

Abstract
α-Ketoglutarate decarboxylase (α-KDE1) is a Krebs cycle enzyme found in the mitochondrion of the procyclic form (PF) of Trypanosoma brucei. The bloodstream form (BF) of T. brucei lacks a functional Krebs cycle and relies exclusively on glycolysis for ATP production. Despite the lack of a functional Krebs cycle, α-KDE1 was expressed in BF T. brucei and RNA interference knockdown of α-KDE1 mRNA resulted in rapid growth arrest and killing. Cell death was preceded by progressive swelling of the flagellar pocket as a consequence of recruitment of both flagellar and plasma membranes into the pocket. BF T. brucei expressing an epitope-tagged copy of α-KDE1 showed localization to glycosomes and not the mitochondrion. We used a cell line transfected with a reporter construct containing the N-terminal sequence of α-KDE1 fused to green fluorescent protein to examine the requirements for glycosome targeting. We found that the N-terminal 18 amino acids of α-KDE1 contain overlapping mitochondrion- and peroxisome-targeting sequences and are sufficient to direct localization to the glycosome in BF T. brucei. These results suggest that α-KDE1 has a novel moonlighting function outside the mitochondrion in BF T. brucei.
INTRODUCTION
The protozoan parasite Trypanosoma brucei causes human African sleeping sickness and the chronic wasting disease nagana in cattle (1–3). T. brucei has a complex life cycle within an insect vector, the tsetse fly (Glossina sp.), and in the blood, lymphatics, and central nervous systems of mammals (4). During development, the parasite undergoes changes in both morphology and metabolism in response, in part, to the carbon source available for energy production. In mammals, bloodstream form (BF) T. brucei has an ample supply of glucose and exclusively utilizes glycolysis for energy production (5, 6). Most of the glycolytic enzymes are localized to the glycosome, a peroxisome-like organelle that catalyzes the conversion of glucose to glyceraldehyde 3-phosphate (7, 8). Consistent with the central role of glycolysis in ATP production, the mitochondrion of BF T. brucei is reduced to a simple, tubular, acristate organelle lacking both respiratory cytochromes and a functional Krebs cycle (4). This developmental stage of T. brucei is unable to carry out mitochondrial oxidative phosphorylation.
In the midgut of the tsetse fly, amino acids from digested blood meals replace glucose as the primary carbon source available to procyclic form (PF) T. brucei. PF T. brucei retains glycosomes, but the role of glycolysis in ATP production is reduced and a large, branched mitochondrion with numerous inner membrane cristae develops shortly after ingestion by the fly (4). Several Krebs cycle enzymes have been shown to be essential for energy metabolism in PF trypanosomes, but an intact Krebs cycle, catalyzing the degradation of glucose and amino acids to CO2, is not operative (9). Rather, internalized amino acids, primarily proline and glutamate, are degraded by the Krebs cycle enzymes α-ketoglutarate dehydrogenase (α-KD) and succinyl coenzyme A (succinyl-CoA) synthetase to succinate (9). Despite the noncyclic nature of the pathway, the Krebs cycle enzymes still provide high-energy electrons, via NADH and FADH2, to the electron transport chain that generates the electrochemical proton gradient necessary for mitochondrial oxidative phosphorylation.
α-KD is a large enzyme complex that catalyzes the conversion of α-ketoglutarate to succinyl-CoA. Multiple copies of the α-ketoglutarate decarboxylase (α-KDE1) (2-oxoglutarate dehydrogenase E1, Tb11.01.1740), dihydrolipoyl succinyltransferase (α-KDE2), and dihydrolipoamide dehydrogenase (α-KDE3) subunits are arranged for efficient substrate transfer between active sites (10). The reaction initiates with α-KDE1-mediated oxidative decarboxylation of α-ketoglutarate and the subsequent release of CO2. Succinyl formed from this step is transferred to CoA by the lipoyl group of α-KDE2; this is followed by the regeneration of the lipoic acid by the reduction of NAD+ via E3 (11). α-KD is a vital component of energy metabolism in most aerobic prokaryotes and eukaryotes. α-KDE1, α-KDE2, and α-KDE3 mRNAs are constitutively expressed in both BF and PF T. brucei trypanosomes, yet a functional Krebs cycle and α-KD activity are present only in PF trypanosomes (12). We showed that α-KDE2 is a bifunctional protein that localized to the mitochondrion of BF T. brucei but was not involved in energy production. Cell fractionation studies showed that α-KDE2 was tightly associated with the trypanosome mitochondrial genome, the kinetoplast DNA (kDNA), and was required for equal segregation of mitochondria and kDNA to daughter cells at cytokinesis (12). Other metabolic enzymes, including the Krebs cycle enzyme aconitase, have been shown to “moonlight,” carrying out multiple functions in other organisms (13, 14).
Here we report that α-KDE1 is also essential to BF T. brucei. RNA interference (RNAi) knockdown of α-KDE1 mRNA levels results in rapid growth arrest, morphological changes, and cell death within 24 h. Following α-KDE1 RNAi induction, the flagellar pocket rapidly swells to eventually occupy much of the cell. Electron microscopy showed that recruitment of both cell surface and flagellar membranes facilitated the formation of the swollen flagellar pocket. Furthermore, we found that α-KDE1 was undetectable in the BF mitochondrion but rather localized to glycosomes, suggesting that this canonical Krebs cycle enzyme can be differentially targeted in BF T. brucei and has a unique, essential function.
MATERIALS AND METHODS
Cell culture.
BF T. brucei strains TREU667 and 427 were grown at 37°C in 5% CO2 in HMI-9 medium containing 10% fetal bovine serum (FBS) (Gemini Bioproducts, West Sacramento, CA) and Serum Plus supplement (SAFC Biosciences, Lenexa, KS). The α-KDE1 RNAi T. brucei cell line was maintained in the same medium but with tetracycline-free FBS (10%).
Construction of cell lines.
Two 580- and 439-bp partial α-KDE1 (Tb11.01.1740) products were amplified from BF T. brucei 9013 genomic DNA with primers 5′-CCCTCGAGTGGCGCAGAGTCACTTATTG-3′ and 5′-CCAAGCTTAATGGGACACTGAAAGGCAC-3′ and primers 5′-CTCGAGGCCCACCGTGTAAATATGGA-3′ and 5′-AAGCTTACACGCGATTCAACGTGATA-3′, respectively, and ligated into the inducible pZJM RNAi vector to produce the α-KDE1 RNAi T. brucei cell lines (15). The construct was linearized with NotI for transfection. For the hemagglutinin (HA)-tagged cell lines, primers 5′-CCCCTCGAGCCGTGAATCCAACAACTGTGG-3′ and 5′-CCCCTCGAGTGAAAATACGCATTCGCAAA-3′ were used to amplify a partial α-KDE1 sequence (301 bp) from BF T. brucei TREU667 genomic DNA that was ligated into the modified pMOTag2H in situ tagging vector (16). The vector was linearized with a unique restriction site and transfected into wild-type BF T. brucei TREU667. The transfections were carried out with the Nucleofector system (Lonza, Walkersville, MD).
Northern analysis.
Total RNA was extracted from cells with TriPure Isolation Reagent (Roche, Indianapolis, IN), and transcripts were separated on a 7% formaldehyde–1% agarose gel. RNAs were transferred to a membrane and hybridized with radiolabeled probes prepared from open reading frames specific for α-KDE1 and β-tubulin with the Prime-It random primer labeling kit (Stratagene, Santa Clara, CA). Radiolabeled probes were hybridized with the trypanosome RNAs in a buffer containing 50% (vol/vol) formamide, 5× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate), 5× Denhardt's solution (Sigma, St. Louis, MO), 1% (wt/vol) SDS, and 100 μg/ml salmon sperm DNA (Life Technologies, Grand Island, NY) at 55°C overnight. Blots were washed three times at 30-min intervals in 0.2× SSC containing 0.1% SDS at 68°C, exposed to a storage phosphor screen (Molecular Dynamics), and analyzed on a STORM-860 PhosphorImager (GE Healthcare).
ConA binding assay.
RNAi T. brucei cells were induced with 1 μg/ml of doxycycline for 6, 12, and 18 h and washed with ice-cold HMI-9 medium without serum proteins. For concanavalin A (ConA)-fluorescein isothiocyanate (FITC) (Sigma) binding, cells were resuspended in 3°C serum-free HMI-9 medium containing 1% bovine serum albumin and 5 μg/ml ConA-FITC and incubated for 15 min. Cells were further incubated on ice for 5 min; washed in ice-cold, serum-free HMI-9 medium; and prepared for fluorescence microscopy.
α-KDE1–eGFP fusion construct and colocalization.
The 17-amino-acid N-terminal signal sequence from α-KDE1 was fused to enhanced green fluorescent protein (eGFP) to test localization. Fusion was carried out with a primer containing the α-KDE1 signal sequence (italicized in the primer sequence) and regions specific for eGFP with forward primer 5′-GATCAAGCTTATGATGCGAAGGCTCAGTCCTGTGAACGGTTCGGTGGTTTCGCCCAATGTCATGAGTAAAGGAGAAGAACTTTTC-3′ and a reverse primer specific for eGFP (5′-GATCGGATCCTTATTTGTATAGTTCATCCATGCC-3′). The α-KDE1–eGFP fusion gene was cloned into the BamHI/HindIII site of the pLew100 vector (17). BF T. brucei 9013 cells were transfected and used for expression and colocalization studies.
Fluorescence microscopy.
BF T. brucei was smeared onto a microscope slide, rapidly air dried, and fixed in methanol (−20°C) for 10 min. Slides were rinsed and blocked with 20% FBS in phosphate-buffered saline (PBS) for 30 min. α-KDE1 RNAi or α-KDE1–HA cells, respectively, were used to localize the RNAi-induced posterior vacuole or to localize α-KDE1. Antibodies against the paraflagellar rod (PFR) protein (1:500) were used in combination with 4′,6-diamidino-2-phenylindole (DAPI) staining to localize the flagellar pocket. The localization of α-KDE1–HA in BF T. brucei was determined by staining with MitoTracker (Life Technologies, Grand Island, NY), antibodies against the HA epitope (1:100; Abcam, Cambridge, MA), and antibodies against the T. brucei aldolase (James Morris, Clemson University) (14). All antibodies were diluted in blocking buffer, and cells were incubated with the primary antibody for 1 h. Slides were then washed with PBS and incubated with the appropriate secondary antibody (1:500) for 30 min in the same blocking buffer. Following incubation with the secondary antibody, slides were washed with PBS and coated with DAPI containing the antifade reagent ProlongGold (Life Technologies). Images were acquired with a Zeiss Axio Observer inverted microscope equipped with an AxioCam HSm and evaluated with AxioVision v4.6 software (Zeiss).
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM).
Induced α-KDE1 RNAi BF T. brucei cells were fixed in 2.5% glutaraldehyde, 2% paraformaldehyde, 2 mM CaCl2, and 100 mM cacodylate (pH 7) in a 1:1 ratio (cell medium to fixative) for 30 min at 25°C. Cells were pelleted twice in serum-free HMI-9 medium, washed for 1 h in a buffer containing 200 mM sucrose and 100 mM cacodylate (pH 7.4), and postfixed with 1% OsO4 in 100 mM cacodylate buffer for 1 h. Fixed cells were washed in distilled H2O and dehydrated through a graded ethanol series. Cell pellets were embedded in Epon resin, and sections were prepared and stained with uranyl acetate and lead citrate. Images were taken with a JEOL-JEM 1210 transmission electron microscope (JEOL). For SEM, 2.5% glutaraldehyde-fixed cells were dehydrated on a 0.22-μm membrane, critical point dried, sputter coated with gold, and viewed with a Zeiss 1450EP scanning electron microscope (Zeiss).
Western blot analysis.
Total cell protein from wild-type and α-KDE1–HA T. brucei was denatured in reducing SDS loading buffer and fractionated by SDS-PAGE. Proteins from the gel were transferred to a membrane, blocked with 5% (wt/vol) milk TBS-T (150 mM NaCl, 10 mM Tris-HCl [pH 8], 0.05% [vol/vol] Tween 20), and incubated overnight with primary antibodies against HA (1:2,000; Abcam) epitopes. The blot was then washed, incubated with a horseradish peroxidase-conjugated secondary antibody (1:5,000) for 1 h, washed again with TBS-T, and developed.
RESULTS
α-KDE1 is essential in BF T. brucei.
Previous analysis of α-KDE1, α-KDE2, and α-KDE3 steady-state mRNA levels showed constitutive expression in both PF and BF T. brucei despite the lack of a functional Krebs cycle in BF developmental stages of this parasite (12, 18). To examine the function of α-KDE1 in BF T. brucei, an inducible RNAi T. brucei cell line was prepared. Treatment with doxycycline resulted in rapid growth arrest, within 6 h, followed by a decrease in cell number, indicating a cytocidal effect of the α-KDE1 RNAi (Fig. 1A). Northern blot analysis revealed a slight decrease in α-KDE1 mRNA at 6 h postinduction and a further reduction, ∼55% of the preinduction levels, after 24 h (Fig. 1B). However, after 24 h of RNAi, approximately 90% of the cells were dead. This lethality was not due to off-target effects of the RNAi, since similar effects were observed when another, nonoverlapping, sequence in the α-KDE1 mRNA was targeted for RNAi silencing (see Fig. S1 and S2 in the supplemental material). These results are consistent with the findings of Alsford et al., who reported that a minimal reduction in α-KDE1 mRNA resulted in a loss of fitness under all BF cell conditions. A minimal reduction of RNA levels resulted in a loss of fitness of ∼70 other BF mRNAs in those experiments (19).
FIG 1.

α-KDE1 is essential in BF T. brucei. Effect of α-KDE1 RNAi knockdown on the growth and morphology of BF T. brucei. (A) Growth of α-KDE1 RNAi T. brucei cells in culture at 37°C in the presence (+) or absence (−) of doxycycline. (B) Northern blot analysis of the levels of α-KDE1 and β-tubulin mRNAs. Total cell RNA was isolated following induction with doxycycline, fractionated on agarose gels, and hybridized with specific radioactively labeled probes for α-KDE1 and tubulin. (C) DIC images taken from videos of α-KDE1 RNAi T. brucei cells following induction with doxycycline for 0, 6, 12, and 18 h. The position of the expanding posterior vacuole is indicated (arrow). (D) SEM of α-KDE1 RNAi T. brucei following treatment with doxycycline. The position of the flagellum (f) is indicated.
Morphological and motility changes in α-KDE1 RNAi T. brucei cells.
Accompanying the RNAi-induced growth arrest was the formation of a 1- to 2-μm vacuole at the posterior end of the cell within 6 h. This vacuole progressively expanded until it occupied much of the cytoplasm of the cells after 18 to 24 h (Fig. 1C; see Fig. S2 in the supplemental material). Live-cell imaging of RNAi-induced cells also showed that α-KDE1 RNAi knockdown resulted in loss of the rapid tumbling motility characteristic of BF T. brucei. Motility was severely restricted by 6 h after RNAi induction, and this correlated with the formation of the posterior vacuole and inclusion of actively moving flagella within the vacuole at 12 to 18 h postinduction (see Fig. S3 in the supplemental material). SEM of α-KDE1 RNAi T. brucei also showed time-dependent changes in the overall morphology with progressive cell swelling originating from the posterior end of the cell (Fig. 1D). Together, these results showed that α-KDE1 was essential for the survival of BF T. brucei and that even small changes in steady-state levels of α-KDE1 mRNA resulted in the rapid arrest of cell growth and dramatic changes in motility and morphology. Since the Krebs cycle is inoperative in BF T. brucei, these results suggested an alternative function for α-KDE1.
In order to better define the morphological changes induced by α-KDE1 RNAi, we used fluorescence microscopy to examine the position of the RNAi-induced vacuole relative to (i) the kDNA, which is located adjacent to the basal bodies at the base of the flagellum; (ii) ConA-reactive mannose residues, which are only found exposed on the T. brucei surface in the flagellar pocket; and (iii) the PFR protein, which is associated with the flagellar axoneme after the flagellum exits the flagellar pocket. Thus, on the basis of these markers, the flagellar pocket can be defined as the area between the kDNA and the PFR protein that binds ConA. At 6 h postinduction, a small posteriorly located vacuole was visible that colocalized with ConA staining and was positioned between the kDNA and the PFR protein-stained portion of the flagellum (Fig. 2A). The enlarged vacuole seen by differential interference contrast (DIC) microscopy at 12 and 18 h postinduction retained its position relative to the kDNA and the PFR protein, but only a small portion of the vacuole stained with ConA (Fig. 2B and C). The nonuniform distribution of the ConA staining over time made it difficult to determine whether the vacuole was the product of the swelling of a single flagellar pocket or rather a collection of closely packed vacuoles.
FIG 2.
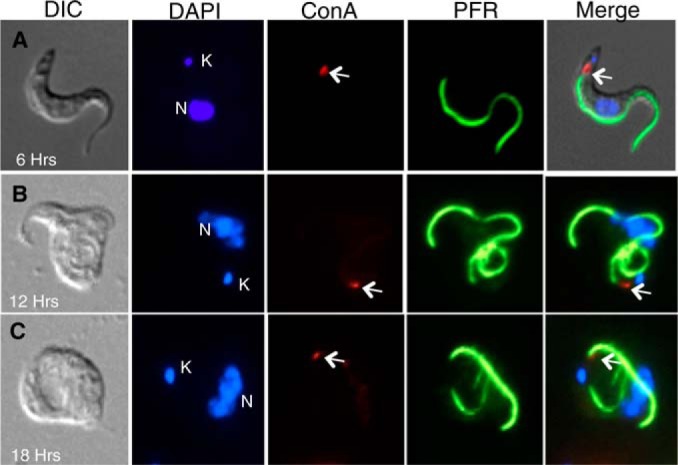
Localization of the α-KDE1 RNAi-induced vacuole. (A to C) Following induction with doxycycline for 6, 12, or 18 h, α-KDE1 RNAi T. brucei cells were incubated at 3°C with ConA-FITC, fixed, incubated with antibodies against the PFR protein, and stained with DAPI. The positions of the DAPI-stained kinetoplast (K) and nucleus (N) are indicated, as is that of bound ConA (arrow).
Depletion of α-KDE1 results in an enlarged flagellar pocket.
Vesicular trafficking in African trypanosomes is highly polarized, with the flagellar pocket serving as the site for all secretion and endocytosis (20, 21). The swollen posterior vacuoles seen in the α-KDE1 RNAi T. brucei cells (Fig. 1C and 2B and C) resembled the swollen flagellar pocket in BF T. brucei following RNAi silencing of genes encoding proteins involved in endocytosis (22–26). However, we observed no change in endocytosis rates of ConA (see Fig. S4A and B in the supplemental material).
We used TEM to examine thin sections of fixed cells taken at 6, 12, 18, and 24 h postinduction in order to determine if the vacuole in α-KDE1 RNAi T. brucei cells was the flagellar pocket. By 18 h, most cells had a prominent cytosolic vacuole (Fig. 3A). During a time course of α-KDE1 RNAi, the vacuole increased in size until it occupied much of the cytoplasm after 18 to 24 h (Fig. 3A to F). Most cells contained a single vacuole, even when it had expanded to occupy much of the cell, and the presence of flagella confirmed that α-KDE1 RNAi resulted in swelling of the flagellar pocket.
FIG 3.

α-KDE1 RNAi causes flagellar-pocket swelling. TEM images of α-KDE1 RNAi BF T. brucei are shown. (A) Low-magnification image of a field of cells 18 h after RNAi induction showing a high percentage of cells having a large intracellular vacuole. α-KDE1 RNAi-treated cells taken at the time of doxycycline induction (B) and after 6 h (C), 12 h (D), 18 h (E), and 24 h (F). The positions of the flagellar pocket (FP), flagellum (F), and kinetoplast (K) are indicated.
Abnormal morphology of α-KDE1 RNAi T. brucei cells.
A girdle of subpellicular microtubules is closely juxtaposed to the cytosolic face of the plasma membrane of trypanosomes. This unusual structure contributes to maintenance of the overall shape and cellular motility. A space in the subpellicular microtubules array corresponds to the opening of the flagellar pocket where the flagellum emerges and leaves the pocket membrane free of subpellicular microtubules. The absence of subpellicular microtubules at the flagellar pocket is an important structural feature of trypanosomes and is likely necessary to allow vesicle transport between the cell and the external environment (27, 28). The mechanism excluding the assembly of subpellicular microtubules at the flagellar pocket is not known; however, in α-KDE1 RNAi T. brucei cells, we found that patches of the expanded flagellar pocket membrane contained subpellicular microtubules, suggesting that plasma membrane, from outside the pocket, may be recruited to the rapidly expanding flagellar pocket upon RNAi induction or that subpellicular microtubules are no longer excluded from this region (Fig. 4A).
FIG 4.

Flagellar and plasma membranes are recruited to form the expanding flagellar pocket. TEM images of BF T. brucei following induction of α-KDE1 RNAi with doxycycline are shown. (A) Plasma membrane-associated pellicular microtubules are found on the membrane of the expanding flagellar pocket. The inset is a higher magnification of a portion of the flagellar membrane with associated subpellicular microtubules (SM). (B) Both axonemes (Ax) and the PFR protein, stripped of flagellar membrane, are displaced to the cytoplasm of α-KDE1 RNAi T. brucei cells. The inset is a higher-magnification view of a stripped axoneme and associated PFR protein in the cytoplasm.
The dynamic changes in the flagellar pocket membrane were accompanied by changes in the overall appearance of the flagellum in α-KDE1 RNAi T. brucei cells. At the light microscope level, the flagellum often appeared to be coiled within the flagellar pocket or associated with the cytoplasm of the trypanosome (Fig. 2; see Fig. S5 in the supplemental material). Several alterations in the flagellum of α-KDE1 RNAi T. brucei were observed by TEM, including the presence of flagellar axonemes, bare of surrounding membranes, in the cytoplasm (Fig. 4B and C). In addition, the cytosolic axonemes often contained associated PFR protein structures, suggesting a selective stripping of the specialized flagellar membrane as the axonemes moved into the cytoplasm (Fig. 4B and C; see Fig. S6A in the supplemental material). Further evidence of dynamic changes at the flagellar membrane in α-KDE1 RNAi T. brucei cells was the presence of a large number of flagella that appeared to contain multiple axonemes and PFR protein complexes (Fig. 4C; see Fig. S6B to E in the supplemental material). The overall recruitment of both plasma and flagellar membranes correlates with the rapid expansion of the flagellar pocket, suggesting that sequestration of membrane components from these contiguous sites may allow rapid expansion of the flagellar pocket.
Association of KDE1 with T. brucei glycosomes.
Electron microscopy of α-KDE1 RNAi T. brucei revealed other unexpected features. We observed that α-KDE1 RNAi T. brucei cells appeared to contain clustered putative glycosomes with a single membrane (Fig. 5; see Fig. S7A to E in the supplemental material). The glycosomes in α-KDE1 RNAi T. brucei were often concentrated near the flagellar pocket (Fig. 5B and C), and many were abnormally elongate and bilobed in structure (Fig. 5A and B). To investigate whether the changes in glycosome abundance and morphology were a direct consequence of α-KDE1 RNAi, we first examined the cellular localization of cells expressing an epitope-tagged copy of α-KDE1–HA. To establish the specificity of the HA antibody, wild-type and α-KDE1–HA total cell lysates were prepared, fractionated by SDS-PAGE, and analyzed by Western blotting with anti-HA antibody (Fig. 6A). The anti-HA antibody did not react with proteins from nontransfected, wild-type T. brucei, and a single 116-kDa immunoreactive band, the expected size for T. brucei α-KDE1–HA, was observed in the transfected cell lysates. The localization of α-KDE1–HA was investigated with Mitotracker to identify the BF T. brucei mitochondrion and immunofluorescence microscopy, with anti-HA antibody, to identify KDE1-HA (Fig. 6B). α-KDE1–HA was distributed throughout the cytoplasm as small punctate structures and did not appear to colocalized with the mitochondrion. This is in contrast to the mitochondrial localization of α-KDE2 in BF T. brucei (12). The punctate cytoplasmic localization of α-KDE1–HA was reminiscent of the distribution of glycosomes in T. brucei (29, 30). Immunofluorescence microscopy with an antibody against the glycolytic enzyme aldolase confirmed that α-KDE1–HA was localized to glycosomes in BF T. brucei (Fig. 6C). These results suggest that α-KDE1 has an unknown function within the glycosome of BF T. brucei. Further, our findings suggest that the trypanocidal effects and morphological changes associated with the RNAi knockdown of α-KDE1 are a consequence of the loss of this function.
FIG 5.
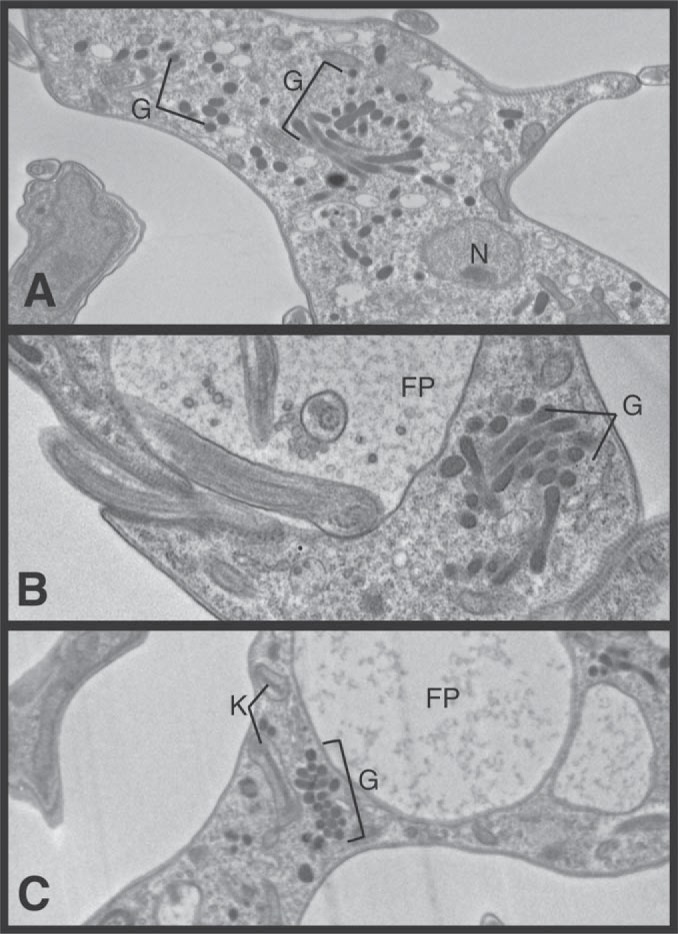
Morphological changes in glycosomes in α-KDE1 RNAi T. brucei. TEM images of BF T. brucei following induction of α-KDE1 RNAi with doxycycline are shown. (A to C) At 18 h after induction of α-KDE1, RNAi revealed clusters of elongated glycosomes throughout the cytoplasm but predominately near the flagellar pocket. The positions of the flagellar pocket (FP), glycosomes (G), kinetoplast (K), and nucleus (N) are indicated.
FIG 6.

Localization of α-KDE1 to the glycosome of BF T. brucei. α-KDE1 was tagged with a C-terminal HA epitope and used to prepare a constitutively expressing α-KDE1–HA cell line. (A) Total cell protein from wild-type (WT) and α-KDE1–HA (E1) cells was fractionated by SDS-PAGE and analyzed by Western blotting. On the left is the Coomassie blue-stained gel, and on the right is the blot following incubation with anti-HA antibody. The values to the left are molecular sizes in kilodaltons. (B, C) Localization of α-KDE1–HA by immunofluorescence microscopy relative to the mitochondrion stained with MitoTracker (B) and aldolase (C). The positions of the nucleus (N) and kinetoplast (K) are indicated.
α-KDE1 contains overlapping N-terminal mitochondrial and glycosomal signal sequences.
The unexpected localization of α-KDE1 to BF T. brucei glycosomes raised the question of how this protein was differentially targeted to mitochondria and glycosomes. Studies of the import of proteins into trypanosome glycosomes and mitochondria have led to the identification of amino acid sequences that can specifically target both organelles. The mitochondrial targeting signals (MTS) are largely, but not exclusively, restricted to N-terminal amino acids that can be as short as five residues in trypanosomes (31–33). T. brucei proteins require either C-terminal peroxisomal targeting signal 1 (PTS1) or N-terminal PTS2 sequences for import into glycosomes (34–37). α-KDE1 has a highly conserved N-terminal MTS (MMRRL) and lacks the characteristic tripeptide C-terminal PTS1 sequence but contains an N-terminal sequence, overlapping the MTS, containing residues conserved in glycosome and peroxisome PTS2 sequences (MMRRLSPVNGSV) with a highly conserved basic amino acid (arginine) at position 4 (in bold) and hydrophobic residues at positions 5, 8, and 12 (also in bold) (38) (Fig. 7A). To determine whether the N-terminal sequence of α-KDE1 functions as a glycosome-targeting sequence, we fused the first 18 amino acids to the coding sequence for the reporter protein eGFP and cloned it into a tetracycline-regulated vector to allow expression in BF T. brucei (Fig. 7B). The localization of α-KDE1–eGFP was determined by fluorescence microscopy in a stable cell line. Consistent with the localization of full-length α-KDE1–HA, α-KDE1–eGFP localized exclusively to glycosomes of BF T. brucei, indicating that the N-terminal 18 amino acids of α-KDE1 contain functional PTS2 (Fig. 7C and D).
FIG 7.

α-KDE1 contains an N-terminal glycosome-targeting signal. (A) Alignment of N-terminal amino acid sequences of α-KDE1, human and yeast peroxisomal, trypanosome glycosomal, and trypanosome mitochondrial proteins. The proposed trypanosome α-KDE1 MTS sequence (red) and PTS2 sequence (green) are shown. The arginine at position 4 and the leucine at position 5 (yellow) overlap in the predicted MTS and PTS2 sequences. Residues highly conserved in all PTS2 sequences are in bold (positions 4, 5, 8, and 12 in α-KDE1). (B) A fusion construct used to produce an α-KDE1–eGFP reporter contains the N-terminal 18 amino acids of α-KDE1 and the coding sequence for eGFP. (C, D) Following induction, eGFP localization was determined by fluorescence microscopy with cells stained with antialdolase antibody (C) and Mitotracker (D). The positions of the nucleus (n) and kinetoplast (k) are indicated.
DISCUSSION
Organisms use a wide array of mechanisms to compensate for a seemingly limitless need for biological diversity in the face of rather limited genetic potential. Generation of functionally distinct proteins from a single gene by genetic recombination, alternative mRNA processing, and posttranslational modifications contributes to changes in all organisms in response to environmental and developmental cues (39–43). In addition, a small but significant number of proteins can have multiple functions without sequence or posttranslation changes. The moonlighting functions of several canonical metabolic enzymes have been described in mammals, fungi, plants, and protozoa (12, 13, 44). Identifying moonlighting activities for essential proteins is difficult since conventional loss-of-function analyses generally cannot distinguish a single activity from multiple activities for a protein. We have begun to investigate potential moonlighting activities of mitochondrial proteins in African trypanosomes. The developmental regulation of mitochondrial carbohydrate metabolism in T. brucei allowed us to initially investigate the function of the enzyme components of the inoperative Krebs cycle in BF T. brucei. We previously reported that α-KDE2 was expressed in BF T. brucei and was associated with the kDNA network and mitochondrial membrane. This protein was essential for the maintenance of the kDNA during cell division (12). The studies reported here show that α-KDE1 is also essential in BF T. brucei since RNAi knockdown resulted in growth arrest and caused death within 24 h. Furthermore, we observed morphological changes in α-KDE1 RNAi T. brucei that included extensive and rapid swelling of the flagellar pocket, which was mediated by sequestering of both flagellar and plasma membranes into the pocket. The function of α-KDE1 in BF T. brucei was addressed by examining the intracellular localization of the protein by immunofluorescence microscopy. Unexpectedly we found that α-KDE1 localized exclusively to glycosomes in BF T. brucei and we showed that the N-terminal 18 amino acids of α-KDE1 contained overlapping mitochondrial and glycosomal targeting sequences. Together, these results showed that α-KDE1 was preferentially targeted to glycosomes in BF T. brucei and that while the function of α-KDE1 in glycosomes is unknown, it is essential.
The knockdown of α-KDE1 mRNA by RNAi resulted in rapid expansion of the flagellar pocket. The resultant cells, after 12 to 18 h of induction, resembled the “big eye” cells that were first observed in clathrin and later in dynamin-like protein RNAi knockdowns (22, 25). In both cases, the expansion of the flagellar pocket was explained by decreased endocytosis since secretion was unaffected. In the α-KDE1 RNAi T. brucei cells, endocytosis was not affected and the expansion of the pocket appeared to result from the recruitment of membrane from both the plasma membrane outside the pocket and the flagellar membrane. While we do not know the role that α-KDE1 plays in the maintenance of the flagellar pocket, our results suggest that even small changes in α-KDE1 mRNA dramatically alter membrane dynamics in these organisms. It is possible that the high fluidity of the BF T. brucei plasma membrane contributes to membrane mobilization in α-KDE1 RNAi T. brucei cells. Rapid lateral mobility of glycosyl-phosphatidylinositol-anchored molecules is necessary to allow clearance of antibodies against the variant surface glycoprotein, preventing early killing of BF trypanosomes (45). It is possible that the high fluidity of BF T. brucei membranes requires positive regulatory mechanisms to maintain functional subdomains within the contiguous membrane systems of the plasma, flagellum, and flagellar pocket. It is difficult to predict the role of α-KDE1 in such a pathway because of the complexity of the metabolic and biosynthetic pathways in glycosomes; however, analysis of α-KDE1-associated proteins in BF T. brucei may provide additional insight.
The localization of α-KDE1 to the BF T. brucei glycosome was explained by the identification of an N-terminal PTS2 consensus sequence (38). Peroxisomal import of both PTS1- and PTS2-containing proteins requires a family of proteins, peroxins (PEX), that recognize PTS1 or PTS2 and allow import (46). Several homologues of the PEX proteins have now been identified in trypanosomes and have been shown to be necessary for protein import into the glycosome (47–50). The exclusive localization of α-KDE1 to the glycosome of BF T. brucei suggests that PTS2 dominates targeting in the BF while the MTS directs localization to PF mitochondria when α-KDE1 assembles into a functional Krebs cycle enzyme complex (12). Dual targeting of peroxisomal proteins has been described for a range of eukaryotes, and the mechanism of targeting to different organelles can be the result of alternative transcription start sites, polyadenylation, or splicing, giving rise to proteins with distinct targeting sequences (51, 52). Proteins that are dually targeted to mitochondria and peroxisomes may have an N-terminal MTS and a C-terminal PTS1. In the case of type II NAD(P)H dehydrogenases (ND) in Arabidopsis, the intracellular distribution of NDs is dependent on the affinity of the NDs for the mitochondrial or peroxisomal receptors (53). Differential phosphorylation at serines near PTS2 can also interfere with peroxisomal targeting (54).
There are several potential mechanisms for the differential targeting of α-KDE1 to the mitochondrion of PF T. brucei and to the glycosomes of BF cells. While lacking cis splicing, all trypanosome mRNAs are processed by the addition of a 39-nucleotide RNA at the 5′ end by trans splicing. Recent studies have shown that alternative trans-spliced mRNAs can be translated to isoforms of proteins that are differentially localized to the mitochondrion, nucleus, or cytosol (18). However, analysis of the transcriptome data for the 5′ ends of PF and BF T. brucei mRNAs did not reveal heterogeneity at the 5′ end of α-KDE1 mRNAs that could alter MTS or PTS2 sequences (18). Rather, it seems likely that the differential localization of α-KDE1 is a consequence of the relative efficiencies of the import of α-KDE1 into the glycosome and mitochondrion of BF and PF T. brucei.
It is tempting to speculate that since BF T. brucei lacks cytochrome-mediated electron transport, the energetic states of the BF and PF may differ and selectively influence protein import. However, the measured mitochondrial membrane potentials of PF and BF T. brucei are nearly identical (130 to 140 mV) and import of proteins into BF T. brucei mitochondria has been shown to be dependent on a membrane potential (5, 55, 56). In contrast to α-KDE1, α-KDE2 remains targeted to the mitochondrion in BF (although it then has a different, moonlighting function in that organelle); thus, the use (expression and routing) of these proteins, which usually are part of a single mitochondrial complex, is uncoupled in BF cells (12). Reduced mitochondrial α-KDE1 was also not a consequence of a deficiency in the general protein import machinery since both mitochondria and glycosomes import a number of proteins constitutively during development. While we do not know the molecular basis for the selective targeting of α-KDE1 to the PF mitochondria and the BF glycosome, an analogous situation has been described for the distribution of catalase A in yeast (55). Catalase A is a peroxisomal protein necessary for the detoxification of oxygen radicals and serves as a scavenger of H2O2 produced by peroxisomal enzymes. However, when cultivated under respiratory growth conditions, where reactive oxygen species accumulate in mitochondria, yeast imports catalase A into both peroxisomes and mitochondria. The changes we have observed in the distribution of α-KDE1 during T. brucei development mirrors the metabolic state of the mitochondrion, suggesting that metabolic sensing may play a role in establishing the cellular distribution of this and other moonlighting proteins.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jim Morris (Clemson University) for antibodies against trypanosome aldolase; Anzio Gartrell, John Shields, and Mary Ard (University of Georgia) for assistance with electron microscopy; and Torsten Ochsenreiter (Bern University) for insight into the mechanisms of protein diversification in trypanosomes. We also thank members of the Hajduk and Sabatini labs for helpful discussion and comments on the manuscript.
This work was supported by NIH grants AI21401 and AI39033.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00214-14.
REFERENCES
- 1.Simarro PP, Diarra A, Postigo JAR, Franco JR, Jannin JG. 2011. The human African trypanosomiasis control and surveillance programme of the World Health Organization 2000-2009: the way forward. PLoS Negl Trop Dis 5:e1007. doi: 10.1371/journal.pntd.0001007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van den Bossche P, de La Rocque S, Hendrickx G, Bouyer JA. 2010. Changing environment and the epidemiology of tsetse-transmitted livestock trypanosomiasis. Trends Parasitol 26:236–243. doi: 10.1016/j.pt.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 3.Wolburg H, Mogk S, Acker S, Frey C, Meinert M, Lazarus M, Urade Y, Kubata BK, Duszenko M. 2012. Late stage infection in sleeping sickness. PLoS One 7:e34304. doi: 10.1371/journal.pone.0034304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matthews KR. 2005. The developmental cell biology of Trypanosoma brucei. J Cell Sci 118:283–290. doi: 10.1242/jcs.01649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bringaud F, Riviere L, Coustou V. 2006. Energy metabolism of trypanosomatids: adaptation to available carbon sources. Mol Biochem Parasitol 149:1–9. doi: 10.1016/j.molbiopara.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 6.Timms MW, van Deursen FJ, Hendricks EF, Matthews KR. 2002. Mitochondrial development during life cycle differentiation of African trypanosomes: evidence kinetoplast-dependent differentiation control point. Mol Biol Cell 13:3747–3759. doi: 10.1091/mbc.E02-05-0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parsons M. 2004. Glycosomes: parasites and the divergence of peroxisomal purpose. Mol Microbiol 53:717–724. doi: 10.1111/j.1365-2958.2004.04203.x. [DOI] [PubMed] [Google Scholar]
- 8.Michels PAM, Bringaud F, Herman M, Hannaert V. 2006. Metabolic functions of glycosomes in trypanosomatids. Biochim Biophys Acta 1763:1463–1477. doi: 10.1016/j.bbamcr.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 9.van Weelden SW, Fast B, Vogt A, van Der Meer P, Saas J, van Hellemond JJ, Tielens AGM, Boshart M. 2003. Procyclic Trypanosoma brucei do not use Krebs cycle activity for energy generation. J Biol Chem 278:12854–12863. doi: 10.1074/jbc.M213190200. [DOI] [PubMed] [Google Scholar]
- 10.Perham RN. 1991. Domains, motifs, and linkers in 2-oxo acid dehydrogenase multienzyme complexes: a paradigm in the design of a multifunctional protein. Biochemistry 30:8501–8512. doi: 10.1021/bi00099a001. [DOI] [PubMed] [Google Scholar]
- 11.Perham RN. 2000. Swinging arms and swinging domains in multifunctional enzymes: catalytic machines for multistep reactions. Annu Rev Biochem 69:961–1004. doi: 10.1146/annurev.biochem.69.1.961. [DOI] [PubMed] [Google Scholar]
- 12.Sykes SE, Hajduk SL. 2013. Dual functions of α-ketoglutarate dehydrogenase E3 in the Krebs cycle and mitochondrial DNA inheritance in Trypanosoma brucei. Eukaryot Cell 12:78–90. doi: 10.1128/EC.00269-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jeffery CJ. 2009. Moonlighting proteins—an update. Mol Biosyst 5:345–350. doi: 10.1039/b900658n. [DOI] [PubMed] [Google Scholar]
- 14.Chen XJ, Wang X, Kaufman BA, Butow RA. 2005. Aconitase couples metabolic regulation to mitochondrial DNA maintenance. Science 307:714–717. doi: 10.1126/science.1106391. [DOI] [PubMed] [Google Scholar]
- 15.Wang Z, Morris JC, Drew ME, Englund PT. 2000. Inhibition of Trypanosoma brucei gene expression by RNA interference using an integratable vector with opposing T7 promoters. J Biol Chem 275:40174–44017. doi: 10.1074/jbc.M008405200. [DOI] [PubMed] [Google Scholar]
- 16.Oberholzer M, Morand S, Kunz S, Seebeck T. 2006. A vector series for rapid PCR-mediated C-terminal in situ tagging of Trypanosoma brucei genes. Mol Biochem Parasitol 145:117–120. doi: 10.1016/j.molbiopara.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 17.Wirtz E, Leal S, Ochatt C, Cross GAM. 1999. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol Biochem Parasitol 99:89–101. doi: 10.1016/S0166-6851(99)00002-X. [DOI] [PubMed] [Google Scholar]
- 18.Nilsson D, Gunasekera K, Mani J, Osteras M, Farinelli L, Baerlocher L, Roditi I, Ochsenreiter T. 2010. Spliced leader trapping reveals widespread alternative splicing patterns in the highly dynamic transcriptome of Trypanosoma brucei. PLoS Pathog 6:e1001037. doi: 10.1371/journal.ppat.1001037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alsford S, Turner DJ, Obado SO, Sanchez-Flores A, Glover L, Berriman M, Hertz-Fowler C, Horn D. 2011. High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res 21:915–924. doi: 10.1101/gr.115089.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Field MC, Carrington M. 2009. The trypanosome flagellar pocket. Nat Rev Microbiol 7:775–786. doi: 10.1038/nrmicro2221. [DOI] [PubMed] [Google Scholar]
- 21.Silverman JS, Bangs JD. 2012. Form and function in the trypanosomal secretory pathway. Curr Opin Microbiol 15:463–468. doi: 10.1016/j.mib.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allen CL, Goulding D, Field MC. 2003. Clathrin-mediated endocytosis in essential in Trypanosoma brucei. EMBO J 22:4991–5002. doi: 10.1093/emboj/cdg481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ali M, Leung KF, Field MC. 2014. The ancient small GTPase Rab21 functions in intermediate endocytic steps in trypanosomes. Eukaryot Cell 13:304–319. doi: 10.1128/EC.00269-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall B, Allen CL, Goulding D, Field MC. 2004. Both of the Rab5 subfamily small GTPases of Trypanosoma brucei are essential and required for endocytosis. Mol Biochem Parasitol 138:67–77. doi: 10.1016/j.molbiopara.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 25.Chanez AL, Hehl AB, Engsler M, Schneider A. 2006. Ablation of the single dynamin of T. brucei blocks mitochondrial fission and endocytosis and leads to precise cytokinesis arrest. J Cell Sci 119:2968–2974. doi: 10.1242/jcs.03023. [DOI] [PubMed] [Google Scholar]
- 26.García-Salcedo J, Perez-Morga D, Gijon P, Dibeck V, Pays E, Nolan DP. 2004. A differential role for actin during the life cycle of Trypanosoma brucei. EMBO J 23:780–789. doi: 10.1038/sj.emboj.7600094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gadelha C, Rothery S, Morhew M, MeIntosh JR, Severs NJ, Gull K. 2009. Membrane domains and flagellar pocket boundaries are influenced by the cytoskeleton in African trypanosomes. Proc Natl Acad Sci U S A 106:17425–17430. doi: 10.1073/pnas.0909289106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balber AE. 1990. The pellicle and membrane of the flagellum, flagellar adhesion zone and flagellar pocket: functionally discrete surface domains in the bloodstream form of African trypanosomes. Crit Rev Immunol 10:177–201. [PubMed] [Google Scholar]
- 29.Clayton CE, Michels P. 1996. Metabolic compartmentation in African trypanosomes. Parasitol Today 12:465–471. doi: 10.1016/S0169-4758(96)10073-9. [DOI] [PubMed] [Google Scholar]
- 30.Guerra-Giraldez C, Quijada L, Clayton CE. 2002. Compartmentation of enzymes in a microbody, the glycosome, is essential in Trypanosoma brucei. J Cell Sci 115:2651–2658. [DOI] [PubMed] [Google Scholar]
- 31.Priest JW, Hajduk SL. 1995. The trypanosomatid Rieske iron-sulfur proteins have a cleaved presequence that may direct mitochondrial import. Biochim Biophys Acta 1269:201–201. doi: 10.1016/0167-4889(95)00154-6. [DOI] [PubMed] [Google Scholar]
- 32.Häusler T, Stierhof YD, Blattner J, Clayton C. 1997. Conservation of mitochondrial targeting sequence function in mitochondrial and hydrogenosomal proteins from the early-branching eukaryotes Crithidia, Trypanosoma and Trichomonas. Eur J Cell Biol 73:240–251. [PubMed] [Google Scholar]
- 33.Schneider A, Bursac D, Lithgow T. 2008. The direct route: a simplified pathway for protein import into the mitochondrion of trypanosomes. Trends Cell Biol 18:12–18. doi: 10.1016/j.tcb.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 34.Gould SJ, Keller GA, Hosken N, Wilkinson J, Subramini S. 1989. A conserved tripeptide sorts proteins to peroxisomes. J Cell Biol 108:1657–1664. doi: 10.1083/jcb.108.5.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsukamoto Hata S, Yokota S, Fujiki Y, Hijkata M, Miyazawa S, Hashimoto T, Osumi T. 1994. Characterization of the signal peptide at the amino terminus of the rat peroxisomal 3-letpacyl-CoA thiolase precursor. J Biol Chem 269:6001–6010. [PubMed] [Google Scholar]
- 36.Colasante C, Ellis M, Ruppert T, Voncken F. 2006. Comparative proteomics of glycosomes from bloodstream form and procyclic culture form Trypanosoma brucei brucei. Proteomics 6:3275–3293. doi: 10.1002/pmic.200500668. [DOI] [PubMed] [Google Scholar]
- 37.Chudzik DM, Michels PA, De Walque S, Hol WGJ. 2000. Structures of type-2 peroxisomal targeting signals in two trypanosomatid aldolases. J Mol Biol 300:697–707. doi: 10.1006/jmbi.2000.3910. [DOI] [PubMed] [Google Scholar]
- 38.Petriv OI, Tang L, Titorenko VI, Rachubinski RA. 2004. A new definition for the consensus sequence of the peroxisome targeting signal type 2. J Mol Biol 341:119–134. doi: 10.1016/j.jmb.2004.05.064. [DOI] [PubMed] [Google Scholar]
- 39.Graveley BR. 2001. Alternative splicing: increasing diversity in the proteomic world. Trends Genet 17:100–107. doi: 10.1016/S0168-9525(00)02176-4. [DOI] [PubMed] [Google Scholar]
- 40.Cooper MD, Alder MN. 2006. The evolution of adaptive immune systems. Cell 124:815–822. doi: 10.1016/j.cell.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 41.Moore MJ, Proudfoot NJ. 2009. Pre-mRNA processing reaches back to transcription and ahead to translation. Cell 136:688–700. doi: 10.1016/j.cell.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 42.Schmucker D. 2007. Molecular diversity of Dscam: recognition of molecular identity in neuronal wiring. Nat Rev Neurosci 8:915–920. doi: 10.1038/nrn2256. [DOI] [PubMed] [Google Scholar]
- 43.Ochsenreiter T, Hajduk SL. 2006. Alternative editing of cytochrome c oxidase III mRNA in trypanosome mitochondria generates protein diversity. EMBO Rep 7:1128–1133. doi: 10.1038/sj.embor.7400817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huberts IJ, van der Klei J. 2010. Moonlighting proteins: an intriguing mode of multitasking. Biochim Biophys Acta 1803:520–525. doi: 10.1016/j.bbamcr.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 45.Engstler M, Pfohl T, Herminghaus S, Boshart M, Wiegertjes G, Heddergott N, Overath P. 2007. Hydrodynamic flow-mediated protein sorting on the cell surface of trypanosomes. Cell 131:505–515. doi: 10.1016/j.cell.2007.08.046. [DOI] [PubMed] [Google Scholar]
- 46.Galland N, Michels PA. 2010. Comparison of the peroxisomal matrix protein import system of different organisms. Exploration of possibilities for developing inhibitors of the import system of trypanosomatids for anti-parasite chemotherapy. Eur J Cell Biol 89:621–637. doi: 10.1016/j.ejcb.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 47.Galland N, Demeure F, Hannaert V, Verplaetse E, Vertommen D, Van der Smissen P, Courtoy PJ, Michels PA. 2007. Characterization of the role of the receptors PEX5 and PEX7 in the import of proteins into glycosomes of Trypanosoma brucei. Biochim Biophys Acta 1773:521–535. doi: 10.1016/j.bbamcr.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 48.Verplaetse E, Rigden DJ, Michels PA. 2009. Identification, characterization and essentiality of the unusual peroxin 13 from Trypanosoma brucei. Biochim Biophys Acta 1793:516–527. doi: 10.1016/j.bbamcr.2008.12.020. [DOI] [PubMed] [Google Scholar]
- 49.Krazy H, Michels PA. 2006. Identification and characterization of three peroxins—PEX6, PEX10 and PEX12—involved in glycosome biogenesis in Trypanosoma brucei. Biochim Biophys Acta 1763:6–17. doi: 10.1016/j.bbamcr.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 50.Kessler PS, Parsons M. 2005. Probing the role of compartmentation of glycolysis in procyclic form Trypanosoma brucei: RNA interference studies of PEX14, hexokinase, and phosphofructokinase. J Biol Chem 280:9030–9036. doi: 10.1074/jbc.M412033200. [DOI] [PubMed] [Google Scholar]
- 51.Ast J, Stiebler AC, Freitag J, Bolker M. 2013. Dual targeting of peroxisomal proteins. Front Physiol 4:297. doi: 10.3389/fphys.2013.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petrova VY, Drescher D, Kujumdzieva AV, Schmitt MJ. 2004. Dual targeting of yeast catalase A to peroxisomes and mitochondria. Biochem J 380:393–400. doi: 10.1042/BJ20040042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carrie C, Murcha MW, Kuehn K, Duncan O, Barthet M, Smith PM. 2008. Type II NAD(P)H dehydrogenases are targeted to mitochondria and chloroplasts or peroxisomes in Arabidopsis thaliana. FEBS Lett 582:3073–3079. doi: 10.1016/j.febslet.2008.07.061. [DOI] [PubMed] [Google Scholar]
- 54.Jung S, Marelli M, Rachubinski RA, Goodlett DR, Aitchinson JD. 2010. Dynamic changes in the subcellular distribution of Gpd1p in response to cell stress. J Biol Chem 285:6739–6749. doi: 10.1074/jbc.M109.058552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vercesi AE, Docampo R, Moreno SNJ. 1992. Energization-dependent Ca2+ accumulation in Trypanosoma brucei bloodstream and procyclic trypomastigotes mitochondria. Mol Biochem Parasitol 56:251–258. doi: 10.1016/0166-6851(92)90174-I. [DOI] [PubMed] [Google Scholar]
- 56.Williams S, Saha L, Singha UK, Chaudhuri M. 2008. Trypanosoma brucei: differential requirement of membrane potential for import of proteins into mitochondria in two developmental stages. Exp Parasitol 118:420–433. doi: 10.1016/j.exppara.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.