produced as previously described28. Bone marrow cells were collected from primary Fhl2+/+ and Fhl2-/- transplantation mice (10 months after transplantation). The Lin+ cells from these BM cells were labeled with antibodies against cell surface markers, including Biotin-Mac-1, Gr-1, CD4, CD8, B220, and Ter119, and depleted with Streptavidin Dynabeads (Life Technologies). The resultant Lin- population was infected with retrovirus as we previously described28. The uninfected cells were killed after 2-day culture with 2.5μg/ml puromycin.
Patient Samples
The existed RNA and DNA samples from the MDS patients were obtained from Huashan hospital of Fudan University, Shanghai, China. The AML patient samples were obtained from the University of Illinois Hospital at Chicago. Informed consent was obtained in accordance with protocols approved by the review boards of participating hospitals.
Statistical analysis
Statistical significance was calculated using the two tailed Student's t test.
Results
Fhl2 loss has limited effects on hematopoiesis under homeostatic conditions
To determine the function of endogenous Fhl2, we conducted a loss of function study. Dr. Ju Chen (UCSD, CA) kindly provided Fhl2-/- mice for this study (previously described 32). Briefly, the Fhl2-/- mice were backcrossed for eight generations into the C57BL/6 background. The deletion of Fhl2 allele was determined by PCR analysis of genomic DNA isolated from the Fhl2-deficient and control wildtype mice (Supplementary Figure S1A). Fhl2-null mice are viable and do not have obvious abnormal hematopoiesis. White blood cell (WBC) counts and hematocrits in cohorts of Fhl2-deficient mice and control wildtype littermates were monitored at regular intervals for signs of hematopoietic abnormalities. At 2 months of age, Fhl2-/- mice revealed normal WBC, platelet, and red blood cell (RBC) counts and hemoglobin (Hb) levels in the peripheral blood (PB) as compared to control littermates. Within the WBC population, there were no differences in absolute lymphocytes, neutrophils, monocytes, basophils, or eosinophils (Supplementary Table S1) between Fhl2-/- mice and control littermates. Analysis of the lineage distributions in bone marrow, spleen and thymus from Fhl2-/- mice and control littermates by flow cytometric analysis revealed that Fhl2-/- mice had a normal frequency of myeloid cells, B cells and T cells in these tissues (Supplementary Figure S1B &Figure S2). This data suggests that loss of Fhl2 does not affect hematopoietic cell differentiation in young mice.
During normal hematopoiesis, the long-term HSCs (LT-HSCs) have a capacity to self-renew with the potential for differentiation to common myeloid progenitor (CMP) and more committed myeloid progenitor cells, including granulocyte-macrophage (GMP) and megakaryocyte-erythroid progenitor (MEP)33. To determine whether FHL2 is involved in the maintenance of HSCs/HPCs in vivo, we characterized the compartments of HSCs and HPCs by flow cytometric analysis. The frequency and total number of the stem cell enriched population LSK (Lin-Sca+c-Kit+) and LT-HSCs (Lin-Sca+c-Kit+CD48-CD150+) were slightly increased in FHL2-/- mice as compared to wildtype mice (Figure 1A&B) whereas there was a statistically significant decrease in the number of HPCs in FHL2-/- mice compared to Fhl2+/+ mice. To determine whether Fhl2 loss affects the differentiation of HPCs, the subpopulations of myeloid progenitors in Fhl2-/- and control mice were analyzed. As shown in Figure 1A&B, both Fhl2-/- and Fhl2+/+ mice had comparable frequencies of CMP, GMP and MEP cells in vivo. However, the total number of GMP and MEP was slightly reduced in Fhl2-/- mice as compared to control mice.
Figure 1. The effects of Fhl2 loss on the number and cell cycle status of HSCs and HPCs in BM.
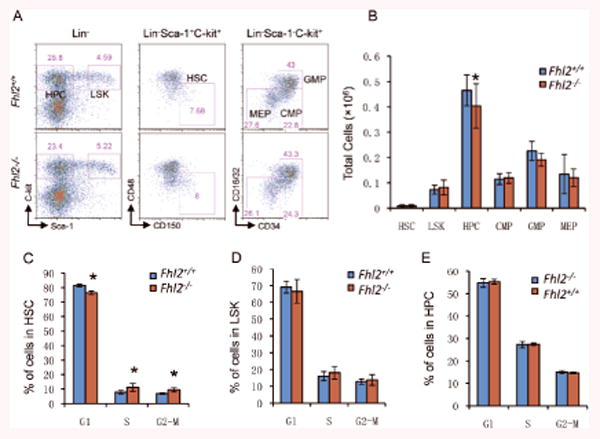
(A-B) The representative flow cytometric analysis of the frequency (A) and average absolute number (B) of HSCs, LSKs, HPCs, CMPs, GMPs and MEPs in BM cells from the control and Fhl2-/- mice (n=5, *, P<0.05). (C-E) The histograms show the distribution of HSCs (C), LSKs (D) and HPCs (E) in G1, S and G2/M phase in BM from control and Fhl2-/- mice (n=5, *, P<0.05). Cells were stained with DAPI. All mice were at age of 2 months. HPCs, LSKs, HSCs are defined as Lin-Sca-1-c-Kit+, Lin-Sca-1+c-Kit+ and LSK, CD150+CD48- respectively. CMPs, GMPs and MEPs are defined as Lin-c-kit+Sca-1- CD34+/loCD16/32int, Lin-c-kit+Sca-1- CD34+CD16/32+, and Lin-c-kit+Sca-1- CD34-CD16/32- respectively.
Given the fact that Fhl2 regulates cell proliferation and apoptosis in various cell types34-37, we examined whether loss of endogenous Fhl2 affects the proliferation and apoptosis of HSCs and HPCs in vivo. As shown in Figure 1C-1E, Fhl2 loss slightly increases proliferation of HSCs but not LSKs and HPCs. However, it did not affect the apoptosis of HSCs and HPCs or the quiescence of HSCs (Supplementary Figure S3&4). Therefore, loss of Fhl2 likely expands the HSCs by increasing the proliferation of HSCs and slightly inhibiting their differentiation into HPCs at steady state, as evidenced by a reduced number of HPCs and a slightly increased number of HSCs in BM cells.
Loss of Fhl2 markedly reduces HSC self-renewal capacity after transplantation
Although loss of Fhl2 affected the HSCs number and proliferation, but it did not significantly disrupt hematopoiesis in homeostatic state. This prompted us to test the role of Fhl2 in the HSCs under stress. BM transplantation exposes HSCs to replicative, inflammatory and oxidative stresses38, 39, eventually leading to loss of HSCs' capacity for self-renewal. We performed competitive repopulation assays, in which the same number of bone marrow (BM) cells (CD45.2) from Fhl2-/- or Fhl2+/+ mice along with equal number of wild-type competitive BM cells (CD45.1CD45.2) were transplanted into lethally-irradiated syngeneic recipients (CD45.1). Thereafter, we performed serial transplantations in which BM cells mixed from 2-3 mice in each group were sequentially transplanted into serial lethally-irradiated syngeneic recipients (Figure 2A), thereby forcing to repeatedly self-renew. The frequency of donor-derived PB cells, which reflects the repopulation capacity of donor HSCs/HPCs, was analyzed every month after transplantation. The ratio of Fhl2-/- derived (CD45.2+) vs. competitor-derived total PB cells (CD45.2+ /CD45.2+) was higher after primary transplantation than after secondary transplantation, and gradually decreased by 4-5-fold as compared to the ratio of Fhl2+/+-derived vs. competitor-derived PB cells after tertiary transplantation (Figure 2B). Interestingly, 4 months after tertiary transplantation, we found the frequency of myeloid cells was increased at expense of B cells in Fhl2-/- derived PB cells as compared to Fhl2+/+ derived PB cells. It is likely that Fhl2-null HSCs were skewed toward myeloid linage differentiation (Figure 2C). Next, we determined the relative ratio of donor-derived vs. competitor-derived cells in Lin-, HPC, LSK, HSC in BM at 4 months after tertiary transplantation in both Fhl2-/- and Fhl2+/+ chimeric mice. Consistent with these results, the Fhl2-/- HSCs generated significantly lower cell numbers within all subsets of primitive hematopoietic cells than did Fhl2+/+ HSCs (Figure 2D). Collectively, these results indicate that Fhl2-/- HSCs have a reduced self-renewal capacity after serial transplantation.
Figure 2. Loss of Fhl2 leads to intrinsic functional defects and impaired long-term engraftment of HSCs.
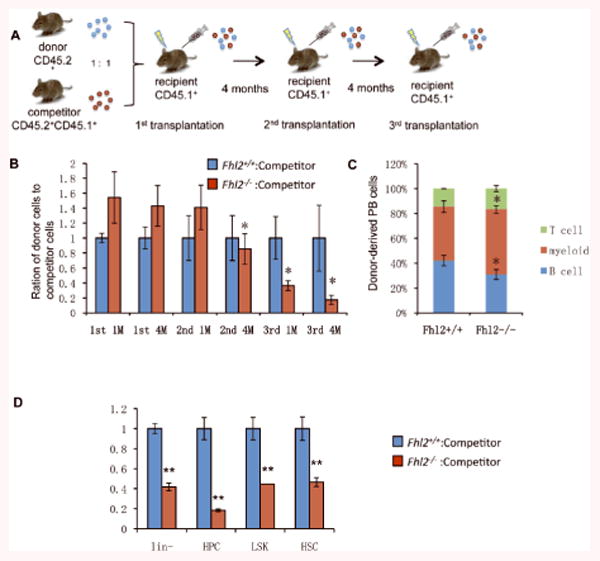
(A) The diagram for the experimental design. (B) Histogram showing the relative ratio of donor (CD45.2+) versus competitor PB cells (CD45.2+ /CD45.2+) at 1 to 4 months after 1st, 2nd and 3rd transplantation(*, P<0.05, n=5). (C)Lineage differentiation in the recipient mice 4 month after 3rd transplantation. (D) The relative ratio of donor (CD45.2+) versus competitor (CD45.2+ /CD45.2+) in Lin-, HPC, LSK, HSC in BM at 4 months after 3rd transplantation (n=5; *, P<0.05; **, P<0.01).
Loss of Fhl2 leads to development of abnormal hematopoiesis in chimeric mice
As wildtype HSCs/HPCs may delay the development of functional defects of Fhl2-null HSCs in vivo in competitive assay, we generated Fhl2-/- and Fhl2+/+ chimeric mice reconstituted with BM cells only from Fhl2-/- or Fhl2+/+ mice. As determined by flow cytometric analysis 6 months post-transplantation (Figure 3A-B), more than 90-97% of BM cells or CD34-LSK HSCs from recipient mice were derived from donor BM cells, indicating a comparable engraftment ability of Fhl2-/- and control BM cells in the recipient mice. We monitored these recipient mice by complete blood cell (CBC) analysis monthly. Interestingly, while both Fhl2-null and control chimeric mice displayed normal hematological parameters at 2 and 3 months post-transplantation (data not shown), the Fhl2-null mice started to develop abnormal hematopoiesis at 6 months post-transplantation, as evidenced by significantly decreased numbers of red blood cells and platelets as well as hemoglobin levels in Fhl2-null chimeric mice as compared to control chimeric mice (Figure 3C).
Figure 3. Fhl2-deficient chimeric mice developed abnormal hematopoiesis.
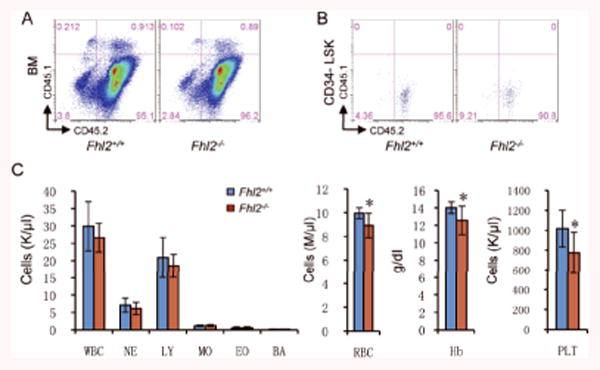
(A-B) The representative flow cytometric analysis of engraftment of CD45.2+ Fhl2-/- and control BM cells in total BM cells (A) and CD34-LSK cells (B) from CD45.1+ wildtype recipient mice (C) The number of white blood cells (WBC), neutrophils (NE), lymphocytes (LY), monocytes (MO), eosinophils (EO), basophils (BA), red blood cells (RBC), platelets (PLT), and hemoglobin (Hb) levels in PB from control and Fhl2-/- chimeric mice (n=7; *, P<0.05).
To determine whether the development of abnormal hematopoiesis in Fhl2-null chimeric mice results from reduced self-renewal capacity of Fhl2-null HSCs in response to regenerative stress, we characterized HSC and HPC compartments in chimeric mice at 7 moths post-transplantation. As shown in Figure 4, the HSCs, LSKs and HPCs were all decreased markedly in Fhl2-null chimeric mice as compared to control chimeric mice, suggesting that Fhl2 loss impaired the repopulation capacity of HSCs in vivo. Consistent with our observation that the number of both red cells and platelets were decreased significantly in Fhl2-null chimeric mice as compared to control mice, the MEPs that eventually develop into megakaryocytes and erythroid cells were significantly reduced in the same mice. Thus, it is likely that Fhl2 loss blocks the differentiation of megakaryocyte and erythroid cells at an early developmental stage under stress.
Figure 4. Decreased BM stem and progenitor cells in transplanted Fhl2-/- mice.
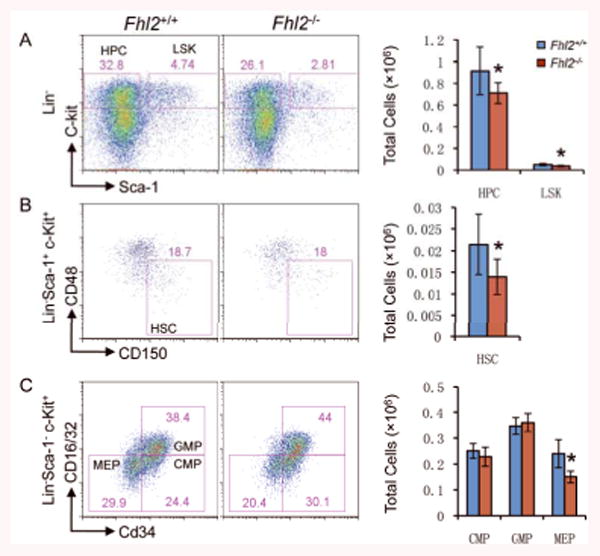
(A-C) Representative FACS analyses (left) and average absolute number (right) of BM cells are shown for control Fhl2+/+ and mutant Fhl2-/- mice. Numbers in the FACS plots indicate percentages in the gated population cells. All BM cells were analyzed from the chimeric mice at 7-8 months post-transplantation (n=5; *, P<0.05).
We also performed in vivo homing assays as we described previously29. The Fhl2 null and wildtype control BM cells had a comparable homing abilities, indicating that Fhl2 loss does not affect the homing ability of BM cells (Supplementary Figure S5).
Fhl2 is required for the maintenance of HSC quiescence under stress
We observed an increase of proliferation of HSCs, but not HPCs, in young Fhl2-null mice. Consistently, the proliferation of HSCs and LSKs, but not HPCs, was increased significantly in Fhl2-null chimeric mice as compared to control mice (Figure 5A-5C). Given the fact that proliferation of HSCs is often associated with exhaustion of HSCs and loss of quiescence 40-41, 42, we next examined the quiescence of HSCs in the chimeric mice. As expected, there were less Fhl2-deficient HSCs in G0 phase than the control HSCs in chimeric mice (Figure 5D-5E). Moreover, the frequency of apoptosis was increased in Fhl2-deficient HSCs as compared to control HSCs in the same chimeric mice (Figure 5F). Therefore, it is likely that an increase in the proliferation of Fhl2-deficient HSCs, augmented by transplantation-induced stress, leads to loss of quiescence and reduced survival of FHL2-deficient HSCs, ultimately disrupting the function of Fhl2-deficient HSCs in vivo.
Figure 5. Fhl2-deficient HSCs had a reduced number of quiescent cells and displayed an increased frequency of proliferation and apoptosis after transplantation.
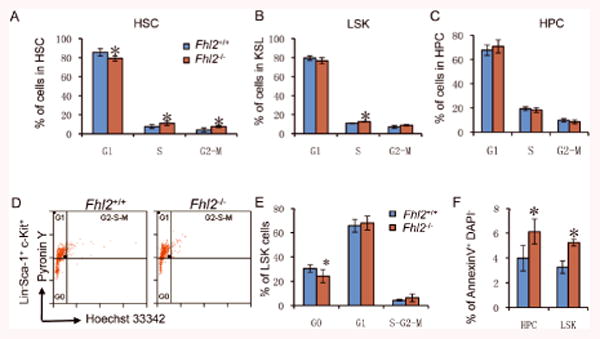
(A-C) The histograms show the distribution of HSCs (A), LSKs (B) and HPCs (C) in G1, S and G2/M phase in BM. Cells were stained with DAPI. (D) Representative flow cytometric analysis of LSK cells stained with Hoechst 33342 and Pyronin. (E) The histograms show the distribution of LSKs in G0, G1 and S-G2-M in BM. (F) The histograms depict the frequency of apoptosis in HPCs and LSKs in BM. All BM cells were isolated from the Fhl2+/+ and Fhl2-/- chimeric mice (n=4-7; *, P<0.05) at 6-7 months post-transplantation.
Fhl2 loss leads to down-regulation of CDK inhibitors in HSC-enriched population
To determine the molecular mechanism underlying the decreased quiescence in the Fhl2-null HSC-enriched population (LSKs), we performed qRT-PCR analysis of critical cell cycle regulators in the LSKs isolated from chimeric mice reconstituted with Fhl2-/- and wildtype BM cells 6 months post-transplantation. As shown in Figure 6A, CDK inhibitors including p21(Cip), p27(Kip1) and p57(Kip2) were all down-regulated significantly in Fhl2-null LSKs as compared to wildtype LSKs, whereas Cdk1, Cdk 2, Cdk3 and Cdk6 were expressed at comparable levels in both Fhl2-null and control LSKs. Cyclin-dependent kinase (CDK) inhibitors include p21(Cip1), p27(Kip1) and p57 (Kip2) are critical regulators of the quiescence and self-renewal of adult HSCs40, 43-45. Interestingly, we found that forced expression of p21 (Cip1) or p27 (Kip1) but not p57 (Kip2) partially rescued the decreased quiescence of Fhl2-deficient LSKs (Figure 6B). These data suggest that down-regulation of CDK inhibitors, including p21 (Cip1) and p27 (Kip1), contributes to the decreased quiescence of LSKs induced by Fhl2 depletion.
Figure 6. Down-regulation of CDK inhibitors in Fhl2-null LSKs.
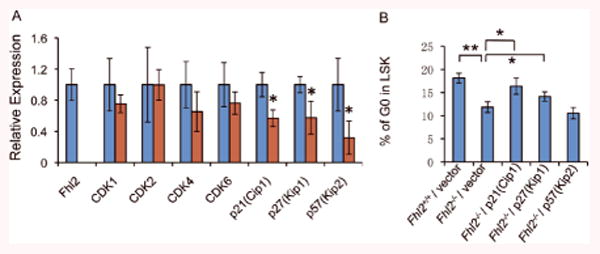
(A) qPCR analysis of the expression of CDKs and CDK inhibitors in Fhl2-null and wildtype control LSKs isolated from chimeric mice 6 months post-transplantation (n=3, p<0.05). LSKs are defined as Lin-Sca+c-Kit+ cells. (B) G0 status of Fhl2-null and wildtype control LSKs with forced expression of p21(Cip1), p27(Kip1) and p57(Kip2). The G0 status of infected cells was determined by flow cytometric analysis. **, P<0.005; *, P<0.05.
Down-regulation of FHL2 in a subset of AML and MDS patients
We found that FHL2 was down-regulated in a subset of t-MDS/t- AML patients31, raising the possibility that FHL2 may also be deregulated in a subset of de novo AML and MDS patients. We performed an analysis of the Affymetrix GeneChip dataset for 542 AML patients and 74 healthy individuals published online (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi/acc=GSE13159) by Haferlach et al46. The expression of FHL2 is significantly downregulated in AML patients compared to the healthy individuals (Figure 7A). We also determined the expression of FHL2 in BM cells from 46 unselected patients with MDS and 10 control individuals without evidence of MDS or hematological malignancies by qRT-PCR analysis. Overall, FHL2 was down-regulated marginally in BM cells from patients with MDS compared to controls (Figure 7B). However, on average, there was about 4-fold down-regulation of FHL2 in approximately 35% of de novo MDS patients as compared to controls. Together, these data indicate that downregulation of FHL2 is detected in AML and MDS patients.
Figure 7. Downregulation of FHL2 in patients with AML and MDS.
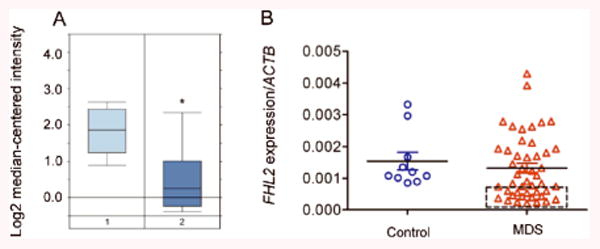
(A) Expression profile of FHL2 transcripts in PB mononuclear cells from 74 healthy individuals (left) and 542 AML patients (right) by microarray analysis 46(Average fold change is -2.288. P=6.28E-23). (B) FHL2 expression level in bone marrow cells from 46 patients with MDS and 10 healthy donors (Control, CTL) by qPCR. ACTB was used as the normalization gene. The square indicates the MDS patients who have a low FHL2 expression level.
FHL2 is regulated in a tissue-specific manner in hematopoietic cells
To examine the expression of FHL2 in CD34+ hematopoietic cells, we performed RT-PCR, and found that the full-length FHL2 transcript is not expressed in CD34+ hematopoietic cells. However, these cells expressed an isoform of FHL2 that does not contain the previously- recognized 5′exon (Figure 8A). 5′-RACE was performed to clone the 5′ end of this isoform of FHL2, as well as the full-length transcript from bone marrow cDNA. The sequence data (GenBank ID: DQ307067) revealed that this isoform of FHL2, designated B-FHL2, encodes the same protein as FHL2, but has a different 5′-UTR. Comparison of B-FHL2 to FHL2 transcripts revealed that FHL2 (gi: 42403584), DRAL (gi:11761688), and B-FHL2 all encode the same peptide, but have unique 5′ flanking sequences (Figure. 8A). A TATA box was identified −813bp upstream of the 5′ sequences of B-FHL2, suggesting that the transcription of B-FHL2 is initiated upstream of the 5′ transcript sequences we cloned, and is regulated by a unique promoter in hematopoietic cells. To determine the tissue distribution of B-FHL2 expression, we performed RT-PCR analysis of a multiple tissue cDNA panel, and found that B-FHL2 is predominantly expressed in bone marrow and testis, is moderately expressed in placenta, and is not expressed in other tissues (Supplementary Figure S6).
Figure 8. Expression of FHL2 is associated with methylation status of the promoter region in myeloid leukemia cells.
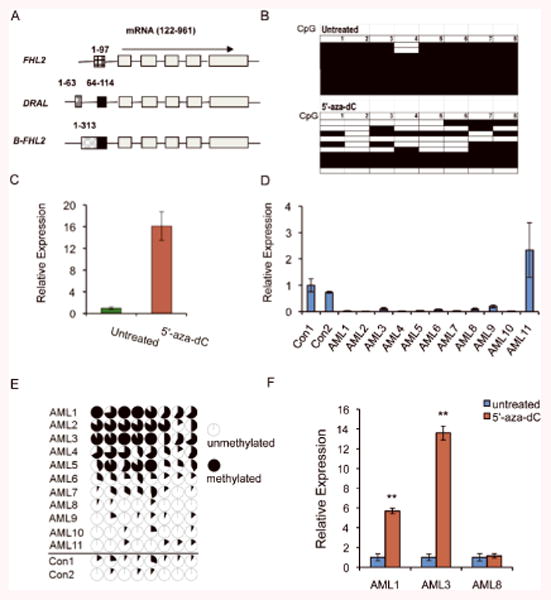
(A) B-FHL2 has a unique promoter region. The boxes represent different exons, and the black line represents intronic sequences. The common exons encoding the FHL2 protein, which are shared among these three transcripts, are shown in grey. The exons encoding the unique 5′ UTR are shown with boxes with different patterns for each transcript. The black boxes within the DRAL and FHL2 transcripts are identical. The numbers indicate the position within the GenBank nucleotide sequence [FHL2 (gi:42403584), DRAL (gi:11761688), and B-FHL2 (DQ307067)]. (B) Bisulfite sequencing of an amplicon from the promoter region of B-FHL2 containing 8 CpG dinucleotides in untreated KG-1 cells or cells treated with 5′-aza-dC. Each row presents a clone. Filled squares indicate methylated CpGs, whereas open squares refer to unmethylated CpGs. (C) Relative expression of B-FHL2 in untreated KG-1 cells, and cells treated with 5′-aza-dC was determined by qRT-PCR. The y-axis represents the ratio of fold-change in gene expression of FHL2 between treated and untreated KG-1 cells. (D) The expression of FHL2 in BM cells from AML patients and healthy individuals, as determined by qRT-PCR. (E) DNA methylation profile of the 8 CpG dinucleotides in the promoter region of B-FHL2 gene in multiple AML samples (AML1-AML11) and 2 control samples (Con1, Con2) from healthy individuals. The cycles represent the average methylation level of a specific CpG dinucleotide. Unmethylated, partially methylated, and fully methylated are indicated by open cycles, partially blacked cycles and blacked cycles respectively. (F) Relative expression of B-FHL2 in primary AML cells before and after treatment with 5′-aza-dC, as determined by qRT-PCR. The y-axis represents the ratio of FHL2 level between treated and untreated cells.
Downregulation of FHL2 in leukemia cells is associated with promoter hypermethylation
Alteration of DNA methylation is associated with aberrant gene silencing in cancer and leukemia 48. FHL2 is under the control of a tissue-specific promoter in bone marrow (Figure 8A). A region containing a CpG island, was identified within the promoter region of B-FHL2 (-1628bp to -1425bp upstream of the 5′ end). qRT-PCR analysis of FHL2 expression showed that FHL2 was expressed at a markedly lower level in KG1 leukemia cells as compared to K562, HL60 and U937 leukemia cells (Supplementary Figure S7). Bisulfite sequencing analysis of the CpG island in the promoter region of B-FHL2 revealed that 99.75% of the CpGs are methylated in untreated KG-1 cells, whereas 52.5% of CpGs are methylated in KG-1 cells treated with the demethylating agent, 5-aza-2′-deoxycytidine (5-aza-dC) (Figure 8B). Thus, this region is highly methylated in untreated KG-1 cells, and can be demethylated significantly after treatment with 5′-aza-dC. To determine if the reduced expression of FHL2 in KG-1 cells is associated with the methylation status of the FHL2 promoter, we performed real-time RT-PCR analysis of FHL2 expression, and found that 5′-aza-dC-treated KG-1 cells have a 15-fold increase in expression of FHL2 as compared to untreated KG-1 cells (Figure 8C). In addition, we also analyzed the FHL2 expression in BM cells from 11 AML patients and 2 healthy individuals. As shown in Figure 8D, FHL2 expression was downregulated in BM cells from the majority of AML patients compared to healthy individuals. However, the promoter region of FHL2 was hypermethylated in BM cells from 6 of 11 AML patients compared to healthy individuals, as determined by bisulfite sequencing (Figure 8E). The hypermethylation of FHL2 promoter was correlated with downregulation of FHL2 in BM cells from these six AML patients. To determine whether hypermethylation of FHL2 promoter directly caused its downregulation, we compared the FHL2 expression in the primary leukemia cells before and after treatment with 5′-aza-dC. We found that 5′-aza-dC markedly induced FHL2 expression in leukemia cells with hypermethylation of FHL2 promoter but not in the cells without FHL2 hypermethylation (Figure 8F). Collectively, these data suggest that the reduced expression of FHL2 in some AMLs is a result of hypermethylation of the promoter region.
Discussion
The role of endogenous Fhl2 in hematopoiesis has not been reported. In this study, we identified a new role for Fhl2 as a critical regulator of HSCs, essential for maintaining HSC quiescence and survival in response to regenerative stress.
Fhl2 has been implicated in tissue regeneration and repair. Deletion of Fhl2 inhibits angiogenic functions of endothelial progenitor cells49 while the lack of Fhl2 perturbed skeletal muscle regeneration by down-regulating the myogenic progenitor cell activity50. Fhl2-deficient mice displayed an impaired intestinal wound healing51 and skin wound healing52 as well as decreased activity of osteroblasts21. We have found that loss of Fhl2 reduces the repopulation capacity of long-term HSC but not short-term HSC and progenitor cells, during hematopoietic regeneration. Competitive repopulation assays also confirmed that Fhl2-null HSCs have a reduced self-renewal capacity. Of note, this study reveals that Fhl2 is involved in hematopoietic regeneration by directly regulating HSC self-renewal capacity. It will be interesting to determine whether Fhl2 also regulates the activity of tissue-specific stem cells during regeneration of nonhematopoietic tissues.
The role of Fhl2 in adult stem cells has not been previously reported. We found that Fhl2-null HSCs had a significantly reduced self-renewal capacity and had engraftment bias toward myeloid cells after serial transplantation, but Fhl2-null HSCs in primary young mice had limited functional defects in proliferation and differentiation, suggesting that Fhl2 is critical for maintaining the self-renewal of HSC in response to regenerative stress but it is dispensable under homeostatic conditions. Quiescence and survival are critical for the maintenance of HSCs. Reduced quiescence of HSCs often results in HSC exhaustion 53. We find that Fhl2 loss does not affect quiescence and survival of HSCs and HPCs in the homeostatic state, but it significantly reduces quiescence and survival of HSCs, but not HPCs, in response to regenerative stress. Thus, it is likely that under stress conditions, Fhl2-loss-induced decrease in quiescence and survival of HSCs under stress leads to the compromised self-renewal capacity of Fhl2-null HSCs. FHL2 is one of the four genes that are most significantly induced in human peripheral blood lymphocytes after irradiation ex vivo54, 55, indicating that FHL2 is responsive to environmental stress. FHL2 is likely to act as a critical regulator of hematopoietic homeostasis in response to various stresses. Previous studies showed that Fhl2 is dispensable for normal cardiac development but modifies responses to certain stress conditions in the adult heart56. Collectively, these data suggest that Fhl2 plays an important role in stress responses in multiple-tissues.
FHL2 is an important regulator of cell cycle and proliferation. Fhl2 loss inhibits the proliferation of embryonic fibroblasts by down-regulating both positive and negative regulators of cell cycle36. In addition, Fhl2 regulates p21 (Cip1) expression in breast cancer cells35, and it acts as a negative regulator of E4F1, a key player in control of cell proliferation in mammalian cells34. Here, we document a role of Fhl2 as an important regulator of the cell cycle of HSCs in response to stress. Of note, all three members of the CIP/KIP family including p21 (Cip1), p27(Kip1) and p57(Kip2), are down-regulated in Fhl2-null LSKs after transplantation. The CIP/KIP family functions as a negative regulator of G1-S progression by interacting with different cyclin-CDK complexes57. p21(Cip1) was initially reported to play a role in regulating HSC quiescence40 whereas recent studies showed that p21(Cip1) is critical for maintaining HSC quiescence under conditions of cellular stress rather than steady state58, 59. p27 (Kip1) deficiency alone has a limited effect on the regulation of HSC cell cycle43. However, absence of p27(Kip1) accelerated the defective quiescence phenotype of p57(Kip2)-null HSCs44, 45. Our results showed that forced expression of p21 (Cip1) or p27 (kip1) but not p57 (kip2) partially rescued the decreased quiescence of Fhl2-deficient LSKs. Therefore, it is likely that downregulation of p21 (Cip1) and p27 (kip1) at least partially mediates Fhl2 deletion-induced quiescence loss in HSC-enriched population. Both p27(Kip1) and p57(Kip2) are direct targets of the TGF-β pathway60, which can be regulated by Fhl217. In addition, a previous study has shown that FHL2 upregulates expression of p21 through the MAPK pathway in breast cancer cells35. Additional studies are necessary to determine whether FHL2 regulates CDK inhibitors through either or both of the MAPK and TGF-β pathways in HSCs.
In this study, we cloned a unique 5′ flanking sequence of the FHL2 transcript in hematopoietic cells. Analysis of the 5′-UTR regions of FHL2, DRAL, and B-FHL2 revealed that the FHL2 isoforms identified from different tissues each contain a unique 5′ UTR region, indicating that the transcription of FHL2 is under the control of different promoters, and is likely to be regulated by different transcriptional regulatory cis- and trans-elements in a variety of tissues. Collectively, transcriptional regulation of FHL2 expression is dependent on cell context, which may be critical for the role of FHL2 in “fine-tuning” a diversity of cellular processes. In addition, a CpG island was identified in the promoter region of B-FHL2. We demonstrated that hypermethylation of the promoter region of FHL2 results in reduced expression of FHL2 in the KG-1 myeloid leukemia cell line and primary AML cells, indicating that FHL2 expression is regulated epigenetically. Our findings indicate that FHL2 is down-regulated in a subgroup of AML and MDS patients. DNA hypermethylation is highly associated with both AML and MDS. It is likely that down-regulation of FHL2 in a subgroup of MDS or AML patients also results from an aberrant DNA methylation.
In summary, our studies revealed that the expression level of FHL2 is critical for maintaining HSC quiescence and survival under stress, and that deregulation of FHL2 may predispose to the development of hematopoietic disorders, and may cooperate with mutations predisposing to leukemogenesis.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health grants RO1 CA140979 (to Z.Q.) We thank the staffs in the UIC flow core facility for their assistance in cell sorting.
Footnotes
Online Supplementary Material: Supplementary Material and Methods and Data can be found with this article online.
Conflict of interest disclosure: The authors declare no competing financial interests
References
- 1.Genini M, Schwalbe P, Scholl FA, Remppis A, Mattei MG, Schafer BW. Subtractive cloning and characterization of DRAL, a novel LIM-domain protein down-regulated in rhabdomyosarcoma. DNA Cell Biol. 1997;16:433–442. doi: 10.1089/dna.1997.16.433. [DOI] [PubMed] [Google Scholar]
- 2.Johannessen M, Moller S, Hansen T, Moens U, Van Ghelue M. The multifunctional roles of the four-and-a-half-LIM only protein FHL2. Cellular and molecular life sciences : CMLS. 2006;63:268–284. doi: 10.1007/s00018-005-5438-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chu PH, Ruiz-Lozano P, Zhou Q, Cai C, Chen J. Expression patterns of FHL/SLIM family members suggest important functional roles in skeletal muscle and cardiovascular system. Mech Dev. 2000;95:259–265. doi: 10.1016/s0925-4773(00)00341-5. [DOI] [PubMed] [Google Scholar]
- 4.Morgan MJ, Madgwick AJ. The fourth member of the FHL family of LIM proteins is expressed exclusively in the testis. Biochem Biophys Res Commun. 1999;255:251–255. doi: 10.1006/bbrc.1999.0180. [DOI] [PubMed] [Google Scholar]
- 5.Morgan MJ, Madgwick AJ. The LIM proteins FHL1 and FHL3 are expressed differently in skeletal muscle. Biochem Biophys Res Commun. 1999;255:245–250. doi: 10.1006/bbrc.1999.0179. [DOI] [PubMed] [Google Scholar]
- 6.Tanahashi H, Tabira T. Alzheimer's disease-associated presenilin 2 interacts with DRAL, an LIM-domain protein. Hum Mol Genet. 2000;9:2281–2289. doi: 10.1093/oxfordjournals.hmg.a018919. [DOI] [PubMed] [Google Scholar]
- 7.Amaar YG, Thompson GR, Linkhart TA, Chen ST, Baylink DJ, Mohan S. Insulin-like growth factor-binding protein 5 (IGFBP-5) interacts with a four and a half LIM protein 2 (FHL2) J Biol Chem. 2002;277:12053–12060. doi: 10.1074/jbc.M110872200. [DOI] [PubMed] [Google Scholar]
- 8.Wixler V, Geerts D, Laplantine E, Westhoff D, Smyth N, Aumailley M, et al. The LIM-only protein DRAL/FHL2 binds to the cytoplasmic domain of several alpha and beta integrin chains and is recruited to adhesion complexes. J Biol Chem. 2000;275:33669–33678. doi: 10.1074/jbc.M002519200. [DOI] [PubMed] [Google Scholar]
- 9.Muller JM, Isele U, Metzger E, Rempel A, Moser M, Pscherer A, et al. FHL2, a novel tissue-specific coactivator of the androgen receptor. Embo J. 2000;19:359–369. doi: 10.1093/emboj/19.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morlon A, Sassone-Corsi P. The LIM-only protein FHL2 is a serum-inducible transcriptional coactivator of AP-1. Proc Natl Acad Sci U S A. 2003;100:3977–3982. doi: 10.1073/pnas.0735923100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fimia GM, De Cesare D, Sassone-Corsi P. A family of LIM-only transcriptional coactivators: tissue-specific expression and selective activation of CREB and CREM. Mol Cell Biol. 2000;20:8613–8622. doi: 10.1128/mcb.20.22.8613-8622.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McLoughlin P, Ehler E, Carlile G, Licht JD, Schafer BW. The LIM-only protein DRAL/FHL2 interacts with and is a corepressor for the promyelocytic leukemia zinc finger protein. J Biol Chem. 2002;277:37045–37053. doi: 10.1074/jbc.M203336200. [DOI] [PubMed] [Google Scholar]
- 13.Chen D, Xu W, Bales E, Colmenares C, Conacci-Sorrell M, Ishii S, et al. SKI activates Wnt/beta-catenin signaling in human melanoma. Cancer Res. 2003;63:6626–6634. [PubMed] [Google Scholar]
- 14.Martin B, Schneider R, Janetzky S, Waibler Z, Pandur P, Kuhl M, et al. The LIM-only protein FHL2 interacts with beta-catenin and promotes differentiation of mouse myoblasts. J Cell Biol. 2002;159:113–122. doi: 10.1083/jcb.200202075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei Y, Renard CA, Labalette C, Wu Y, Levy L, Neuveut C, et al. Identification of the LIM protein FHL2 as a coactivator of beta-catenin. J Biol Chem. 2003;278:5188–5194. doi: 10.1074/jbc.M207216200. [DOI] [PubMed] [Google Scholar]
- 16.Purcell NH, Darwis D, Bueno OF, Muller JM, Schule R, Molkentin JD. Extracellular signal-regulated kinase 2 interacts with and is negatively regulated by the LIM-only protein FHL2 in cardiomyocytes. Mol Cell Biol. 2004;24:1081–1095. doi: 10.1128/MCB.24.3.1081-1095.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding L, Wang Z, Yan J, Yang X, Liu A, Qiu W, et al. Human four-and-a-half LIM family members suppress tumor cell growth through a TGF-beta-like signaling pathway. J Clin Invest. 2009;119:349–361. doi: 10.1172/JCI35930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi X, Bowlin KM, Garry DJ. Fhl2 interacts with Foxk1 and corepresses Foxo4 activity in myogenic progenitors. Stem Cells. 28:462–469. doi: 10.1002/stem.274. [DOI] [PubMed] [Google Scholar]
- 19.Du X, Hublitz P, Gunther T, Wilhelm D, Englert C, Schule R. The LIM-only coactivator FHL2 modulates WT1 transcriptional activity during gonadal differentiation. Biochim Biophys Acta. 2002;1577:93–101. doi: 10.1016/s0167-4781(02)00414-1. [DOI] [PubMed] [Google Scholar]
- 20.Bai S, Kitaura H, Zhao H, Chen J, Muller JM, Schule R, et al. FHL2 inhibits the activated osteoclast in a TRAF6-dependent manner. J Clin Invest. 2005;115:2742–2751. doi: 10.1172/JCI24921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gunther T, Poli C, Muller JM, Catala-Lehnen P, Schinke T, Yin N, et al. Fhl2 deficiency results in osteopenia due to decreased activity of osteoblasts. Embo J. 2005;24:3049–3056. doi: 10.1038/sj.emboj.7600773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Philippar U, Schratt G, Dieterich C, Muller JM, Galgoczy P, Engel FB, et al. The SRF target gene Fhl2 antagonizes RhoA/MAL-dependent activation of SRF. Mol Cell. 2004;16:867–880. doi: 10.1016/j.molcel.2004.11.039. [DOI] [PubMed] [Google Scholar]
- 23.Stilo R, Leonardi A, Formisano L, Di Jeso B, Vito P, Liguoro D. TUCAN/CARDINAL and DRAL participate in a common pathway for modulation of NF-kappaB activation. FEBS Lett. 2002;521:165–169. doi: 10.1016/s0014-5793(02)02869-7. [DOI] [PubMed] [Google Scholar]
- 24.Kinoshita M, Nakagawa T, Shimizu A, Katsuoka Y. Differently regulated androgen receptor transcriptional complex in prostate cancer compared with normal prostate. International journal of urology : official journal of the Japanese Urological Association. 2005;12:390–397. doi: 10.1111/j.1442-2042.2005.01093.x. [DOI] [PubMed] [Google Scholar]
- 25.Gabriel B, Mildenberger S, Weisser CW, Metzger E, Gitsch G, Schule R, et al. Focal adhesion kinase interacts with the transcriptional coactivator FHL2 and both are overexpressed in epithelial ovarian cancer. Anticancer Res. 2004;24:921–927. [PubMed] [Google Scholar]
- 26.Yan J, Zhu J, Zhong H, Lu Q, Huang C, Ye Q. BRCA1 interacts with FHL2 and enhances FHL2 transactivation function. FEBS Lett. 2003;553:183–189. doi: 10.1016/s0014-5793(03)00978-5. [DOI] [PubMed] [Google Scholar]
- 27.Dahan J, Nouet Y, Jouvion G, Levillayer F, Adib-Conquy M, Cassard-Doulcier AM, et al. LIM-Only Protein FHL2 Activates NF-kappaB Signaling in the Control of Liver Regeneration and Hepatocarcinogenesis. Mol Cell Biol. 2013;33:3299–3308. doi: 10.1128/MCB.00105-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qian Z, Mao L, Fernald AA, Yu H, Luo R, Jiang Y, et al. Enhanced expression of FHL2 leads to abnormal myelopoiesis in vivo. Leukemia. 2009;23:1650–1657. doi: 10.1038/leu.2009.78. [DOI] [PubMed] [Google Scholar]
- 29.Qian Z, Chen L, Fernald AA, Williams BO, Le Beau MM. A critical role for Apc in hematopoietic stem and progenitor cell survival. J Exp Med. 2008;205:2163–2175. doi: 10.1084/jem.20080578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Foudi A, Jarrier P, Zhang Y, Wittner M, Geay JF, Lecluse Y, et al. Reduced retention of radioprotective hematopoietic cells within the bone marrow microenvironment in CXCR4-/- chimeric mice. Blood. 2006;107:2243–2251. doi: 10.1182/blood-2005-02-0581. [DOI] [PubMed] [Google Scholar]
- 31.Qian Z, Fernald AA, Godley LA, Larson RA, Le Beau MM. Expression profiling of CD34+ hematopoietic stem/ progenitor cells reveals distinct subtypes of therapy-related acute myeloid leukemia. Proc Natl Acad Sci U S A. 2002;99:14925–14930. doi: 10.1073/pnas.222491799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chu PH, Bardwell WM, Gu Y, Ross J, Jr, Chen J. FHL2 (SLIM3) is not essential for cardiac development and function. Mol Cell Biol. 2000;20:7460–7462. doi: 10.1128/mcb.20.20.7460-7462.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–197. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- 34.Paul C, Lacroix M, Iankova I, Julien E, Schafer BW, Labalette C, et al. The LIM-only protein FHL2 is a negative regulator of E4F1. Oncogene. 2006;25:5475–5484. doi: 10.1038/sj.onc.1209567. [DOI] [PubMed] [Google Scholar]
- 35.Martin BT, Kleiber K, Wixler V, Raab M, Zimmer B, Kaufmann M, et al. FHL2 regulates cell cycle-dependent and doxorubicin-induced p21Cip1/Waf1 expression in breast cancer cells. Cell Cycle. 2007;6:1779–1788. doi: 10.4161/cc.6.14.4448. [DOI] [PubMed] [Google Scholar]
- 36.Labalette C, Nouet Y, Sobczak-Thepot J, Armengol C, Levillayer F, Gendron MC, et al. The LIM-only protein FHL2 regulates cyclin D1 expression and cell proliferation. J Biol Chem. 2008;283:15201–15208. doi: 10.1074/jbc.M800708200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brun J, Fromigue O, Dieudonne FX, Marty C, Chen J, Dahan J, et al. The LIM-only protein FHL2 controls mesenchymal cell osteogenic differentiation and bone formation through Wnt5a and Wnt10b. Bone. 2013;53:6–12. doi: 10.1016/j.bone.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 38.Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12:446–451. doi: 10.1038/nm1388. [DOI] [PubMed] [Google Scholar]
- 39.Harrison DE, Stone M, Astle CM. Effects of transplantation on the primitive immunohematopoietic stem cell. J Exp Med. 1990;172:431–437. doi: 10.1084/jem.172.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 41.Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 42.Miyamoto K, Araki KY, Naka K, Arai F, Takubo K, Yamazaki S, et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 2007;1:101–112. doi: 10.1016/j.stem.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 43.Cheng T, Rodrigues N, Dombkowski D, Stier S, Scadden DT. Stem cell repopulation efficiency but not pool size is governed by p27(kip1) Nat Med. 2000;6:1235–1240. doi: 10.1038/81335. [DOI] [PubMed] [Google Scholar]
- 44.Matsumoto A, Takeishi S, Kanie T, Susaki E, Onoyama I, Tateishi Y, et al. p57 is required for quiescence and maintenance of adult hematopoietic stem cells. Cell Stem Cell. 2011;9:262–271. doi: 10.1016/j.stem.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 45.Zou P, Yoshihara H, Hosokawa K, Tai I, Shinmyozu K, Tsukahara F, et al. p57(Kip2) and p27(Kip1) cooperate to maintain hematopoietic stem cell quiescence through interactions with Hsc70. Cell Stem Cell. 2011;9:247–261. doi: 10.1016/j.stem.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 46.Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Bene MC, et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol. 2010;28:2529–2537. doi: 10.1200/JCO.2009.23.4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilson CS, Davidson GS, Martin SB, Andries E, Potter J, Harvey R, et al. Gene expression profiling of adult acute myeloid leukemia identifies novel biologic clusters for risk classification and outcome prediction. Blood. 2006 doi: 10.1182/blood-2004-12-4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bhalla KN. Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. J Clin Oncol. 2005;23:3971–3993. doi: 10.1200/JCO.2005.16.600. [DOI] [PubMed] [Google Scholar]
- 49.Huang PH, Chen CY, Lin CP, Wang CH, Tsai HY, Lo WY, et al. Deletion of FHL2 gene impaired ischemia-induced blood flow recovery by modulating circulating proangiogenic cells. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:709–717. doi: 10.1161/ATVBAHA.112.300318. [DOI] [PubMed] [Google Scholar]
- 50.Shi X, Bowlin KM, Garry DJ. Fhl2 interacts with Foxk1 and corepresses Foxo4 activity in myogenic progenitors. Stem Cells. 2010;28:462–469. doi: 10.1002/stem.274. [DOI] [PubMed] [Google Scholar]
- 51.Kirfel J, Pantelis D, Kabba M, Kahl P, Roper A, Kalff JC, et al. Impaired intestinal wound healing in Fhl2-deficient mice is due to disturbed collagen metabolism. Experimental cell research. 2008;314:3684–3691. doi: 10.1016/j.yexcr.2008.09.023. [DOI] [PubMed] [Google Scholar]
- 52.Wixler V, Hirner S, Muller JM, Gullotti L, Will C, Kirfel J, et al. Deficiency in the LIM-only protein Fhl2 impairs skin wound healing. J Cell Biol. 2007;177:163–172. doi: 10.1083/jcb.200606043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pietras EM, Warr MR, Passegue E. Cell cycle regulation in hematopoietic stem cells. J Cell Biol. 2011;195:709–720. doi: 10.1083/jcb.201102131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kang CM, Park KP, Song JE, Jeoung DI, Cho CK, Kim TH, et al. Possible biomarkers for ionizing radiation exposure in human peripheral blood lymphocytes. Radiat Res. 2003;159:312–319. doi: 10.1667/0033-7587(2003)159[0312:pbfire]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 55.Paul S, Amundson SA. Gene expression signatures of radiation exposure in peripheral white blood cells of smokers and non-smokers. International journal of radiation biology. 2011;87:791–801. doi: 10.3109/09553002.2011.568574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kong Y, Shelton JM, Rothermel B, Li X, Richardson JA, Bassel-Duby R, et al. Cardiac-specific LIM protein FHL2 modifies the hypertrophic response to beta-adrenergic stimulation. Circulation. 2001;103:2731–2738. doi: 10.1161/01.cir.103.22.2731. [DOI] [PubMed] [Google Scholar]
- 57.Denicourt C, Dowdy SF. Cip/Kip proteins: more than just CDKs inhibitors. Genes Dev. 2004;18:851–855. doi: 10.1101/gad.1205304. [DOI] [PubMed] [Google Scholar]
- 58.van Os R, Kamminga LM, Ausema A, Bystrykh LV, Draijer DP, van Pelt K, et al. A Limited role for p21Cip1/Waf1 in maintaining normal hematopoietic stem cell functioning. Stem Cells. 2007;25:836–843. doi: 10.1634/stemcells.2006-0631. [DOI] [PubMed] [Google Scholar]
- 59.Foudi A, Hochedlinger K, Van Buren D, Schindler JW, Jaenisch R, Carey V, et al. Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat Biotechnol. 2009;27:84–90. doi: 10.1038/nbt.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Scandura JM, Boccuni P, Massague J, Nimer SD. Transforming growth factor beta-induced cell cycle arrest of human hematopoietic cells requires p57KIP2 up-regulation. Proc Natl Acad Sci U S A. 2004;101:15231–15236. doi: 10.1073/pnas.0406771101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.