

Abstract
The molecular mechanisms of membrane merger during somatic cell fusion in eukaryotic species are poorly understood. In the filamentous fungus Neurospora crassa, somatic cell fusion occurs between genetically identical germinated asexual spores (germlings) and between hyphae to form the interconnected network characteristic of a filamentous fungal colony. In N. crassa, two proteins have been identified to function at the step of membrane fusion during somatic cell fusion: PRM1 and LFD-1. The absence of either one of these two proteins results in an increase of germling pairs arrested during cell fusion with tightly appressed plasma membranes and an increase in the frequency of cell lysis of adhered germlings. The level of cell lysis in ΔPrm1 or Δlfd-1 germlings is dependent on the extracellular calcium concentration. An available transcriptional profile data set was used to identify genes encoding predicted transmembrane proteins that showed reduced expression levels in germlings cultured in the absence of extracellular calcium. From these analyses, we identified a mutant (lfd-2, for late fusion defect-2) that showed a calcium-dependent cell lysis phenotype. lfd-2 encodes a protein with a Fringe domain and showed endoplasmic reticulum and Golgi membrane localization. The deletion of an additional gene predicted to encode a low-affinity calcium transporter, fig1, also resulted in a strain that showed a calcium-dependent cell lysis phenotype. Genetic analyses showed that LFD-2 and FIG1 likely function in separate pathways to regulate aspects of membrane merger and repair during cell fusion.
INTRODUCTION
Membrane fusion plays an important role in intracellular trafficking of vesicles, exo- and endocytosis, fertilization, the entry of enveloped animal viruses into their host cell, and syncytium formation during developmental processes (1–3). In intracellular vesicle trafficking, SNARE proteins facilitate membrane merger by forming coiled-coil interactions between v-SNARES on vesicles and t-SNARES on target membranes to bring membranes into close proximity (1, 4). The mechanisms that direct cell-cell fusion are less well understood, although genetic screens have identified candidate molecules, termed fusogens, which are membrane-incorporated proteins that facilitate membrane fusion. For example, in Caenorhabditis elegans, two fusogenic glycoproteins, EFF-1 and AFF-1, are necessary and sufficient to fuse cells (5, 6), but their conservation is restricted to nematodes and related organisms (7).
In the yeast Saccharomyces cerevisiae, a number of proteins that are involved in mating cell fusion have been identified (8–14). Mating pairs between strains bearing deletions of Prm1p or Fig1p show tightly appressed plasma membranes, indicating a defect in membrane fusion (10, 12). Mating pairs of prm1Δ or fig1Δ strains also show increased cell lysis, the frequency of which is influenced by the concentration of extracellular calcium (12, 15). However, Prm1p and Fig1p are not fusogens, as neither is necessary or sufficient for membrane merger. In Schizosaccharomyces pombe, prm1 also plays a role in mating cell fusion. However, unlike S. cerevisiae prm1Δ mutants, S. pombe prm1Δ mutants are almost completely blocked in cell fusion and display an abnormal distribution of plasma membrane and cell wall in the area of cell-cell interaction (16).
In the filamentous fungus Neurospora crassa, cell-cell fusion plays an essential role during both sexual and vegetative development. During vegetative growth, cell fusion can occur between genetically identical germinated asexual spores (germlings) as well as between hyphae within a single colony. Cell-cell fusion allows the formation of the syncytial hyphal network that is characteristic of this group of organisms (17–20). Two proteins in N. crassa that play a role in membrane fusion have been identified: PRM1-like (PRM1) and LFD-1 (for late fusion defect-1). PRM1 is the ortholog of S. cerevisiae Prm1p, and N. crassa ΔPrm1 mutants have a defect in membrane fusion similar to that observed for S. cerevisiae prm1Δ mutants (21). LFD-1 was recently identified as playing a role in membrane merger by screening for genes regulated by PP-1, a transcription factor that regulates genes associated with cell fusion in N. crassa (22, 23). In addition to defects in membrane merger, the absence of PRM1 or LFD-1 also led to significant cell lysis of adhered germlings. The cell lysis defect in ΔPrm1 and Δlfd-1 cells was suppressed by the addition of extracellular calcium and was shown to be dependent upon SYT1, a synaptotagmin homolog involved in calcium-mediated membrane damage repair (22). These observations suggest that membrane damage is associated with membrane merger and that PRM1 and LFD-1 play a role in preventing catastrophic membrane damage during cell fusion in N. crassa.
The observation that calcium plays an important role in cell lysis during fusion in N. crassa prompted us to look for new genes involved in membrane fusion. Our strategy relied on the identification of genes encoding predicted transmembrane proteins and which showed a significant decrease in expression level in the absence of extracellular calcium (24). We screened 29 strains carrying deletions of candidate genes for defects in membrane merger and cell lysis during germling fusion. We identified 2 genes that, when deleted, resulted in strains that showed increased lysis during germling fusion. One of them, encoding a single-pass transmembrane protein, LFD-2, showed calcium-dependent cell lysis and contains a Fringe domain. Fringe has been implicated in the activation of epidermal growth factor receptors, such as Notch, in metazoan species (25). The Δlfd-2 germling fusion phenotype, LFD-2 localization, and genetic interactions of lfd-2 with Prm1, lfd-1, and a predicted low-affinity calcium transporter, fig1, were evaluated. Our study indicates that the process of membrane merger during germling fusion is a complex process requiring many proteins that play a role in mediating the fidelity of the cell fusion process.
MATERIALS AND METHODS
Membrane fusion and cell lysis screening.
The deletion mutants used in this study were generated by the Neurospora Genome Project (26, 27) (Table 1) and were obtained from the Fungal Genetics Stock Center (FGSC) (28). Strains were grown on Vogel's minimal media (VMM) (29) slant tubes for 4 to 6 days or until significant conidiation occurred. Conidial suspensions were prepared as described in Palma-Guerrero et al. (22). Homokaryotic deletion strains were obtained via microconidiation selection (Table 1) (26, 27). Conidia were diluted to a concentration of 3 × 107 conidia/ml, and 300 μl of conidial suspension was spread onto VMM plates. Plates were incubated for 5 h at 30°C, and 1-cm squares were excised and treated with 4 μM FM4-64 for screening for membrane fusion defects and cell lysis. Samples were observed with a Zeiss Axioskop 2 using a 403 Plan-Neofluar oil immersion objective. A rhodamine filter set (excitation at 543 nm and emission at 590 nm) was used for detecting FM4-64 fluorescence. Images were captured using a Hamamatsu digital camera C8484 (Hamamatsu, Japan) on a Zeiss Axio Imager microscope and analyzed using iVision Mac4.5 software.
TABLE 1.
Strains used in this study
| Strain | Genotype | Origin or reference |
|---|---|---|
| FGSC2489 | A | FGSC |
| FGSC21584 | ΔNCU10610::hph+ A | FGSC |
| FGSC15199 | ΔNCU01382::hph+ a | FGSC |
| FGSC14575 | ΔNCU01697::hph+ A | FGSC |
| FGSC20488 | ΔNCU07802::hph+ A | FGSC |
| FGSC11423 | ΔNCU08447::hph+ A | FGSC |
| FGSC21874 | ΔNCU09562::hph+ a | FGSC |
| FGSC17749 | ΔNCU06034::hph+ A | FGSC |
| FGSC14335 | ΔNCU07235::hph+ A | FGSC |
| FGSC17420 | ΔNCU03863::hph+ A | FGSC |
| FGSC14419 | ΔNCU06130::hph+ A | FGSC |
| FGSC18043 | ΔNCU04872::hph+ a (heterokaryon) | FGSC |
| 4872 | ΔNCU04872::hph+ a (homokaryon) | This study |
| FGSC14037 | ΔNCU03132::hph+ a | FGSC |
| FGSC13071 | ΔNCU04736::hph+ A | FGSC |
| FGSC22512 | ΔNCU09955::hph+ A (heterokaryon) | FGSC |
| 9955 | ΔNCU09955::hph+ A (homokaryon) | This study |
| FGSC17821 | ΔNCU05915::hph+ A | FGSC |
| FGSC17765 | ΔNCU05798::hph+ A | FGSC |
| FGSC11249 | ΔNCU07075::hph+ A | FGSC |
| FGSC19479 | ΔNCU08870::hph+ A | FGSC |
| FGSC11148 | ΔNCU07769::hph+ a | FGSC |
| FGSC16642 | ΔNCU00832::hph+ A | FGSC |
| FGSC13466 | ΔNCU01289::hph+ A | FGSC |
| FGSC19337 | ΔNCU08524::hph+ a | FGSC |
| FGSC14995 | ΔNCU06116::hph+ a | FGSC |
| FGSC21689 | ΔNCU07582::hph+ A | FGSC |
| FGSC19267 | ΔNCU02191::hph+ A | FGSC |
| FGSC15048 | ΔNCU03192::hph+ a | FGSC |
| FGSC18602 | ΔNCU09852::hph+ A | FGSC |
| FGSC17778 | ΔNCU05826::hph+ A | FGSC |
| FGSC 15811 | ΔNCU06328::hph+ A | FGSC |
| FGSC17683 | ΔNCU05629::hph+ a | FGSC |
| FGSC12093 | ΔNCU05681::hph+ A | FGSC |
| FGSC12948 | ΔNCU06912::hph+ A | FGSC |
| FGSC6103 | his-3 A | FGSC |
| JPG6 | Δlfd-1::hph+ a | 22 |
| A32 | ΔPrm1::hph+ A | 21 |
| A9 | his-3+::Pccg1-gfp A | 21, 67 |
| A10 | his-3+::Pccg1-dsRed A | 67 |
| A7 | his-3+::Pccg1-gfp; ΔPrm1::hph+; Δmus-51::bar A | 67 |
| A8 | his-3+::Pccg1-dsRed; ΔPrm1::hph+; Δmus-51::bar A | 67 |
| JPG7 | Δlfd-1::hph+ his-3+::Pccg1-gfp a | 22 |
| JPG8 | Δlfd-1::hph+ his-3+::Pccg1-mCherry a | 22 |
| JPG26 | Δlfd-1::hph+ his-3+::Pccg1-gfp; ΔPrm1::hph+ a | 22 |
| JPG27 | Δlfd-1::hph+ his-3+::Pccg1-mCherry; ΔPrm1::hph+ a | 22 |
| Δlfd-2 his-3 | his-3 Δlfd-2::hph+A | This study |
| Δlfd-2 GFP | his-3+::Pccg1-gfp Δlfd-2::hph+ A | This study |
| Δlfd-2 Cherry | his-3+::Pccg1-mCherry Δlfd-2::hph+ A | This study |
| Δlfd-1Δlfd-2 GFP | Δlfd-1::hph+ his-3+::Pccg1-gfp; Δlfd-2::hph+ a | This study |
| Δlfd-1Δlfd-2 Cherry | Δlfd-1::hph+ his-3+::Pccg1-mCherry; Δlfd-2::hph+ a | This study |
| ΔPrm1Δlfd-2 GFP | his-3+::Pccg1-gfp ΔPrm1::hph+; Δlfd-2::hph+ a | This study |
| ΔPrm1Δlfd-2 Cherry | his-3+::Pccg1-mCherry ΔPrm1::hph+; Δlfd-2::hph+ a | This study |
| FGSC17273 | Δfig1::hph+ a | FGSC |
| Δfig1 his3 | his-3 Δfig1::hph+ A | This study |
| Δfig1 GFP | his-3+::Pccg1-gfp Δfig1::hph+ A | This study |
| Δfig1 Cherry | his-3+::Pccg1-mCherry Δfig1::hph+ A | This study |
| Δlfd-1Δfig1 GFP | Δlfd-1::hph+ his-3+::Pccg1-gfp; Δfig1::hph+ a | This study |
| Δlfd-1Δfig1 mCherry | Δlfd-1::hph+ his-3+::Pccg1-mCherry; Δfig1::hph+ a | This study |
| ΔPrm1Δfig1 GFP | Δlfd-1::hph+ his-3+::Pccg1-gfp; Δfig1::hph+ a | This study |
| ΔPrm1Δfig1 mCherry | his-3+::Pccg1-mCherry; ΔPrm-1::hph+; Δfig1::hph+ a | This study |
| Δlfd-1ΔPrm1Δfig1 GFP | Δlfd-1::hph+ his-3+::Pccg1-gfp; ΔPrm1::hph+; Δfig1::hph+ a | This study |
| Δlfd-1ΔPrm1Δfig1 mCherry | Δlfd-1::hph+ his-3+::Pccg1-mCherry; ΔPrm1::hph+; Δfig1::hph+ a | This study |
| tef1-lfd-2-gfp | Δlfd-2::hph+ his-3+::PTef1-lfd-2-gfp A | This study |
| Sec61-mCherry | his-3+::Pccg1-sec61-mCherry A | This study |
| TSF314 | ΔNCU01342::hph+ csr+1::Pgpd-ncu01342-mCherry-6xHis A | This study |
| RFP-VAM-3 | his-3+::Pccg1-rfp-vam-3 A | 36 |
| RFP-VPS-52 | his-3+::Pccg1-rfp-vps-52 A | 36 |
For lysis experiments in reduced calcium, VMM containing a 0.34 mM final concentration of CaCl2 · 2H2, which is half of the normal CaCl2 · 2H2O concentration in the VMM, was used (29). Germlings were prepared and examined as described above, using 0.002% methylene blue to differentiate lysed from nonlysed cells. The same procedure was used to evaluate lysis under increased calcium concentrations (2× = 1.28 mM and 5× = 3.2 mM). For testing cell lysis under osmotic stabilization, 1 M sorbitol was added to the growth media, and germling pairs were examined as described above.
For examining lysis during hyphal fusion, VMM plates were inoculated in the center with conidia and incubated for 16 to 24 h. One-cm squares were excised from the area between the center of the colony and the tips, and the agar squares were treated with 0.002% methylene blue. Images were captured using a Qimaging Micropublisher 5.0 digital camera (Qimaging, Canada) on a Zeiss Axio Imager microscope and analyzed using iVision Mac4.5 software.
Strain and plasmid construction.
Strains constructed for this study are listed in Table 1. To obtain Δlfd-2 strains expressing cytoplasmic markers, the his-3 A strain (FGSC 6103) was used as the female in crosses with Δlfd-2 a. Progeny carrying the deletion mutation and the his-3 marker were used for transformation, with either pMF272 (30, 31) or the modified version of pMF272 containing mCherry instead of green fluorescent protein (GFP), as described in Palma-Guerrero et al. (22). Macroconidia were transformed by electroporation as described by Margolin et al. (32).
To obtain double mutants with ΔPrm1, crosses were performed with a ΔPrm1 strain complemented at the his-3 locus with ccg1-Prm1-gfp (A24) (Table 1). ΔPrm1 mutants are sterile both as a female and as a male due to dominant postfertilization defects in the ΔPrm1 mutant (21). The Δfig1 mutant is female sterile (33); therefore, it was used as a male in crosses.
For Δlfd-2 complementation and GFP tagging of LFD-2 for localization studies, the open reading frame (ORF) of NCU02191 was amplified by PCR, as annotated by the N. crassa database (http://www.broadinstitute.org/annotation/genome/neurospora/MultiHome.html), using genomic DNA with primers that incorporated XbaI/PacI sites. Due to the low expression levels of lfd-2 in germlings and hyphae, we constructed an lfd-2 allele under the regulation of the tef1 promoter by replacing Prm1 with lfd-2 in the tef1-Prm1-GFP plasmid (provided by A. Fleissner, Technische Universitat Braunschweig, Germany) using the XbaI/PacI sites.
The plasmid containing sec61-mCherry under the ccg-1 promoter was constructed by introducing the ORF of sec61 (NCU08897) into the modified version of pMF272 containing mCherry instead of GFP, as described in Palma-Guerrero et al. (22), using the XbaI/PacI sites.
A strain expressing the mCherry-tagged endoplasmic reticulum (ER) protein ERV25 (NCU01342) was generated by targeting a constitutively expressed version of ERV25-mCherry-6× His to the CSR1 locus of a mutant strain lacking the erv25 open reading frame. To achieve this, the pCSR1 vector (34) was modified to contain promoter and terminator elements, as well as an expanded multiple cloning site, into which mCherry-6× His and erv25 fragments were sequentially cloned. Primer pair OTS336/OTS337 (AAACTGCAGAGCGCATTTCCGACGTTAAG and AAAGCGGCCGCTTTGATT TCTGTGATGTG), incorporating PstI and NotI restriction sites, respectively, was used to amplify a 1-kb promoter fragment immediately upstream of the predicted open reading frame of the gene encoding glyceraldehyde 3-phosphate dehydrogenase (GPD) from Myceliophthora thermophila genomic DNA (nucleotides 1,538,499 to 1,539,504 of chromosome 7). Insertion of this promoter fragment into the pCSR1 vector resulted in plasmid pCSR1-GPD. This plasmid was further modified to incorporate a new multiple cloning site (MCS) and the trpC terminator from Aspergillus nidulans. Primer pair OTS340/OTS341 (AAAGCGGCCGCCCTGCAGGAATCGATCCTAGGTGTACAGCATGCTTAATTAATTTAATAGCTCCATGTCAACAA and AAACCGCGGGGAGCATTCACTAGGCAACCATGGT) and template plasmid pMFP26 (35) were used to generate a multiple cloning site (MCS)/terminator fragment with NotI and SacII sites at the 5′ and 3′ ends, respectively. The new MCS, appended onto primer OTS340, included NotI, SbfI, ClaI, AvrII, BsrGI, SphI, and PacI restriction sites. Cloning of the MCS/terminator fragment into pCSR1-GPD yielded plasmid pTSL126B. To create a version of this plasmid containing an mCherry-6× His tag, primer pair OTS415/OTS416 (AAAGCATGCGTGAGCAAGGGCGAGGAGGATAAC and CGCTTAATTAACTAGTGGTGGTGGTGGTGGTGGCTGCCCTTGTACAGCTCGTCCATGCC), incorporating SphI and PacI sites, respectively, was used to amplify a Neurospora codon-optimized mCherry-6× His fragment from plasmid pMFP26. Cloning of this fragment resulted in plasmid pTS184J. Subsequently, primer pair OTS387/418 (AAAGCGGCCGCATGGCATCCCTAAAGTCGCTG and AAACCTAGGAATAAGATGCTTAGATCTAAGCAG), incorporating NotI and AvrII restriction sites, respectively, was used to amplify the erv25 open reading frame for insertion into pTS184J, resulting in plasmid pTSL190A. Plasmid pTSL190A was linearized and transformed into the Δerv25 deletion strain FGSC 18824. Transformants were selected on medium containing cyclosporine.
Protein localization.
Protein localization was analyzed in germlings prepared as described above or in hyphal samples growing on VMM agar plates. Strains containing the vacuolar marker red fluorescent protein (RFP)-VAM3 and the Golgi membrane marker RFP-VPS52 (36) were used for colocalization studies. Heterokaryotic cells were obtained by mixing equal amounts of the two strains expressing the different proteins on VMM slant tubes. After conidiation, conidial suspensions were prepared and examined by confocal microscopy using a Leica SD6000 microscope with a 100×, 1.4-numeric-aperture oil immersion objective equipped with a Yokogawa CSU-X1 spinning disk head and 488-nm and 561-nm lasers controlled by Metamorph software (Molecular Devices).
Phylogenetic analyses.
The N. crassa LFD-2 protein sequence was used to search the JGI (http://jgi.doe.gov/) and FungiDB (http://fungidb.org/fungidb/) databases. JGI blast searches were conducted using the default settings (e value of <1e−5) with the scoring matrix set to BLOSUM62. FungiDB blast parameters matched those used for JGI database searches. Sequences with significant similarity (more than 33% amino acid identity) to LFD-2 (NCU02191) were selected for further analysis, with the exception of NCU07762 and Batrachochytrium dendrobatidis (GP3.005680), which were used as outgroups. Protein sequences were aligned using MAFFT (37), and gaps were trimmed by TrimAl (38) and further improved by Gblock (39) to remove highly divergent regions. Mview (40) was used to view alignments, and percent identities in alignment were calculated. Phylogenetic trees were built by PhyML V 3.0 (41) using LG mode (42), and the number of relative substitution rate categories was set to 16. The trees were visualized and converted with Figtree V 1.40 (http://tree.bio.ed.ac.uk/software/figtree/).
Protein domain prediction.
Pfam, InterPro, and SMART (43–45) were used to predict conserved protein domains. Predicted domains supported by all databases are shown. TMHMM Server v. 2.0 (46) was used for transmembrane domain prediction. Signal peptides were detected by SignalP 4.1 (47). N-linked glycosylation sites were predicted by NetNGlyc (http://www.cbs.dtu.dk/services/NetNGlyc/), and O-glycosylation sites were predicted by YinOYang, DictyOGly, and NetOGlyc (48–50).
Statistical analyses.
Data are presented as means ± standard deviations (SD), averaged for a minimum of three replicates of the experiments. Data were analyzed with Student's t test. A P value of less than 0.05 was accepted as indicating a statistically significant difference compared with controls.
RESULTS
Identification of new genes involved in germling fusion.
Calcium plays an important role during membrane fusion in N. crassa, as cell lysis frequency in ΔPrm1 or Δlfd-1 germling pairs is affected by the extracellular calcium concentration (22, 51). These observations prompted us to examine the transcriptional profile of N. crassa germlings grown in the absence of calcium in the external medium (http://www.ncbi.nlm.nih.gov/geo/; series record: GSE53013) (24). We narrowed our search to genes that showed decreased expression levels in the absence of calcium and encoded proteins containing transmembrane domains; 50 genes were identified that met these criteria (see Table S1 in the supplemental material). Among this gene set were Prm1 (NCU09337) and lfd-1 (NCU09307). Mutants for 7 of the 50 downregulated genes were not available at the time of this study, three other mutants showed conidiation defects (therefore, germling fusion frequencies could not be ascertained), and nine additional deletion mutants were eliminated, because the annotation of their corresponding gene was predicted to encode a metabolic enzyme or transporter.
Deletion mutants for two (ΔNCU04872 and ΔNCU09955) of the remaining 29 genes were available only as heterokaryons (Table 1); homokaryotic strains were isolated via microconidial isolation (see Materials and Methods). Strains containing deletions of all 29 genes were screened for membrane fusion defects and cell lysis of adhered germlings by using the plasma membrane dye FM4-64 (22, 52) (Fig. 1A). This initial screening showed that 10 of the 29 mutants had significantly higher frequencies of cell lysis of adhered germling pairs than wild-type (WT) germling pairs (P < 0.05) (see Table S1 in the supplemental material). All 10 mutants were crossed to the wild type to assess segregation of the germling lysis phenotype with the hygromycin marker. Crosses with one mutant (NCU09955) failed to produce progeny. In 2 of the 9 strains, clear segregation of the germling lysis phenotype and the hygromycin marker was observed (ΔNCU01697 and ΔNCU02191). Cell lysis levels in progeny in crosses with the remaining mutants were variable and could not be definitively correlated with the hygromycin marker, suggesting modifiers in the backgrounds of these strains that affected cell lysis frequency.
FIG 1.

Identification and phenotype of deletion mutants that show increased cell lysis of adhered germlings. Mutants showed cell lysis of adhered germlings during vegetative cell fusion. (A) Adhered germlings treated with FM4-64. Arrows point at lysed germlings showing intense FM4-64 staining, either across the entire germling (left) or at the point of cell fusion (right). Asterisks indicate normal fusion events. BF indicates bright-field images, and FM4-64 indicates FM4-64 fluorescence images. Scale bar, 5 μm. (B) Lysis frequencies of adhered germlings for strains carrying deletions for two candidate genes identified from RNA-seq analyses (24) compared to the WT and known fusion mutants ΔPrm1 and Δlfd-1. Gray bars indicate lysis values in normal VMM (29), white bars indicate lysis values in a reduced concentration of extracellular calcium (Ca2+), and patterned bars indicate lysis in 1 M sorbitol. Asterisks indicate a significant increase of cell lysis (P < 0.05) compared to WT values. Diamonds indicate a significant increase of cell lysis in reduced calcium concentration with respect to the same strain in normal VMM (P < 0.05). No significant difference was observed in the frequency of cell lysis between normal and 1 M sorbitol conditions in any of the strains. Bars indicate standard deviations.
Lysis of ΔNCU01697 or ΔNCU02191 adhered germlings resulted in cell death of one or both germlings and occurred at higher levels than those for the WT (Fig. 1). Cell lysis was never detected in germlings that were not adhered, indicating that lysis is the result of a defect during plasma membrane merger or cell wall deconstruction, which occurs during the process of cell-cell fusion. Strong FM4-64 staining was observed in the contact area of lysed germling pairs of ΔNCU01697 and ΔNCU02191, suggesting that plasma membrane permeabilization in the contact area allowed greater accumulation of FM4-64 in membranes near damaged areas (Fig. 1A, arrows).
Lysis is due to defects in plasma membrane integrity.
The lysis phenotype of adhered germlings in the ΔNCU01697 and ΔNCU02191 mutants could be due to defects in cell wall remodeling, which occurs at the point of adherence of germlings and is required for subsequent membrane fusion, or to defects in plasma membrane integrity during membrane merger. In S. cerevisiae, the growth defect of cell wall remodeling mutants is remediated by the addition of an osmotic stabilizer, such as sorbitol, to the growth medium but does not suppress the cell lysis phenotype of membrane fusion mutants, such as the prm1Δ mutant (15). To differentiate whether cell lysis during germling fusion is due to a defect in plasma membrane integrity versus cell wall integrity, we performed fusion assays in VMM (29) supplemented with 1 M sorbitol. As shown in Fig. 1B, osmotic stabilization failed to suppress cell lysis observed in VMM in both ΔPrm1 and Δlfd-1 mutants. In the two new mutants, the addition of sorbitol also failed to suppress the cell lysis phenotype of adhered germlings, suggesting that lysis is due to a defect in membrane integrity in these mutants and not a defect in cell wall integrity.
Characterization of the lfd-2 mutant.
The N. crassa ΔPrm1 and Δlfd-1 mutants show increased cell lysis of adhered germling pairs under conditions of reduced extracellular calcium (22). Therefore, we tested whether the frequency of cell lysis increased under reduced calcium conditions in the two deletion strains that showed lysis during cell-cell fusion. As shown in Fig. 1B, only the strain carrying a deletion of NCU02191 showed a significant increase in lysis of germling pairs under reduced extracellular calcium concentrations. Due to the similarity in germling fusion phenotype between ΔPrm1, Δlfd-1, and ΔNCU02191 strains, we focused on NCU02191, which we named lfd-2 for late fusion defect-2.
To obtain quantitative data for cell fusion events, we constructed duplicate strains where one Δlfd-2 strain carried a cytoplasmic GFP marker and another Δlfd-2 strain carried a cytoplasmic mCherry marker. Five hours postgermination, the fusion phenotype between green and red fluorescent Δlfd-2 germling pairs was analyzed. Whereas 100% of the adhered wild-type germling pairs fused and showed mixed green and red fluorescence, fusion between adhered Δlfd-2 germlings was reduced to 81% (Fig. 2). The reduction in fusion frequency in Δlfd-2 germling pairs was due to a significant increase in cell lysis (14% of the fusion pairs) rather than an increase in the class of adhered but nonfused germlings, as was observed for both ΔPrm1 and Δlfd-1 strains (Fig. 2B and C). However, similar to ΔPrm1 and Δlfd-1 mutants (22), increasing the calcium concentration suppressed the lysis phenotype of adhered lfd-2 germlings (Fig. 2A).
FIG 2.

Characterization of cell lysis frequencies of adhered germlings in Δlfd-2, Δlfd-1; Δlfd-2, and ΔPrm1; Δlfd-2 mutants under normal and altered calcium conditions. (A) Fusion (black bars), lysis (white bars), and blocked fusion (nonfused; gray bars) frequencies of adhered Δlfd-2 germlings at different calcium (Ca2+) concentrations. B) Fusion (black bars), lysis (white bars), and blocked fusion (gray bars) for single (Δlfd-1 or Δlfd-2) and double (Δlfd-1; Δlfd-2) mutants under different calcium concentrations. (C) Fusion (black bars), lysis (white bars), and blocked fusion (gray bars) of adhered germlings from single mutants (ΔPrm1 or Δlfd-2) versus the double mutant (ΔPrm1; Δlfd-2) under different calcium (Ca2+) concentrations. Bars indicate standard deviations.
The similarity in lysis phenotype between Δlfd-2, ΔPrm1, and Δlfd-1 mutants suggested that these proteins function in a similar process during cell fusion. To assess this hypothesis, we performed an epistasis test by constructing Δlfd-1 Δlfd-2 and ΔPrm1 Δlfd-2 double mutants. Compared with the cell lysis frequency in Δlfd-1 or Δlfd-2 adhered germlings (8% and 14%, respectively), the Δlfd-1 Δlfd-2 germlings showed an additive increase in lysis frequency (23%) (Fig. 2B, white bars). This lysis value increased significantly under reduced calcium concentrations (31%). The deletion of lfd-2 reduced the adhered, nonfused germling phenotype of Δlfd-1 mutants, with a concomitant increase in cell lysis, but did not affect the frequency of the class of fused germlings (Fig. 2B).
N. crassa germlings lacking Prm1 showed a high percentage of adhered but unfused germlings (∼44%) (Fig. 2C) (21, 22), of which ∼11% showed a lysis phenotype. An additive cell lysis phenotype also was observed in the ΔPrm1 Δlfd-2 germlings (26%) compared to ΔPrm1 or Δlfd-2 germlings alone (11% and 14%, respectively) (Fig. 3B, white bars). The cell lysis frequency of the ΔPrm1 Δlfd-2 germlings also increased significantly (34%) under conditions of reduced calcium. However, the frequency of germlings that underwent productive cell fusion in the ΔPrm1 Δlfd-2 germlings was not significantly different from that of ΔPrm1 germlings alone (Fig. 2C). Similar to the Δlfd-1 Δlfd-2 germlings, the absence of lfd-2 from ΔPrm1 germlings resulted in a substantial reduction in the adhered but nonfused germling class, which was replaced by an increase in the class of germlings that showed cell lysis.
FIG 3.

Characterization of cell lysis frequencies of adhered germlings in Δfig1, ΔPrm1; Δfig1, Δfig1; Δlfd-1, and ΔPrm1; Δfig1; Δldf-1 mutants under normal and altered calcium conditions. (A) Frequency of fusion (black bars), lysis (white bars), or nonfusion (where adhered germlings are blocked at cell fusion; gray bars) of Δfig1 germlings under different calcium concentrations. (B) Frequency of fusion (black bars), lysis (white bars), or nonfusion (where adhered germlings are blocked at cell fusion; gray bars) of ΔPrm1; Δfig1, Δfig1; Δlfd-1, and ΔPrm1; Δlfd-1; Δfig1 germlings. Bars indicate standard deviations.
By transmission electron microscopy analyses, it was previously shown that a significant portion of ΔPrm1 and Δlfd-1 adhered germlings show stable appressed plasma membranes, which often display cellular invaginations (21, 22). Our data suggest that in the absence of LFD-2, the appressed membranes representative of the adhered, nonfused class of ΔPrm1 and Δlfd-1 germlings are destabilized, resulting in an increase in cell lysis in the ΔPrm1 Δlfd-2 and Δlfd-1 Δlfd-2 germling pairs. This result is different from that observed for ΔPrm1 Δlfd-1 mutants, where the increase in cell lysis occurs as a result of a decrease in cell fusion frequencies but the percentage of adhered, nonfused germlings was unaffected (22) (Fig. 3B).
The N. crassa fig1 mutant displays a germling lysis phenotype.
In S. cerevisiae, Fig1p is a plasma membrane protein that controls pheromone-induced calcium influx and is a component of the low-affinity calcium uptake system (LACS) (53). fig1Δ mating pairs show an ∼25% reduction in cell fusion and show calcium-dependent cell lysis (12). A FIG1-like homolog recently was identified in N. crassa, and a strain bearing a deletion of fig1 displays a mating type-specific defect in the production of female reproductive structures (33). Due to its mating cell fusion defect and cell lysis phenotype in S. cerevisiae, we examined the role of fig1 during germling fusion in N. crassa. The fig1 locus (NCU02219) encodes a 267-amino-acid (aa) protein with 4 transmembrane domains. To obtain quantitative data for cell fusion events, we constructed Δfig1 strains carrying GFP or mCherry cytoplasmic markers. Δfig1 germlings showed normal chemotropic behavior and adherence. However, adhered Δfig1 germlings showed significant cell lysis (12.7%) (Fig. 3A), a phenotype very similar to that of Δlfd-2 mutants. As for fig1Δ mutants in S. cerevisiae and for ΔPrm1, Δlfd-1, and Δlfd-2 mutants in N. crassa, increasing extracellular calcium suppressed the cell lysis defect of Δfig1 germlings (Fig. 3A). Similar to the Δlfd-2 mutant, the frequency of adhered but nonfused Δfig1 germlings was not significantly different from that of the wild type. Complementation of Δfig1 with the fig1 open reading frame under the regulation of the tef1 promoter recovered WT levels of cell lysis (4.6% ± 1.2%), confirming that the lysis phenotype detected in the mutant resulted from the absence of FIG1.
To assess epistasis interactions between Prm1, lfd-1, and fig1, we constructed double and triple mutants and quantitatively assessed fusion and lysis in adhered germlings. The ΔPrm1 Δfig1 mutant showed a significant increase in cell lysis (∼40%) relative to ΔPrm1 or Δfig1 germlings (∼11% and ∼12.7%, respectively), indicating a synergistic effect of the deletion of both genes. The adhered germling class, and particularly the fused germling class, were reduced in the ΔPrm1 Δfig1 germlings. In contrast, the Δfig1 Δlfd-1 mutants showed an additive effect on cell lysis (∼22%) (Fig. 3B). As with the Δlfd-1 Δlfd-2 germlings, the increase in cell lysis of the Δfig1 Δlfd-1 germlings was due to a decrease in the class of adhered, nonfused germlings, but not in the frequency of germlings that underwent productive cell fusion (Fig. 3B). The triple mutant (ΔPrm1 Δlfd-1 Δfig1) showed a phenotype similar to that of ΔPrm1 Δlfd-1 germlings (Fig. 3B).
To determine whether cell lysis and death observed during germling fusion also occurs during hyphal fusion in an N. crassa colony, we assessed lysis in a mature colony by staining with the vital dye methylene blue. In a wild-type colony, no staining by methylene blue was observed in fungal compartments at hyphal fusion junctions (Fig. 4A; circles indicate fusion junctions). However, in a ΔPrm1 colony, cell lysis at approximately half of the hyphal fusion points was observed (Fig. 4B, red circles). The vital dye staining showed compartmentalization at the fusion point, suggesting that the septal pores surrounding the fusion point were blocked in these cells. For Δlfd-1 and Δlfd-2 colonies (Fig. 4C and D) as well as for Δfig1 and ΔNCU01697, no lysis at hyphal fusion points within a colony was detected.
FIG 4.

Evaluation of lysis and compartmentation during hyphal fusion in a wild-type colony versus ΔPrm1, Δlfd-1, and Δlfd-2 mutant colonies. (A) Lysis at hyphal fusion points in a wild-type colony was assessed by staining with the vital dye methylene blue; black circles show productive fusion events (no cell lysis). (B) Lysis at hyphal fusion points in a ΔPrm1 colony was assessed by staining with the vital dye methylene blue. Red circles show cell lysis that occurred at fusion points; black circles show productive fusion events. (C) Lysis at hyphal fusion points in a Δlfd-1 colony was assessed by staining with the vital dye methylene blue. Black circles show productive fusion events. (D) Lysis at hyphal fusion points in a Δlfd-2 colony was assessed by staining with the vital dye methylene blue. Black circles indicate productive fusion events. Scale bars, 50 μm.
LFD-2 localizes to the ER and Golgi membrane.
lfd-2 is predicted to encode a protein of 502 aa containing one transmembrane domain (aa 26 to 43) (Fig. 5A). Although a signal peptide is not predicted (47), it is possible that the transmembrane domain functions to target LFD-2 to the ER given its proximity to the N terminus. TMHMM and PSORT (46, 54) support placement of the C terminus of LFD-2 on the exterior of the cell or in the interior of the lumen of the ER/Golgi membrane. Three N-glycosylation sites (N147, N387, and N499) and six O-glycosylation sites (S4, S60, S83, T406, S501, and S502) were predicted (48–50). A Fringe domain (aa 202 to 262) also was identified in LFD-2 by Pfam InterPro and SMART analyses (43–45) (Fig. 5A). Fringe domains are associated with proteins having glycosyltransferase activity, such as Fringe, which is an O-fucosyltransferase involved in the glycosylation of Notch (55, 56). Fringe contains a DxD motif, which is found in most glycosyltransferases that use nucleoside diphosphate sugars. A DAD motif is present within the predicted Fringe domain of LFD-2 (aa 214 to 216). Inactivation of Fringe in metazoan species gives rise to Notch signaling defects (57).
FIG 5.
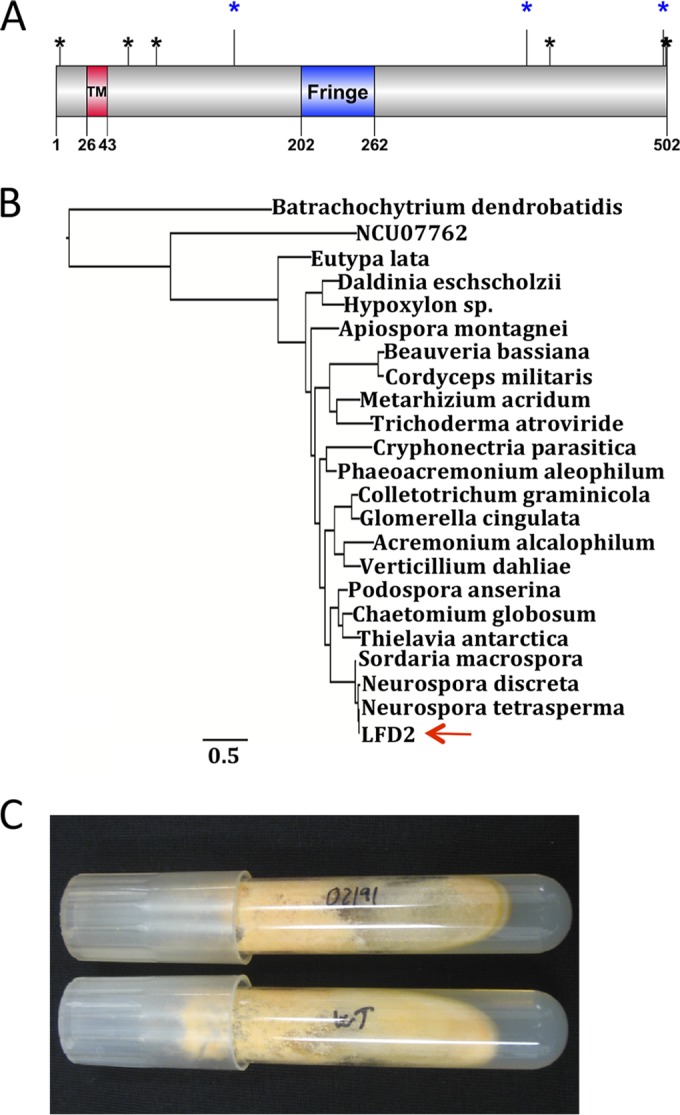
Structure, phylogenetic, and phenotypic analyses of LFD-2. (A) Cartoon of domain structure of LFD-2. The red box shows the transmembrane domain, and the blue box shows the Fringe domain. The blue asterisks indicate predicted N-linked glycosylation sites, and black asterisks indicate predicted O-glycosylation sites (see Materials and Methods). The cartoon was made by DOG (68). (B) Phylogenetic tree for LFD-2 (NCU02191) in the genomes of species from Sordariomycetes: Acremonium alcalophilum (Acral1_c3a.estExt_fgenesh2_pg.C_50864), Apiospora montagnei (e_gw1.92.82.1), Beauveria bassiana (BBA_03605m.01), Chaetomium globosum (CHGT_02214), Colletotrichum graminicola (GLRG_06237T0), Cordyceps militaris (CCM_08528m.01), Cryphonectria parasitica (Crypa1.estExt_Genewise1Plus.C_20699), Daldinia eschscholzii (MIX4991_8_43), Eutypa lata (UCREL1_7331m.01), Glomerella cingulata (e_gw1.9.114.1), Hypoxylon sp. strain CI-4A (gm1.9432_g), Metarhizium acridum (MAC_03592m.01), Neurospora discreta (estExt_Genewise1Plus.C_90445), Neurospora tetrasperma (e_gw1.5.373.1), Phaeoacremonium aleophilum (UCRPA7_6658m.01), Podospora anserina (Pa_5_2960), Thielavia antarctica (e_gw1.19.390.1), Trichoderma atroviride (Triat1.e_gw1.1.1843.1), Verticillium dahliae (VDAG_07638T0), and Sordaria macrospora (SMAC_03630). The LFD-2 homologous sequence in Batrachochytrium dendrobatidis (GP3.005680) and the putative paralog of LFD-2 in N. crassa (NCU07762) were used as outgroups. Arrow points at LFD-2 (NCU02191) from N. crassa. The scale bar indicates the number of substitutions per amino acid. (C) Wild-type culture (bottom) and Δlfd-2 culture (upper) growing on VMM agar.
BLAST searches of the N. crassa genome identified a paralog of lfd-2 (NCU07762) that has ∼17% aa identity to LFD-2. Other filamentous fungal species within the Eurotiomycetes, Dothidiomycetes, and Sordariomycetes also have a variable number of LFD-2 homologs (from 1 to 9 homologs within a single genome). The majority of these homologs show ∼18 to 25% aa identity to LFD-2. However, within the Sordariomycetes, homologs of LFD-2 were identified that showed high identity (>32% aa identity; E value of <1−100) (Fig. 5B). In contrast, homologs of LFD-2 were not identified in the Schizosaccharomycetes. Homologs to lfd-2 also were absent from most species in the Saccharomycotina, with the exception of Yarrowia lipolytica (∼21% aa identity) and Lipomyces starkeyi (4 homologs ranging from 16 to 27% aa identity). lfd-2 homologs also were identified in a few basidiomycete species, including Ustilago maydis (∼18% aa identity), and even in more distantly related species, such as in the chytrid Batrachochytrium dendrobatidis (one lfd-2 homolog; 22% aa identity).
Macroscopically, the Δlfd-2 mutant was indistinguishable from the wild type (Fig. 5C) and showed normal growth, conidiation, female fertility, and crossing behavior. To determine the cellular location of LFD-2, an lfd-2-gfp allele, regulated by the tef1 promoter and integrated into the his-3 locus of a his-3; Δlfd-2 strain, was constructed. Full complementation of germling fusion defects was observed, including abrogating cell lysis during germling fusion to wild-type levels (5.3% ± 2.06%). LFD-2-GFP localized to endomembranes during germling fusion (Fig. 6A to C) and was not observed at the plasma membrane or at the point of contact between germlings. In hyphae, LFD-2-GFP localization was detected distal to the peripheral hyphal tips and within the inner section of the colony. LFD-2-GFP localized to the nuclear ER as well as with the plasma membrane, suggesting both nuclear and cortical ER localization (Fig. 6F to I). LFD-2-GFP localization to the ER was never observed near or at hyphal tips (Fig. 6D and E), and LFD-2-GFP was not observed at hyphal fusion points (Fig. 6 and 7).
FIG 6.

Localization of LDF-2-GFP in germlings and in hyphae. (A to C) LFD-2-GFP localization in germlings undergoing chemotropic interactions and cell fusion. Arrows indicate the point at which germlings are adhered. (D to F) Cellular localization of LFD-2-GFP in apical hyphae and in hyphae more distant from the colony periphery. Note localization to punctae in apical hyphae and hyphae near the tips (D and E) but localization to nuclear ER and membrane structures in hyphae further from the periphery of a colony (F). (G to I) Localization of LFD-2-GFP to fusion hyphae, as well as hyphae within the interior of a colony. Arrows indicate the point of contact between fusion hyphae. Note the localization of LFD-2-GFP to nuclear ER, puncta, and cortical ER or plasma membrane in these hyphae. Scale bars, 5 μm.
FIG 7.

Colocalization of LDF-2-GFP and different cellular markers in germlings and hyphae undergoing cell fusion. (A) Colocalization of LFD-2-GFP with the vacuolar marker VAM3-RFP in adhered germlings undergoing cell fusion. (B) Colocalization of LFD-2-GFP with the endoplasmic reticulum (ER) protein SEC61-mCherry in adhered hyphae undergoing cell fusion. (C) Colocalization of LFD-2-GFP with the ER protein ERV25-mCherry in adhered hyphae undergoing cell fusion. (D) Colocalization of LFD-2-GFP with the Golgi protein VPS52-RFP in adhered hyphae undergoing cell fusion. White arrows indicate fusion points. Orange arrows indicate colocalization of GFP- and mCherry (or RFP)-tagged proteins. Scale bars, 5 μm.
In germlings, colocalization of LFD-2-GFP with the vacuolar marker RFP-VAM3 also was observed (Fig. 7A). To assess colocalization of LFD-2-GFP with ER markers in hyphae, we constructed a strain expressing SEC61-mCherry. Sec61p is part of the Sec61 complex, which is the major component of a channel-forming translocon complex that mediates protein translocation across the ER; Sec61 localizes both at nuclear and cortical ER in S. cerevisiae (58). In a localization pattern identical to that observed in S. cerevisiae, in N. crassa germlings, Sec61-mCherry localized to the nuclear membrane (nuclear ER) and to foci at the plasma membrane (cortical ER) (see Fig. S1 in the supplemental material). In addition, similar to LFD-2 (Fig. 7A), SEC61 also localized to vacuoles in conidia (see Fig. S1). In hyphae, LFD-2-GFP and SEC61-mCherry showed colocalization to nuclear ER (Fig. 7B). For further confirmation of LFD-2 localization, we used a strain expressing ERV25-mCherry, an ortholog of the yeast ER cargo adaptor protein Erv25p, which is an ER membrane protein involved in ER-to-Golgi membrane transport (59). We observed clear ERV25-mCherry localization to both nuclear and cortical ER in hyphae; LFD-2-GFP colocalized with ERV25-mCherry at both ER locations (Fig. 7C).
Foci of LFD-2-GFP were observed in hyphae which were not labeled with Sec61-mCherry or Erv25-mCherry. To test if the localization of LFD-2-GFP corresponded to the Golgi membrane, we performed colocalization with the Golgi membrane marker RFP-VPS52 (36); colocalization of LFD-2-GFP and RFP-VPS52 was observed for some foci (Fig. 7D), indicating that LFD-2-GFP also localized to Golgi membrane.
DISCUSSION
The process of membrane merger, whether between cells or between membrane compartments in the cell, is an inherently perilous process. Cell lysis can be a significant side reaction of the fusion process in membrane merger during mating in S. cerevisiae, fusion of vegetative germlings in N. crassa, and homotypic fusion of vacuoles in S. cerevisiae (12, 22, 60, 61). Cell lysis frequencies can be affected by membrane composition as well as by the disruption of the protein machinery and regulatory factors required for the fusion process (for example, an excess of SNARE proteins [62]). Previously, only two proteins were known to be involved in membrane merger during cell fusion in N. crassa: Prm1 and lfd-1 (21, 22). The absence of either of these proteins results in both membrane fusion defects and calcium-dependent cell lysis during germling fusion. With this work, we expanded the list of genes involved in cell lysis during germling fusion and describe a new gene, lfd-2, whose deletion results in a strain that also shows calcium-dependent lysis during germling fusion. LFD-2 localized to endomembranes in N. crassa germlings, while in a colony, LFD-2 localized mainly to the ER and Golgi membrane. LFD-2 was not observed at the fusion point between germlings or between fusion hyphae. The localization of LFD-2 to ER and to Golgi membrane suggests that it plays a role in secretion or in the modification of a component needed for proper membrane fusion.
LFD-2 is predicted to contain a Fringe domain. In metazoans, Fringe is a Golgi membrane-localized glycosyltransferase involved in the transfer of N-acetylglucosamine to fucose on extracellular domains of epidermal growth factors, such as NOTCH receptors (55, 56, 63). Other glycosyltransferases involved in glycolipid synthesis also are necessary for Notch signaling and may play a role in the appropriate organization of Notch receptors in the plasma membrane. The paralog of lfd-2 in N. crassa (NCU07762; gt31-2) is annotated as a member of glycosyltransferase family 31. In metazoans, Notch receptors are cell surface glycoproteins that are activated by specific ligands to regulate cell fate decisions (25, 57). Activation of Notch by ligand binding results in proteolysis and translocation of the cytoplasmic portion of Notch to the nucleus and, via interaction with other proteins, mediates transcriptional activation of target genes involved in cell fate determination. Disruption of O-fucose glycan synthesis, such as in Fringe mutants, leads to Notch signaling defects. Notch is also a calcium binding protein which is thought to play a role in the stability of EFG repeats (64). It is tempting to speculate that LFD-2 plays a role in the modification of a protein involved in proper regulation of membrane merger during germling fusion in N. crassa and that its dysfunction results in calcium-dependent cell lysis. However, the identity of such a receptor/protein in N. crassa remains elusive, as components of Notch signaling are highly conserved only among metazoan species (65).
The cell lysis phenotype observed in adhered mating cells in S. cerevisiae in the absence of proteins such as Prm1p and Fig1p has been proposed to be the consequence of the engagement of defective membrane fusion machinery (12). Cell lysis of N. crassa ΔPrm1 or Δlfd-1 strains bearing a deletion of a synaptotagmin ortholog, syt-1, which is involved in membrane repair in other species, show increased frequencies of lysis of adhered germlings and is not affected by the concentration of external calcium. These observations suggest that engagement of the membrane merger machinery can have two outcomes: membrane fusion or cell lysis. Here, we show that strains bearing deletions of either lfd-2 or fig1 showed calcium concentration-dependent cell lysis during germling fusion. These data suggest that the true extent of cell lysis phenotypes of the ΔPrm1, Δlfd-1, Δlfd-2, or Δfig1 mutants are suppressed by calcium-dependent membrane repair mechanisms.
FIG1 in N. crassa is a component of the low-affinity calcium uptake system (33), with a predicted role similar to that of Fig1p in S. cerevisiae, which is involved in calcium uptake in response to pheromone during mating (53). In S. cerevisiae, fig1 mutants show a 25% reduction in cell fusion with a concomitant increase in failure to undergo membrane merger (12). Here, we show that the N. crassa Δfig1 mutant did not show an increase of adhered germlings pairs blocked at membrane merger (nonfused) but showed a calcium-dependent cell lysis phenotype. This cell fusion phenotype was very similar to that observed for the Δlfd-2 strain. However, our epistasis experiments indicate that FIG1 and LFD2 function in different pathways. For example, in both ΔPrm1; Δlfd-2 and Δlfd-1; Δlfd-2 germlings, the increase in cell lysis occurred exclusively within the class of germlings that were adhered (Fig. 4). In contrast, ΔPrm1; Δfig1 germlings showed a similar frequency of adhered germlings but a significant reduction in the frequency of fused germlings, with a concomitant increase in the frequency of lysed germlings. These observations suggest that PRM1 and FIG1 function early in membrane merger, perhaps in conjunction with initial steps in the assembly of the fusion machinery, while LFD-1 and LFD-2 play an important function later, perhaps ensuring the fidelity of membrane pore formation.
In addition to lfd-2, we identified another mutant (NCU01697) that showed significant lysis of adhered germlings but for which alterations in calcium concentration had no effect on the frequency of cell lysis during germling fusion. One possible explanation for cell lysis during germling fusion observed in this mutant is that the protein encoded by this gene plays a role in membrane organization and/or lipid composition. This hypothesis is supported by the fact that NCU01697 contains a predicted acyltransferase domain (5.1e−04); proteins containing acyltransferase domains are involved in phospholipid biosynthesis (66). The identification of this additional mutant that showed lysis of adhered germlings but which was calcium concentration insensitive shows the complexity of the membrane merger/cell fusion process. Since membrane merger is a ubiquitous phenomenon that occurs between cells during mating; during syncytial development of muscle cells, osteoclasts, and the placenta (1, 3); and during intracellular vesicle trafficking, elucidation of this process using the genetic and molecular resources of N. crassa may provide information on mechanisms of membrane merger and repair that are extendable to other systems. Further characterization of PRM1, FIG1, LFD-1, LFD-2, and NCU01697 provides a rich resource to dissect the cell membrane merger pathway and to investigate biochemical functions of these proteins during the cell fusion process.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Andre Fleissner (Technische Universitat Braunschweig, Germany) for the tef-1 promoter vector and Barry Bowman (University of California, Santa Cruz) for the RFP-VPS52 and RFP-VAM3 strains.
This work was supported by a National Science Foundation grant (MCB 1121311) and a National Institutes of Health grant (R01GM060468) to N.L.G. We acknowledge the use of materials generated by NIH program project grant P01GM068087, functional analysis of a model filamentous fungus. A.P.G. was the recipient of a fellowship from Fundação Calouste Gulbenkian (104210) and a short-term fellowship from EMBO (329-2012).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00233-14.
REFERENCES
- 1.Jahn R, Lang T, Sudhof TC. 2003. Membrane fusion. Cell 112:519–533. doi: 10.1016/S0092-8674(03)00112-0. [DOI] [PubMed] [Google Scholar]
- 2.Oren-Suissa MO, Podbilewicz B. 2010. Evolution of programmed cell fusion: common mechanisms and distinct functions. Dev Dynamics 239:1515–1528. doi: 10.1002/dvdy.22284. [DOI] [PubMed] [Google Scholar]
- 3.Aguilar PS, Baylies MK, Fleissner A, Helming L, Inoue N, Podbilewicz B, Wang H, Wong M. 2013. Genetic basis of cell-cell fusion mechanisms. Trends Genet 29:427–437. doi: 10.1016/j.tig.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sollner TH. 2004. Intracellular and viral membrane fusion: a uniting mechanism. Curr Opin Cell Biol 16:429–435. doi: 10.1016/j.ceb.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 5.Mohler WA, Shemer G, del Campo JJ, Valansi C, Opoku-Serebuoh E, Scranton V, Assaf N, White JG, Podbilewicz B. 2002. The type I membrane protein EFF-1 is essential for developmental cell fusion. Dev Cell 2:355–362. doi: 10.1016/S1534-5807(02)00129-6. [DOI] [PubMed] [Google Scholar]
- 6.Sapir A, Choi J, Leikina E, Avinoam O, Valansi C, Chernomordik LV, Newman AP, Podbilewicz B. 2007. AFF-1, a FOS-1-regulated fusogen, mediates fusion of the anchor cell in C. elegans. Dev Cell 12:683–698. doi: 10.1016/j.devcel.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Avinoam O, Fridman K, Valansi C, Abutbul I, Zeev-Ben-Mordehai T, Maurer UE, Sapir A, Danino D, Grunewald K, White JM, Podbilewicz B. 2011. Conserved eukaryotic fusogens can fuse viral envelopes to cells. Science 332:589–592. doi: 10.1126/science.1202333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elion EA, Trueheart J, Fink GR. 1995. Fus2 localizes near the site of cell fusion and is required for both cell fusion and nuclear alignment during zygote formation. J Cell Biol 130:1283–1296. doi: 10.1083/jcb.130.6.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Erdman S, Lin L, Malczynski M, Snyder M. 1998. Pheromone-regulated genes required for yeast mating differentiation. J Cell Biol 140:461–483. doi: 10.1083/jcb.140.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heiman MG, Walter P. 2000. Prm1p, a pheromone-regulated multispanning membrane protein, facilitates plasma membrane fusion during yeast mating. J Cell Biol 151:719–730. doi: 10.1083/jcb.151.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Proszynski TJ, Klemm R, Bagnat M, Gaus K, Simons K. 2006. Plasma membrane polarization during mating in yeast cells. J Cell Biol 173:861–866. doi: 10.1083/jcb.200602007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aguilar PS, Engel A, Walter P. 2007. The plasma membrane proteins Prm1 and Fig1 ascertain fidelity of membrane fusion during yeast mating. Mol Biol Cell 18:547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paterson JM, Ydenberg CA, Rose MD. 2008. Dynamic localization of yeast Fus2p to an expanding ring at the cell fusion junction during mating. J Cell Biol 181:697–709. doi: 10.1083/jcb.200801101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heiman MG, Engel A, Walter P. 2007. The Golgi-resident protease Kex2 acts in conjunction with Prm1 to facilitate cell fusion during yeast mating. J Cell Biol 176:209–222. doi: 10.1083/jcb.200609182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin H, Carlile C, Nolan S, Grote E. 2004. Prm1 prevents contact-dependent lysis of yeast mating pairs. Eukaryot Cell 3:1664–1673. doi: 10.1128/EC.3.6.1664-1673.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Curto MA, Sharifmoghadam MR, Calpena E, De Leon N, Hoya M, Doncel C, Leatherwood J, Valdivieso MH. 2014. Membrane organization and cell fusion during mating in fission yeast requires multipass membrane protein Prm1. Genetics 196:1059–1076. doi: 10.1534/genetics.113.159558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simonin A, Palma-Guerrero J, Fricker M, Glass NL. 2012. Physiological significance of network organization in fungi. Eukaryot Cell 11:1345–1352. doi: 10.1128/EC.00213-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hickey PC, Jacobson D, Read ND, Louise Glass NL. 2002. Live-cell imaging of vegetative hyphal fusion in Neurospora crassa. Fungal Genet Biol 37:109–119. doi: 10.1016/S1087-1845(02)00035-X. [DOI] [PubMed] [Google Scholar]
- 19.Read ND, Fleißner A, Roca MG, Glass NL. 2010. Hyphal fusion, p 260–273. In Borkovich KA, Ebbole D (ed), Cellular and molecular biology of filamentous fungi. ASM Press, Washington, DC. [Google Scholar]
- 20.Roper M, Simonin A, Hickey PC, Leeder A, Glass NL. 2013. Nuclear dynamics in a fungal chimera. Proc Natl Acad Sci U S A 110:12875–12880. doi: 10.1073/pnas.1220842110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fleissner A, Diamond S, Glass NL. 2009. The Saccharomyces cerevisiae PRM1 homolog in Neurospora crassa is involved in vegetative and sexual cell fusion events but also has post-fertilization functions. Genetics 181:497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palma-Guerrero J, Leeder AC, Welch J, Glass NL. 2014. Identification and characterization of LFD1, a novel protein involved in membrane merger during cell fusion in Neurospora crassa. Mol Microbiol 92:164–182. doi: 10.1111/mmi.12545. [DOI] [PubMed] [Google Scholar]
- 23.Leeder AC, Jonkers W, Li J, Glass NL. 2013. Early colony establishment in Neurospora crassa requires a MAP kinase regulatory network. Genetics 195:883–898. doi: 10.1534/genetics.113.156984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goncalves AP, Monteiro J, Lucchi C, Kowbel DJ, Cordeiro JM, Correia-de-Sa P, Rigden CJ, Glass NL, Videira A. 2014. Extracellular calcium triggers unique transcriptional programs and modulates staurosporine-induced cell death in Neurospora crassa. Microb Cell 1:289–302. doi: 10.15698/mic2014.09.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takeuchi H, Haltiwanger RS. 2014. Significance of glycosylation in Notch signaling. Biochem Biophys Res Commun 453:235–242. doi: 10.1016/j.bbrc.2014.05.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dunlap JC, Borkovich KA, Henn MR, Turner GE, Sachs MS, Glass NL, McCluskey K, Plamann M, Galagan JE, Birren BW, Weiss RL, Townsend JP, Loros JJ, Nelson MA, Lambreghts R, Colot HV, Park G, Collopy P, Ringelberg C, Crew C, Litvinkova L, DeCaprio D, Hood HM, Curilla S, Shi M, Crawford M, Koerhsen M, Montgomery P, Larson L, Pearson M, Kasuga T, Tian C, Basturkmen M, Altamirano L, Xu J. 2007. Enabling a community to dissect an organism: overview of the Neurospora functional genomics project. Adv Genet 57:49–96. doi: 10.1016/S0065-2660(06)57002-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Colot HV, Park G, Turner GE, Ringelberg C, Crew CM, Litvinkova L, Weiss RL, Borkovich KA, Dunlap JC. 2006. A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc Natl Acad Sci U S A 103:10352–10357. doi: 10.1073/pnas.0601456103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCluskey K. 2003. The Fungal Genetics Stock Center: from molds to molecules. Adv Appl Microbiol 52:245–262. doi: 10.1016/S0065-2164(03)01010-4. [DOI] [PubMed] [Google Scholar]
- 29.Vogel H. 1956. A convenient growth medium for Neurospora. Microbial Genetics Bulletin 13:42–46. [Google Scholar]
- 30.Freitag M, Hickey PC, Raju NB, Selker EU, Read ND. 2004. GFP as a tool to analyze the organization, dynamics and function of nuclei and microtubules in Neurospora crassa. Fungal Genet Biol 41:897–910. doi: 10.1016/j.fgb.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 31.Freitag M, Selker EU. 2005. Expression and visualization of red fluorescent protein (RFP) in Neurospora crassa. Fungal Genet Newsl 52:14–17. [Google Scholar]
- 32.Margolin BS, Freitag M, Selker EU. 1997. Improved plasmids for gene targeting at the his-3 locus of Neurospora crassa by electroporation. Fungal Genet Newsl 44:34–36. [Google Scholar]
- 33.Cavinder B, Trail F. 2012. Role of Fig1, a component of the low-affinity calcium uptake system, in growth and sexual development of filamentous fungi. Eukaryot Cell 11:978–988. doi: 10.1128/EC.00007-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bardiya N, Shiu PK. 2007. Cyclosporin A-resistance based gene placement system for Neurospora crassa. Fungal Genet Biol 44:307–314. doi: 10.1016/j.fgb.2006.12.011. [DOI] [PubMed] [Google Scholar]
- 35.Castro-Longoria E, Ferry M, Bartnicki-Garcia S, Hasty J, Brody S. 2010. Circadian rhythms in Neurospora crassa: dynamics of the clock component frequency visualized using a fluorescent reporter. Fungal Genet Biol 47:332–341. doi: 10.1016/j.fgb.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bowman BJ, Draskovic M, Freitag M, Bowman EJ. 2009. Structure and distribution of organelles and cellular location of calcium transporters in Neurospora crassa. Eukaryot Cell 8:1845–1855. doi: 10.1128/EC.00174-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Talavera G, Castresana J. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56:564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- 40.Brown NP, Leroy C, Sander C. 1998. MView: a web-compatible database search or multiple alignment viewer. Bioinformatics 14:380–381. doi: 10.1093/bioinformatics/14.4.380. [DOI] [PubMed] [Google Scholar]
- 41.Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, Claverie JM, Gascuel O. 2008. Phylogeny.fr: robust phylogenetic analysis for the nonspecialist. Nucleic Acids Res 36:W465–W469. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Le SQ, Gascuel O. 2008. An improved general amino acid replacement matrix. Mol Biol Evol 25:1307–1320. doi: 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- 43.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer EL, Tate J, Punta M. 2014. Pfam: the protein families database. Nucleic Acids Res 42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, Bateman A, Bernard T, Binns D, Bork P, Burge S, de Castro E, Coggill P, Corbett M, Das U, Daugherty L, Duquenne L, Finn RD, Fraser M, Gough J, Haft D, Hulo N, Kahn D, Kelly E, Letunic I, Lonsdale D, Lopez R, Madera M, Maslen J, McAnulla C, McDowall J, McMenamin C, Mi H, Mutowo-Muellenet P, Mulder N, Natale D, Orengo C, Pesseat S, Punta M, Quinn AF, Rivoire C, Sangrador-Vegas A, Selengut JD, Sigrist CJ, Scheremetjew M, Tate J, Thimmajanarthanan M, Thomas PD, Wu CH, Yeats C, Yong SY. 2012. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res 40:D306–D312. doi: 10.1093/nar/gkr948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schultz J, Copley RR, Doerks T, Ponting CP, Bork P. 2000. SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Res 28:231–234. doi: 10.1093/nar/28.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 47.Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 48.Steentoft C, Vakhrushev SY, Joshi HJ, Kong Y, Vester-Christensen MB, Schjoldager KT, Lavrsen K, Dabelsteen S, Pedersen NB, Marcos-Silva L, Gupta R, Bennett EP, Mandel U, Brunak S, Wandall HH, Levery SB, Clausen H. 2013. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J 32:1478–1488. doi: 10.1038/emboj.2013.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gupta R, Jung E, Gooley AA, Williams KL, Brunak S, Hansen J. 1999. Scanning the available Dictyostelium discoideum proteome for O-linked GlcNAc glycosylation sites using neural networks. Glycobiology 9:1009–1022. doi: 10.1093/glycob/9.10.1009. [DOI] [PubMed] [Google Scholar]
- 50.Gupta R, Brunak S. 2002. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac Symp Biocomput 2002:310–322. [PubMed] [Google Scholar]
- 51.Palma-Guerrero J, Hall CR, Kowbel D, Welch J, Taylor JW, Brem RB, Glass NL. 2013. Genome wide association identifies novel loci involved in fungal communication. PLoS Genet 9:e1003669. doi: 10.1371/journal.pgen.1003669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Palma-Guerrero J, Huang IC, Jansson HB, Salinas J, Lopez-Llorca LV, Read ND. 2009. Chitosan permeabilizes the plasma membrane and kills cells of Neurospora crassa in an energy dependent manner. Fungal Genet Biol 46:585–594. doi: 10.1016/j.fgb.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 53.Muller EM, Mackin NA, Erdman SE, Cunningham KW. 2003. Fig1p facilitates Ca2+ influx and cell fusion during mating of Saccharomyces cerevisiae. J Biol Chem 278:38461–38469. doi: 10.1074/jbc.M304089200. [DOI] [PubMed] [Google Scholar]
- 54.Horton P, Park KJ, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, Nakai K. 2007. WoLF PSORT: protein localization predictor. Nucleic Acids Res 35:W585–W587. doi: 10.1093/nar/gkm259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moloney DJ, Panin VM, Johnston SH, Chen J, Shao L, Wilson R, Wang Y, Stanley P, Irvine KD, Haltiwanger RS, Vogt TF. 2000. Fringe is a glycosyltransferase that modifies Notch. Nature 406:369–375. doi: 10.1038/35019000. [DOI] [PubMed] [Google Scholar]
- 56.Bruckner K, Perez L, Clausen H, Cohen S. 2000. Glycosyltransferase activity of Fringe modulates Notch-Delta interactions. Nature 406:411–415. doi: 10.1038/35019075. [DOI] [PubMed] [Google Scholar]
- 57.Stanley P. 2007. Regulation of Notch signaling by glycosylation. Curr Opin Struct Biol 17:530–535. doi: 10.1016/j.sbi.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Manford AG, Stefan CJ, Yuan HL, Macgurn JA, Emr SD. 2012. ER-to-plasma membrane tethering proteins regulate cell signaling and ER morphology. Dev Cell 23:1129–1140. doi: 10.1016/j.devcel.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 59.Belden WJ, Barlowe C. 1996. Erv25p, a component of COPII-coated vesicles, forms a complex with Emp24p that is required for efficient endoplasmic reticulum to Golgi transport. J Biol Chem 271:26939–26946. doi: 10.1074/jbc.271.43.26939. [DOI] [PubMed] [Google Scholar]
- 60.Zucchi PC, Zick M. 2011. Membrane fusion catalyzed by a Rab, SNAREs, and SNARE chaperones is accompanied by enhanced permeability to small molecules and by lysis. Mol Biol Cell 22:4635–4646. doi: 10.1091/mbc.E11-08-0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Engel A, Walter P. 2008. Membrane lysis during biological membrane fusion: collateral damage by misregulated fusion machines. J Cell Biol 183:181–186. doi: 10.1083/jcb.200805182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Starai VJ, Jun Y, Wickner W. 2007. Excess vacuolar SNAREs drive lysis and Rab bypass fusion. Proc Natl Acad Sci U S A 104:13551–13558. doi: 10.1073/pnas.0704741104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Munro S, Freeman M. 2000. The notch signalling regulator fringe acts in the Golgi apparatus and requires the glycosyltransferase signature motif DXD. Curr Biol 10:813–820. doi: 10.1016/S0960-9822(00)00578-9. [DOI] [PubMed] [Google Scholar]
- 64.Hambleton S, Valeyev NV, Muranyi A, Knott V, Werner JM, McMichael AJ, Handford PA, Downing AK. 2004. Structural and functional properties of the human notch-1 ligand binding region. Structure 12:2173–2183. doi: 10.1016/j.str.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 65.Gazave E, Lapebie P, Richards GS, Brunet F, Ereskovsky AV, Degnan BM, Borchiellini C, Vervoort M, Renard E. 2009. Origin and evolution of the Notch signalling pathway: an overview from eukaryotic genomes. BMC Evol Biol 9:249. doi: 10.1186/1471-2148-9-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Neuwald AF. 1997. Barth syndrome may be due to an acyltransferase deficiency. Curr Biol 7:R465–R466. [DOI] [PubMed] [Google Scholar]
- 67.Fleissner A, Leeder AC, Roca MG, Read ND, Glass NL. 2009. Oscillatory recruitment of signaling proteins to cell tips promotes coordinated behavior during cell fusion. Proc Natl Acad Sci U S A 106:19387–19392. doi: 10.1073/pnas.0907039106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ren J, Wen L, Gao X, Jin C, Xue Y, Yao X. 2009. DOG 1.0: illustrator of protein domain structures. Cell Res 19:271–273. doi: 10.1038/cr.2009.6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.