

Abstract
Human-infecting microbial pathogens all face a serious problem of elimination by the host immune response. Antigenic variation is an effective immune evasion mechanism where the pathogen regularly switches its major surface antigen. In many cases, the major surface antigen is encoded by genes from the same gene family, and its expression is strictly monoallelic. Among pathogens that undergo antigenic variation, Trypanosoma brucei (a kinetoplastid), which causes human African trypanosomiasis, Plasmodium falciparum (an apicomplexan), which causes malaria, Pneumocystis jirovecii (a fungus), which causes pneumonia, and Borrelia burgdorferi (a bacterium), which causes Lyme disease, also express their major surface antigens from loci next to the telomere. Except for Plasmodium, DNA recombination-mediated gene conversion is a major pathway for surface antigen switching in these pathogens. In the last decade, more sophisticated molecular and genetic tools have been developed in T. brucei, and our knowledge of functions of DNA recombination in antigenic variation has been greatly advanced. VSG is the major surface antigen in T. brucei. In subtelomeric VSG expression sites (ESs), VSG genes invariably are flanked by a long stretch of upstream 70-bp repeats. Recent studies have shown that DNA double-strand breaks (DSBs), particularly those in 70-bp repeats in the active ES, are a natural potent trigger for antigenic variation in T. brucei. In addition, telomere proteins can influence VSG switching by reducing the DSB amount at subtelomeric regions. These findings will be summarized and their implications will be discussed in this review.
INTRODUCTION
Trypanosoma brucei is a protozoan parasite that causes human African trypanosomiasis and is transmitted by the tsetse fly (Glossina spp.). The bloodstream form of T. brucei stays in extracellular spaces in its mammalian host and is constantly exposed to host immune surveillance. To evade elimination by its mammalian host immune response, T. brucei undergoes antigenic variation and regularly switches its major surface antigen, variant surface glycoprotein (VSG), through elaborated mechanisms that often involve DNA recombination (1).
ANTIGENIC VARIATION IN T. BRUCEI
The T. brucei genome (2) has a large VSG gene pool. Recent deep-sequencing analysis of the Lister 427 strain identified more than 2,500 VSG genes and pseudogenes (3). Most of these are in gene arrays located at subtelomeric regions of the 11 pairs of megabase chromosomes (Fig. 1A) (4). Individual VSG genes are found at approximately one-third of all subtelomeres on ∼100 minichromosomes (Fig. 1B) (3), which contain terminal telomere repeats and central 177-bp repeats (5). Normally, VSG gene arrays and minichromosome VSG genes are not transcribed but serve as a large VSG gene pool for VSG switching. VSGs are transcribed by RNA polymerase I exclusively from subtelomeric VSG expression sites (ESs) located on megabase chromosomes and intermediate chromosomes (Fig. 1C) (6, 7). Each ES contains a number of ES-associated genes (ESAG), and ES promoters usually are 40 to 60 kb upstream of the VSG gene (8, 9), which is the last gene in any ES and is located within 2 kb of the telomere repeats (10, 11). It is noteworthy that about half of the annotated VSG genes have upstream 70-bp repeats (3). In the assembled ES sequences, 70-bp repeats are 0.2 to 7.1 kb long (11). However, sequencing and assembly of repetitive sequences are not completely reliable. The 70-bp repeats are underrepresented and can be several tens of kb long in ESs. The 70-bp repeats upstream of individual VSG genes and pseudogenes in VSG gene arrays generally are much shorter and of only a few copies (12, 13). The 70-bp repeats upstream of VSG genes presumably provide homologous sequences for efficient DNA recombination in VSG switching (14–17) (see below).
FIG 1.
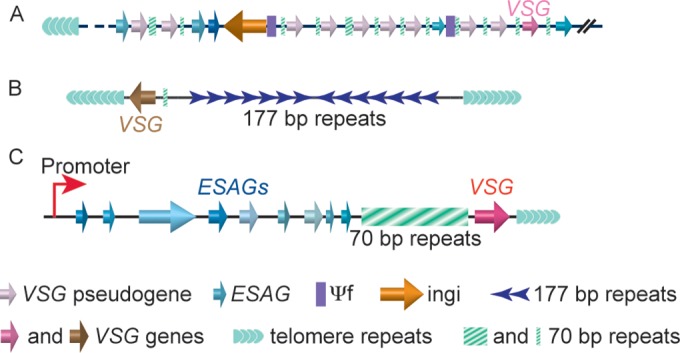
VSG genes are located mostly at subtelomeric regions in the T. brucei genome. (A) Large subtelomeric VSG arrays, including both VSG genes and pseudogenes. (B) Individual VSG genes often are found on minichromosomes at subtelomeric regions. (C) A typical VSG expression site (ES). VSG is the last gene in any ES and is located within 2 kb of the telomere repeats. A long stretch of 70-bp repeats is upstream of the VSG gene. ESs also contain a number of ESAG genes, which are upstream of the 70-bp repeats. The ES promoter is often 40 to 60 kb upstream of the VSG gene. ESs are located on megabase and intermediate chromosomes.
There are multiple ESs in the T. brucei genome (e.g., 15 ESs in the Lister 427 strain) (11, 18, 19). Although different ESs usually contain different VSG genes, ES promoter sequences are highly conserved (20–22). Different ESs also have very similar gene organizations and exhibit ∼90% sequence identity (11). However, at any moment, only one ES promoter is fully active, resulting in a single type of VSG being expressed (23). This monoallelic VSG expression ensures that after a VSG switching event, the originally active VSG no longer is expressed on the cell surface. Several mechanisms of VSG expression regulation have been identified, including specialized localization of the active ES at an extranucleolar ES body (ESB) that is enriched with RNA polymerase I (24), regulated transcription elongation along ESs (25, 26), modulation of ES chromatin structure (27–32), modulation of ES promoter activities (33–37), and telomere protein-mediated telomeric silencing (38, 39). VSG expression regulation has been reviewed elsewhere recently (40, 41) and will not be discussed here in detail.
VSG switching occurs through two major pathways. One involves a transcriptional switch and the other DNA recombination (Fig. 2). During an in situ switch, the originally active ES is silenced while an originally silent ES is expressed (Fig. 2A) (19, 42, 43), but no gene rearrangement is involved. In situ switch can be the most frequent switching event, such as when the active VSG mRNA is depleted by RNA interference (RNAi) (44), when T. brucei ORC1 (TbORC1) is depleted (45), and when switchers were selected from infected mice (46).
FIG 2.

Major VSG switching pathways. (A) In situ switch. The originally active ES is silenced, while an originally silent ES is expressed. (B) In telomere exchange/crossover (TE/CO) switches, the active VSG and a silent VSG exchange places. A silent ES is depicted to participate in CO. However, a VSG gene at a minichromosome subtelomere theoretically can be involved in a TE/CO event as well. (C) In gene conversion (GC) switches, the originally active VSG gene is lost and an originally silent VSG gene is copied into the active ES. Top right, a silent ES-linked VSG serves as the GC donor; bottom left, a silent VSG gene at a minichromosome subtelomere serves as the GC donor; bottom right, one or several VSG gene(s) in a VSG gene array serve(s) as the GC donor. Both a break-induced replication (BIR) event that copies the whole telomeric region downstream of the VSG donor and a true GC event can occur when a silent ES-linked or a minichromosome subtelomeric VSG gene serves as the GC donor. When a VSG gene array serves as the donor, a mosaic VSG can be built from several silent VSG genes. TE/CO and GC switches are proposed to be initiated with breaks in the 70-bp repeats (shown as a red lightning bolt). Long red arrow, active ES promoter; short blue arrow, silent ES promoter; red, orange, purple, and pink three-dimensional (3D) arrows, VSG genes; blue 3D arrows, ESAG genes; green boxes with diagonal bars, 70-bp repeats; arrays of green arrowheads, telomere repeats; arrays of dark blue arrowheads, 177-bp repeats.
Essentially all VSG genes have an invariant 14-bp (GATATATTTTAACA) motif in their 3′ untranslated region (UTR) (3). Fifty-four (3) to 92% (47) of VSG genes also are associated with upstream 70-bp repeats. In addition, ES-linked and minichromosome VSG genes are flanked with downstream telomere repeats. Such organization apparently facilitates DNA recombination between different VSG genes. DNA recombination-mediated VSG switching can occur in several ways. First, the active VSG gene can exchange places with a silent VSG gene in a different ES, resulting in telomere exchange (TE) (also referred to as crossover, or CO) switches (Fig. 2B). In TE/CO switches, the upstream recombination site is located mostly in the 70-bp repeats. However, because all ES sequences are highly homologous (11), the recombination site can be upstream of the 70-bp repeats.
Gene conversion (GC) is more frequent than CO in VSG switching events (48). In this case, a silent VSG gene is copied into the active ES to replace the originally active VSG, which is lost after the switch, while the newly expressed VSG gene is duplicated (Fig. 2C). The VSG donor in GC switches can originate from a silent ES, a minichromosome subtelomere, or a VSG gene array. However, ES-linked VSG genes appear to be preferably copied (48). In this case, the upstream boundary of GC can be within the 70-bp repeats so that only the VSG gene and its adjacent sequences are involved, which is often referred to as VSG GC. GC also can involve a much larger portion of the ES, including markers upstream of the 70-bp repeats (49), markers immediately downstream of the ES promoter (50–53), and sometimes the ES promoter itself (11), which are termed ES GC. On the other hand, when a VSG gene on a minichromosome or in a gene array acts as the GC donor, the upstream boundary of GC is almost always in the 70-bp repeats. When the GC donor is from a VSG gene array, the downstream boundary of GC can extend to the 3′ coding or noncoding parts of the VSG gene (Fig. 2C) (54). The downstream boundary of GC is less clear if an ES-linked or a minichromosome VSG is used as the GC donor. Because no telomere terminal marker is available, it is not known how often GC VSG switching is a true gene conversion event, in which only a short fragment downstream of the donor VSG gene is duplicated, and how often it is actually a break-induced replication (BIR) event in which all of the terminal portion of the chromosome downstream of the VSG donor is replicated (Fig. 2C). GC appears to be the preferred mechanism of VSG switching (55), particularly in several recent studies when the Lister 427 strain is used in in vitro VSG switching analysis (17, 45, 50–53, 56, 57).
Several more complicated VSG switching events also have been observed. In one type of switch, the originally active ES is lost and a different ES is expressed, resulting in an ES loss coupled to an in situ switch. This event has been observed in several recent studies and appears to be quite frequent (45, 50–53). In a similar situation, the originally active VSG gene (possibly with its adjacent sequences) is lost while a silent ES is expressed, resulting in a VSG loss coupled with in situ switch. Although this is observed in in vitro studies (52), it appears to be a relatively rare event. In addition, usually at late stages of a T. brucei infection, segments of different VSG genes can be copied into the active ES, resulting in a novel mosaic VSG gene being expressed (Fig. 2C, bottom right) (58, 59). Most TE/CO and GC VSG switches rely on TbRAD51-mediated homologous recombination (HR) (46). However, RAD51-independent microhomology-mediated end joining (MMEJ) also has been suggested to contribute to VSG switching (60).
DSBs, DNA RECOMBINATION, AND VSG SWITCHING
DNA double-strand breaks (DSBs) are the most deleterious DNA damages, and they usually result from DNA replication fork stalling/collapse and ionizing irradiation, etc. (61, 62). It is well known that two major pathways are involved in repair of DSBs: HR and nonhomologous end joining (NHEJ). HR-mediated DSB repair is more accurate but requires a donor with homologous sequence, such as the sister chromatid, after DNA replication. NHEJ is more error prone but more prevalent when homologous sequences are not available. HR appears to be much more frequent than NHEJ in yeast, but the reverse is true in most mammalian cells (63). In T. brucei, no NHEJ events have been reported. However, MMEJ, an alternative NHEJ pathway, has been identified (60, 64–66), but HR is much more efficient and frequent than MMEJ (67).
When HR is necessary for proper chiasmata formation between homologous chromosomes during meiosis, DSBs are induced by the Spo11 nuclease (68). A brief review of HR, NHEJ, and MMEJ mechanisms will show why DSBs are required for these DNA recombination processes (see below). In T. brucei, recent studies have revealed that DSBs are a natural trigger for VSG switching (17), and the location of the DSB influences the choice of VSG switching mechanisms (69). Apparently, induction and regulation of DSBs at subtelomeric regions in T. brucei are critical for proper VSG switching.
(i) HR and its roles in VSG switching.
In HR-mediated DSB repair, the 5′ end of the broken DNA first is cleaved by MRE11-RAD50-XRS2 (yeast)/NBS1 (mammal) (70, 71) and Sae2 (yeast)/CtIP (mammal) and then processed more extensively by the 5′-3′ exonuclease ExoI or the combined helicase/nuclease activities of Sgs1/Dna2 (72–75) (Fig. 3A and Table 1). The exposed 3′ single-stranded DNA then is bound by RPA (replication protein A) that removes DNA secondary structures (76), and a number of mediators are necessary to displace RPA to promote subsequent binding of RAD51, a DNA-dependent ATPase, onto the 3′ single-stranded DNA (ssDNA) to form nucleoprotein filaments (77) (Fig. 3A). In mammalian cells, BRCA2 is an important RAD51 mediator (78, 79), while in yeast, RAD52 mediates most of the loading of RAD51 (80) (Table 1). The nucleoprotein filament then searches for homologous sequences, and RAD51 catalyzes the strand exchange (81). The extended strand invasion intermediate has many potential outcomes, eventually resulting in NCO or CO (Fig. 3A) (61).
FIG 3.

Schematic diagram of HR and MMEJ pathways. (A) Mechanisms of HR. DNA 5′ ends at a DSB site initially are processed by the MRX complex and Sae2 nucleases, followed by further resection by ExoI and Sgs1/Dna2. The resulting single-stranded 3′ ends then are bound by RPA. With the help of RAD51 mediators, RAD51 displaces RPA on the single-stranded DNA. Subsequently, RAD51 mediates homology search, strand invasion, and D-loop formation steps. (Bottom left) Synthesis-dependent strand annealing leads to noncrossover products. (Bottom middle) Double Holliday junction (dHJ) can lead to either noncrossover or crossover products depending on resolvase cleavage sites (shown as red arrowheads). (Bottom right) Branch migration mediated by the BLM-Topo3α-RMI complex also can resolve dHJ into noncrossover products. (B) A current model of MMEJ. DNA ends at the DSB site also are processed by MRX and Sae2 nucleases in MMEJ. Subsequently, Rad52 or Rad59 help DNA ends search and anneal at preexisting microhomologies. Ligase 3 finishes the ligation of the broken ends in MMEJ. Yeast and mammalian homologues of different nucleases and Rad51 mediators are listed in Table 1.
TABLE 1.
List of yeast and mammalian homologs of HR players
| Category | Homolog(s) in: |
|
|---|---|---|
| Yeast | Mammal | |
| 5′ to 3′ nucleases | MRX | MRN |
| Mre11, Rad50, Xrs2 | Mre11, Rad50, Nbs1 | |
| Sae2 | CtIP | |
| RecQ helicases | SgsI | BLM, WRN |
| RAD51 mediators | RAD52 | BRCA2, RAD51 paralogs |
In mitotic cells, a primary pathway to generate NCOs is synthesis-dependent strand annealing (SDSA), where the newly synthesized DNA strand (according to the homologous sequence as a result from the strand invasion event) dissociates from the D-loop to anneal to the other DNA end (Fig. 3A) (82–84). Alternatively, the second end of the processed DSB can be captured by the D loop to form a double Holliday junction (dHJ), an important HR intermediate (85). dHJ can be resolved to form NCO or CO depending on how the DNA strands are cleaved by resolvases (Fig. 3A, bottom) (86–89). In mitotic cells, dHJs also can be dissolved by the branch migration and topoisomerase activity of the BLM (Sgs1)/TOP3α/RMI complex, which leads to NCO (Fig. 3A) (90).
Homologous recombination is highly efficient in T. brucei (91–94). It has been shown that a minimal 42 bp of homology is sufficient for HR-mediated DNA integration in insect-stage T. brucei cells (95). In bloodstream-form TbRAD51 wild-type (WT) cells, as little as 24 bp of homology is sufficient for HR-mediated targeting, although the efficiency (2.5 × 10−7) is 4- to 5-fold lower than targeting with homologous sequence of 200 to 300 bp (64).
Several RAD51 paralogs have been identified in vertebrate cells, including RAD51B, RAD51C, RAD51D, XRCC2, and XRCC3, which are bona fide HR factors and are required for HR-mediated DNA damage repair (96). Sequence identities between RAD51 and its paralogs are 20 to 30%, and the conserved sequences are primarily in the Walker A and B domains that are essential for their DNA binding activities (97). The functions of these paralogs are not well known, but they appear to function as RAD51 mediators and facilitate RAD51 assembly at DSB sites, as their deficiency prevents RAD51 focus formation even after ionizing radiation (96).
(ii) MMEJ may be an important mechanism of VSG switching.
NHEJ includes the classical NHEJ (cNHEJ) and MMEJ (also known as alternative NHEJ, or aNHEJ) (98, 99). NHEJ is the preferred pathway to HR in vertebrate cells throughout the cell cycle (63). The Ku70/80 dimer, DNA-PKcs, and the DNA ligase complex XRCC4-ligase IV-XLF (XRCC4-like factor) are the core components of cNHEJ (98).
cNHEJ often causes short deletions and insertions at the junctions, while MMEJ appears to be more error prone than cNHEJ and often leads to chromosome translocations (100). As a result of MMEJ, DNA junctions often have large deletions, microhomologies, or occasional insertions of large DNA segments of unknown origin, although none of these features is invariably present (98). In MMEJ, MRE11 and CtIP are involved in end resection (101–105), Rad52 or Rad59 is involved in annealing of DNA ends with microhomologies (106, 107), and DNA ligase III appears to promote the DNA ligation (108–110) (Fig. 3B). Recent studies also suggest the existence of an additional alternative end-joining pathway that relies on ligase I and is independent of preexisting microhomologies in mammalian cells (99). However, whether this pathway is conserved in all eukaryotic cells is unknown.
In T. brucei, after introducing a chromosomal DSB, most DNA damage repairs were mediated by allelic HR (85% of all repaired events) and the rest can occur through ectopic HR and MMEJ (66). In contrast, cNHEJ events have never been reported in T. brucei. Although T. brucei has Ku70/80 homologues (111, 112), DNA ligase IV and XRCC4 homologs are missing (65), indicating that cNHEJ is either absent or mechanistically divergent in T. brucei.
In T. brucei, MMEJ initially was observed as a subsidiary TbRAD51-independent pathway that mediates the integration of transformed DNA into the genome (64). As short as 3 to 7 bp of sequence homology with base mismatches, insertions, and deletions is enough to mediate MMEJ, and 11 to 74 bp of sequences was lost during the integration (64). MMEJ events also were observed when T. brucei cell extract was used to join ends of linear DNA molecules in vitro, which is mediated by a microhomology of 6 to 16 bp long, often with at least one mismatched base (65). MMEJ is independent of either TbRAD51 or Ku (65), indicating that it is similar to the MMEJ observed in yeast and mammalian cells (98).
By targeting an I-SceI site into the T. brucei genome and inducing I-SceI expression, Glover et al. examined repair products of a specific chromosomal DSB and identified that a small fraction (∼10%) of DNA damage repair occurred through MMEJ (11 to 13 bp of homology), which resulted exclusively in intrachromosomal joining (66). Using a negative selection, Glover et al. enriched the MMEJ-mediated DNA damage repair products (60), among which both intra- and interchromosomal gene conversion products were identified. Careful examination of MMEJ products suggests that these resemble the micro-single-strand annealing (SSA) events (60), which is the same as those observed in yeast (107). Occasionally, products with one end repaired by HR and the other by MMEJ were observed which appear to be TbRAD51 dependent (60). Importantly, MMEJ has been observed in a subtelomeric ES, contributing to 25% of DSB repair events, while MMEJ represents only 5% of DSB repair at a chromosome internal locus (60). Since VSG ESs are at subtelomeric regions that often lack allelic homologous sequences on the corresponding homologous chromosomes (2), MMEJ has been proposed to be an important pathway for VSG switching (60).
(iii) HR proteins that influence VSG switching in T. brucei.
A number of DNA repair proteins have been examined for their functions in VSG switching. So far, TbRAD51 (46), TbRAD51-3 (a RAD51 paralog) (113), TbBRCA2 (114), and the TOPO3α/RMI1 complex (50, 51) are required for normal VSG switching.
TbRAD51 is not essential, but TbRAD51 null cells are sensitive to the DNA damage reagent MMS (46). Infecting preimmunized mice with a T. brucei strain containing antibiotic resistance markers in the active ES can yield switched trypanosome cells and allow estimation of VSG switching frequency. When TbRAD51 double-knockout (dko), single-knockout (sko), and WT cells were analyzed using this method, the VSG switching frequency was much lower (6- to 50-fold) in TbRAD51 dko cells than in TbRAD51 sko and WT cells, indicating that TbRAD51 is an important player for VSG switching. Since TbRAD51 is required for DNA recombination, it was expected that deletion of TbRAD51 would mostly reduce HR-mediated VSG switching events. However, loss of TbRAD51 did not change the distribution of VSG switchers that arose from different pathways. In WT cells, ∼60% of switchers arose from in situ switch, while the rest arose from HR-mediated gene conversion. In TbRAD51 null cells, ∼51% of switchers were in situ switchers, while the rest were gene conversion products (46). Since TbRAD51 deletion did not abolish HR, it is speculated that a TbRAD51-independent pathway exists in T. brucei. In yeast, RAD51 and RAD50 mediate different HR events, particularly at the subtelomeric regions (115). A TbRAD50 homolog has been identified in the T. brucei genome, although no detailed characterization of this gene has been reported. It is possible that TbRAD50 can mediate some TbRAD51-independent DNA recombination events. In addition, MMEJ has been found to be TbRAD51 independent (65), which has been proposed to contribute to VSG switching (60).
Besides TbRAD51, the T. brucei genome encodes five other RAD51-related proteins: DMC1, TbRAD51-3, TbRAD51-4, TbRAD51-5, and TbRAD51-6 (113). Among these, TbRAD51-3 and TbRAD51-5 are involved in DNA damage repair and HR and are required for DNA damage-induced TbRAD51 subnuclear foci. This is similar to the situation in mammalian cells, where RAD51 paralogs are required for RAD51 focus formation in response to DNA damage (116). However, only TbRAD51-3 is involved in VSG switching, and TbRAD51-3 null cells have ∼10-fold lower VSG switching frequency than WT cells (113). Similarly, TbBRCA2, a mediator for RAD51 filament formation, also plays an important role in HR, and deletion of TbBRCA2 leads to a 10-fold decrease in VSG switching frequency (114). In particular, TbBRCA2 has multiple BRC repeats at its N terminus, which are required for DNA damage-induced TbRAD51 subnuclear foci (114).
In yeast, the RecQ helicase Sgs1 forms a complex with a type IA topoisomerase, Top3, and RecQ-mediated genome instability 1 (RMI1) (117, 118). In human cells, a conserved complex also exists containing BLM, Topo3α, and BLAP75/18 (RMI1/2) (90, 119). This RTR complex plays an important function in dissolution of the HR intermediates and double Holliday junctions, and it suppresses crossover in HR (120). Using a T. brucei strain in which the active ES is marked with a thymidine kinase gene immediately upstream of the active VSG gene, Kim and Cross were able to negatively select VSG switchers by ganciclovir (GCV), a nucleoside analog, because TK-expressing T. brucei cells are sensitive to GCV (50). The same strain also carries a Blasticidin resistance (BSD) marker immediately downstream of the active ES promoter. Therefore, by examination of antibiotic resistance phenotypes and genotypes of BSD and the originally active VSG gene, it is possible to determine the VSG switching pathway. Using this method, it was shown that T. brucei Topo3α deletion led to a more than 10-fold increase in VSG switching frequency, which is dependent on TbRAD51 (50). In addition, VSG GC events were most frequent in Topo3α null cells (50). Similarly, deletion of TbRMI1 also leads to a 5-fold increase in VSG switching frequency, and most VSG switchers arose through VSG GC (51).
Deletion of mismatch repair proteins does not affect VSG switching (67). However, it is surprising that deletion of TbMRE11 does not influence VSG switching frequency (121), as the MRN complex is required for processing broken DNA ends in both HR (70, 71) and MMEJ (101, 103, 104). It is possible that additional nucleases are available in T. brucei for DNA end processing.
(iv) DSBs are a key for initiation of VSG switching.
HR-mediated VSG switching is the most frequent event in many switching assays (17, 45, 48, 50–53, 55), and HR initiates with DSBs. Therefore, DSBs in ES have long been proposed to be the first step of VSG switching (122, 123). Indeed, inducing an I-SceI-generated DSB adjacent to the 70-bp repeats and immediately upstream of the active VSG gene leads to an ∼250-fold increase in VSG switching frequency (17). In addition, DSBs can be detected in subtelomeric regions in both the active and silent ESs in WT cells (17, 52, 69). In particular, using ligation-mediated PCR (LMPCR) analysis, more DSBs with staggered ends were detected in the 70-bp repeats in the active ES than in a silent one (17). These observations suggest that VSG switching initiates with DSBs in the active ES.
Although DSBs are required for HR-mediated VSG switching, not all DSBs in ESs induce VSG switching with equal efficiency, and not all DSBs leads to DNA recombination-mediated VSG switching (69). Specifically, very few survivors (8%) were VSG switchers if the DSB is at the active ES promoter region, and only 28% of the survivors were VSG switchers if the DSB is downstream of the active VSG gene (69). Particularly in the latter case, many switchers have lost their originally active ES and survived after an in situ switch, indicating that DSBs also can induce transcription-mediated VSG switching.
In contrast, DSBs introduced immediately upstream of the active VSG gene and next to the 70-bp repeats is most efficient at inducing VSG switching; all survivors are VSG switchers (69). Therefore, the 70-bp repeats upstream of the active VSG gene appear to be a hot spot for VSG switching-inducing DSBs. Several possibilities have been proposed as to why 70-bp repeats frequently have DSBs: they consist of a large number of TTA repeats that are known to be unstable for plasmids (59), and transcription through these repeats makes the active ES unstable; the repetitive sequence may form an unusual structure that is difficult to be replicated, and they may be digested by a special T. brucei endonuclease (1, 17).
TELOMERES INFLUENCE VSG SWITCHING
Since a considerable amount of DSBs can be detected at subtelomeric regions, it has been proposed that subtelomeres are fragile sites in T. brucei (69), which presumably facilitates VSG switching and contributes positively to antigenic variation. However, T. brucei, like any other eukaryotic organism, needs to maintain a stable genome. Therefore, it is a delicate task to balance between telomere/subtelomere stability and plasticity in T. brucei.
It is well known that telomere proteins play important roles in maintaining chromosome stability and genome integrity (124). Our recent studies showed that TbTIF2, a telomeric protein, indeed is essential for maintaining subtelomere integrity and reducing DSB amounts at subtelomeres (52). Consequently, a transient depletion of TbTIF2 led to increased VSG switching frequency, with the majority of switchers arising through ES GC or ES loss coupled with in situ switches (52). The TbTIF2 deficiency-induced DSBs appear to be repaired by TbRAD51, as deletion of TbRAD51 and depletion of TbTIF2 concurrently resulted in a much higher level of DSBs. Most interestingly, deletion of TbRAD51 increased the TbTIF2 deficiency-induced DSBs in the active ES much more strongly than in silent ESs, suggesting that WT TbRAD51 preferably repairs DSBs in the active ES (52). This may explain why introducing DSBs in silent ESs seldom leads to VSG switching (69). It is possible that the ends of DSBs in silent ESs are not processed, so fewer DSBs with staggered ends are detected in silent 70-bp repeats than in active ones (17). Why are DSBs in the active ES and silent ones not treated the same? One possibility is that the chromatin structure in the two types of ESs is very different: the active ES is largely depleted of nucleosomes, while the silent ones are packed with nucleosomes (27, 28).
How does a protein associating with the telomere influence switching of VSGs at subtelomeres? We found that TbTIF2 influences VSG switching by reducing the amount of DSBs in subtelomeric regions, including both active and silent ESs (52). What could be the underlying mechanism of TbTIF2 in subtelomere integrity maintenance? We anticipate two most likely possibilities. First, telomere proteins have been shown to be important for telomere DNA replication in yeast and vertebrate cells (125–128). Loss of TbTIF2 may induce more replication fork stalling at telomeric and subtelomeric regions. The increased topological stress may lead to elevated DSBs in subtelomeric ESs. Second, several telomere proteins, including the TbTIF2 homolog TIN2, are important for telomere cohesion and sister telomere pairing (129, 130). TbTIF2 may have a similar function, and loss of TbTIF2 may lead to premature dissociation of sister telomeres. In this case, subtelomeric DSBs may not be efficiently repaired when the sister homolog is not available.
Independent of TbTIF2's function in subtelomere integrity maintenance, it is possible that loss of TbTIF2 destabilizes the telomere structure and leads to chromosome end-to-end fusions, similar to that observed in mammalian cells with telomere dysfunctions (131). In support of this idea, transient depletion of the duplex telomere DNA binding factor, TbTRF, also led to a significant increase in VSG switching frequency (53). In addition, the DNA binding activity of TbTRF is required for its role in suppression of VSG switching (53). Loss of mammalian TRF2, the homolog of TbTRF, led to chromosome end-to-end fusions (132). Therefore, it is possible that loss of TbTRF results in similar defects. Dicentric chromosome-induced breakage-fusion-bridge cycles often result in the loss of large regions of terminal chromosomes (133), which can lead to increased VSG switching (69). Although chromosome end fusions have not been identified in T. brucei, this could be simply because of the insensitivity of currently available tools. NHEJ events have not been observed in T. brucei, and XRCC4 and ligase IV homologs are absent from the T. brucei genome (65); however, it is possible that telomere fusion occurs through MMEJ. Interestingly, TbTIF2 and TbTRF interact strongly (52), and transient induction of TbTRF and TbTIF2 RNAi lead to similarly increased VSG switching frequency, with most switchers arising from ES GC and ES loss coupled with in situ switches (52, 53). Therefore, it is possible that both proteins function in the same pathway in suppression of VSG switching. However, whether depletion of TbTRF also leads to increased subtelomeric DSB amounts is unknown, and more genetic analysis is necessary before a conclusion is drawn on whether TbTIF2 and TbTRF function in the same genetic pathway in VSG switching regulation.
CONCLUSIONS AND PERSPECTIVES
Recent studies clearly showed that DSBs in the active ES, particularly those in and near 70-bp repeats, are a key factor that induces efficient VSG switching (17, 69). In addition, we have identified at least one factor that influences the subtelomere DSB amount: telomere-associated TbTIF2 (52). However, exactly how TbTIF2, as a telomere-specific protein, regulates the subtelomere integrity is still unknown. We have found that a second telomere protein, TbTRF, also is important for suppressing VSG switching (53). However, the relationship between the functions of TbTIF2 and TbTRF still is unclear.
Importantly, we learned that DSBs in 70-bp repeats of the active ES induce efficient VSG switching (17, 69), and DSBs naturally occur more frequently at subtelomeres than chromosome internal regions (69). However, whether telomeres and transcription through the active ES are the only factors contributing to subtelomeric DSBs is not clear. Although DSBs in 70-bp repeats are critical for initiation of antigenic variation, maintaining 70-bp repeat stability also is important for maintaining a relatively stable genome and a VSG gene pool in T. brucei. However, how are 70-bp repeats maintained is completely unknown to us. Does any protein specifically bind the 70-bp repeats? The answer to this question no doubt would contribute greatly to our understanding of VSG switching regulation.
New molecular tools recently developed for T. brucei allowed us to examine in greater detail the HR and MMEJ events, their underlying mechanisms and players, their roles in VSG switching, and their regulation. Our knowledge about players in HR has been improved. However, MMEJ in T. brucei appears not to be completely conserved with that in vertebrates, and the key players in T. brucei MMEJ still are unknown. Since MMEJ has been proposed to be an important mechanism of VSG switching (60), identifying key players in this pathway will contribute to our better understanding of VSG switching regulation.
We have shown that the telomere structure and telomere-associated proteins play important roles in VSG switching regulation (52, 53). Similar to T. brucei, antigenic variation in several other microbial pathogens also relies on HR-mediated gene conversion, such as switching of the major surface glycoprotein (MSG) in Pneumocystis jirovecii that causes pneumonia (134, 135) and switching of the VlsE variant surface protein in Borrelia burgdorferi that causes Lyme disease (136, 137). However, molecular tools for studying both P. jirovecii and B. burgdorferi still are very limited. Therefore, studying DNA recombination-mediated antigenic variation and its regulation by telomeres in T. brucei also serves as a good model for understanding similar processes in other microbial pathogens.
REFERENCES
- 1.Barry JD, McCulloch R. 2001. Antigenic variation in trypanosomes: enhanced phenotypic variation in a eukaryotic parasite. Adv Parasitol 49:1–70. doi: 10.1016/S0065-308X(01)49037-3. [DOI] [PubMed] [Google Scholar]
- 2.Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, Bartholomeu DC, Lennard NJ, Caler E, Hamlin NE, Haas B, Bohme U, Hannick L, Aslett MA, Shallom J, Marcello L, Hou L, Wickstead B, Alsmark UC, Arrowsmith C, Atkin RJ, Barron AJ, Bringaud F, Brooks K, Carrington M, Cherevach I, Chillingworth TJ, Churcher C, Clark LN, Corton CH, Cronin A, Davies RM, Doggett J, Djikeng A, Feldblyum T, Field MC, Fraser A, Goodhead I, Hance Z, Harper D, Harris BR, Hauser H, Hostetler J, Ivens A, Jagels K, Johnson D, Johnson J, Jones K, Kerhornou AX, Koo H, Larke N, Landfear S, Larkin C, Leech V, Line A, Lord A, Macleod A, Mooney PJ, Moule S, Martin DM, Morgan GW, Mungall K, Norbertczak H, Ormond D, Pai G, Peacock CS, Peterson J, Quail MA, Rabbinowitsch E, Rajandream MA, Reitter C, Salzberg SL, Sanders M, Schobel S, Sharp S, Simmonds M, Simpson AJ, Tallon L, Turner CM, Tait A, Tivey AR, Van Aken S, Walker D, Wanless D, Wang S, White B, White O, Whitehead S, Woodward J, Wortman J, Adams MD, Embley TM, Gull K, Ullu E, Barry JD, Fairlamb AH, Opperdoes F, Barrell BG, Donelson JE, Hall N, Fraser CM, Melville SE, El-Sayed NM. 2005. The genome of the African trypanosome Trypanosoma brucei. Science 309:416–422. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- 3.Cross GAM, Kim HS, Wickstead B. 2014. Capturing the variant surface glycoprotein repertoire (the VSGnome) of Trypanosoma brucei Lister 427. Mol Biochem Parasitol 195:59–73. doi: 10.1016/j.molbiopara.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 4.Horn D, Barry JD. 2005. The central roles of telomeres and subtelomeres in antigenic variation in African trypanosomes. Chromosome Res 13:525–533. doi: 10.1007/s10577-005-0991-8. [DOI] [PubMed] [Google Scholar]
- 5.Wickstead B, Ersfeld K, Gull K. 2004. The small chromosomes of Trypanosoma brucei involved in antigenic variation are constructed around repetitive palindromes. Genome Res 14:1014–1024. doi: 10.1101/gr.2227704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gunzl A, Bruderer T, Laufer G, Schimanski B, Tu LC, Chung HM, Lee PT, Lee MG. 2003. RNA polymerase I transcribes procyclin genes and variant surface glycoprotein gene expression sites in Trypanosoma brucei. Eukaryot Cell 2:542–551. doi: 10.1128/EC.2.3.542-551.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Lange T, Borst P. 1982. Genomic environment of the expression-linked extra copies of genes for surface antigens of Trypanosoma brucei resembles the end of a chromosome. Nature 299:451–453. doi: 10.1038/299451a0. [DOI] [PubMed] [Google Scholar]
- 8.Kooter JM, van der Spek HJ, Wagter R, d'Oliveira CE, van der Hoeven F, Johnson PJ, Borst P. 1987. The anatomy and transcription of a telomeric expression site for variant-specific surface antigens in T. brucei. Cell 51:261–272. doi: 10.1016/0092-8674(87)90153-X. [DOI] [PubMed] [Google Scholar]
- 9.Johnson PJ, Kooter JM, Borst P. 1987. Inactivation of transcription by UV irradiation of T. brucei provides evidence for a multicistronic transcription unit including a VSG gene. Cell 51:273–281. doi: 10.1016/0092-8674(87)90154-1. [DOI] [PubMed] [Google Scholar]
- 10.Berriman M, Hall N, Sheader K, Bringaud F, Tiwari B, Isobe T, Bowman S, Corton C, Clark L, Cross GA, Hoek M, Zanders T, Berberof M, Borst P, Rudenko G. 2002. The architecture of variant surface glycoprotein gene expression sites in Trypanosoma brucei. Mol Biochem Parasitol 122:131–140. doi: 10.1016/S0166-6851(02)00092-0. [DOI] [PubMed] [Google Scholar]
- 11.Hertz-Fowler C, Figueiredo LM, Quail MA, Becker M, Jackson A, Bason N, Brooks K, Churcher C, Fahkro S, Goodhead I, Heath P, Kartvelishvili M, Mungall K, Harris D, Hauser H, Sanders M, Saunders D, Seeger K, Sharp S, Taylor JE, Walker D, White B, Young R, Cross GAM, Rudenko G, Barry JD, Louis EJ, Berriman M. 2008. Telomeric expression sites are highly conserved in Trypanosoma brucei. PLoS One 3:e3527. doi: 10.1371/journal.pone.0003527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pays E, Steinert S. 1988. Control of antigen gene expression in African trypanosomes. Annu Rev Genet 22:107–126. doi: 10.1146/annurev.ge.22.120188.000543. [DOI] [PubMed] [Google Scholar]
- 13.Aline RF Jr, MacDonald G, Brown E, Allison J, Myler P, Rothwell V, Stuart K. 1985. (TAA)n within sequences flanking several intrachromosomal variant surface glycoprotein genes in Trypanosoma brucei. Nucleic Acids Res 13:3161–3177. doi: 10.1093/nar/13.9.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aline RF Jr, Scholler JK, Nelson RG, Agabian N, Stuart K. 1985. Preferential activation of telomeric variant surface glycoprotein genes in Trypanosoma brucei. Mol Biochem Parasitol 17:311–321. doi: 10.1016/0166-6851(85)90005-2. [DOI] [PubMed] [Google Scholar]
- 15.Timmers HT, T de Lange Kooter JM, Borst P. 1987. Coincident multiple activations of the same surface antigen gene in Trypanosoma brucei. J Mol Biol 194:81–90. doi: 10.1016/0022-2836(87)90717-0. [DOI] [PubMed] [Google Scholar]
- 16.Michels PAM, van der Ploeg LHT, Liu AYC, Borst P. 1984. The inactivation and reactivation of an expression-linked gene copy for a variant surface glycoprotein in Trypanosoma brucei. EMBO J 3:1345–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boothroyd CE, Dreesen O, Leonova T, Ly KI, Figueiredo LM, Cross GAM, Papavasiliou FN. 2009. A yeast-endonuclease-generated DNA break induces antigenic switching in Trypanosoma brucei. Nature 459:278–281. doi: 10.1038/nature07982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cully DF, Ip HS, Cross GAM. 1985. Coordinate transcription of variant surface glycoprotein genes and an expression site associated gene family in Trypanosoma brucei. Cell 42:173–182. doi: 10.1016/S0092-8674(85)80113-6. [DOI] [PubMed] [Google Scholar]
- 19.Zomerdijk JC, Ouellete M, ten Asbroek AL, Kieft R, Bommer AM, Clayton CE, Borst P. 1990. The promoter for a variant surface glycoprotein gene expression site in Trypanosoma brucei. EMBO J 9:2791–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gottesdiener K, Chung H-M, Brown SD, Lee MG-S, van der Ploeg LHT. 1991. Characterization of VSG gene expression site promoters and promoter-associated DNA rearrangement events. Mol Cell Biol 11:2467–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pays E, Coquelet H, Tebabi P, Pays A, Jefferies D, Steinert M, Koenig E, Williams RO, Roditi I. 1990. Trypanosoma brucei: constitutive activity of the VSG and procyclin gene promoters. EMBO J 9:3145–3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zomerdijk JCBM, Kieft R, Shiels PG, Borst P. 1991. Alpha-amanitin-resistant transcription units in trypanosomes: a comparison of promoter sequences for a VSG gene expression site and for the ribosomal RNA genes. Nucleic Acids Res 19:5153–5158. doi: 10.1093/nar/19.19.5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cross GAM. 1975. Identification, purification and properties of clone-specific glycoprotein antigens constituting the surface coat of Trypanosoma brucei. Parasitology 71:393–417. doi: 10.1017/S003118200004717X. [DOI] [PubMed] [Google Scholar]
- 24.Navarro M, Gull K. 2001. A pol I transcriptional body associated with VSG mono-allelic expression in Trypanosoma brucei. Nature 414:759–763. doi: 10.1038/414759a. [DOI] [PubMed] [Google Scholar]
- 25.Vanhamme L, Poelvoorde P, Pays A, Tebabi P, Van Xong H, Pays E. 2000. Differential RNA elongation controls the variant surface glycoprotein gene expression sites of Trypanosoma brucei. Mol Microbiol 36:328–340. doi: 10.1046/j.1365-2958.2000.01844.x. [DOI] [PubMed] [Google Scholar]
- 26.Kassem A, Pays E, Vanhamme L. 2014. Transcription is initiated on silent variant surface glycoprotein expression sites despite monoallelic expression in Trypanosoma brucei. Proc Natl Acad Sci U S A 111:8943–8948. doi: 10.1073/pnas.1404873111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stanne TM, Rudenko G. 2010. Active VSG expression sites in Trypanosoma brucei are depleted of nucleosomes. Eukaryot Cell 9:136–147. doi: 10.1128/EC.00281-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Figueiredo LM, Cross GAM. 2010. Nucleosomes are depleted at the VSG expression site transcribed by RNA polymerase I in African trypanosomes. Eukaryot Cell 9:148–154. doi: 10.1128/EC.00282-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Povelones ML, Gluenz E, Dembek M, Gull K, Rudenko G. 2012. Histone H1 plays a role in heterochromatin formation and VSG expression site silencing in Trypanosoma brucei. PLoS Pathog 8:e1003010. doi: 10.1371/journal.ppat.1003010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pena AC, Pimentel MR, Manso H, Vaz-Drago R, Pinto-Neves D, Aresta-Branco F, Rijo-Ferreira F, Guegan F, Pedro Coelho L, Carmo-Fonseca M, Barbosa-Morais NL, Figueiredo LM. 2014. Trypanosoma brucei histone H1 inhibits RNA polymerase I transcription and is important for parasite fitness in vivo. Mol Microbiol 93:645–663. doi: 10.1111/mmi.12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Batram C, Jones NG, Janzen CJ, Markert SM, Engstler M. 2014. Expression site attenuation mechanistically links antigenic variation and development in Trypanosoma brucei. eLife 3:e02324. doi: 10.7554/eLife.02324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alsford S, Horn D. 2012. Cell-cycle-regulated control of VSG expression site silencing by histones and histone chaperones ASF1A and CAF-1b in Trypanosoma brucei. Nucleic Acids Res 40:10150–10160. doi: 10.1093/nar/gks813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hughes K, Wand M, Foulston L, Young R, Harley K, Terry S, Ersfeld K, Rudenko G. 2007. A novel ISWI is involved in VSG expression site downregulation in African trypanosomes. EMBO J 26:2400–2410. doi: 10.1038/sj.emboj.7601678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Narayanan MS, Kushwaha M, Ersfeld K, Fullbrook A, Stanne TM, Rudenko G. 2011. NLP is a novel transcription regulator involved in VSG expression site control in Trypanosoma brucei. Nucleic Acids Res 39:2018–2031. doi: 10.1093/nar/gkq950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Denninger V, Fullbrook A, Bessat M, Ersfeld K, Rudenko G. 2010. The FACT subunit TbSpt16 is involved in cell cycle specific control of VSG expression sites in Trypanosoma brucei. Mol Microbiol 78:459–474. doi: 10.1111/j.1365-2958.2010.07350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nguyen TN, Muller LS, Park SH, Siegel TN, Gunzl A. 2014. Promoter occupancy of the basal class I transcription factor A differs strongly between active and silent VSG expression sites in Trypanosoma brucei. Nucleic Acids Res 42:3164–3176. doi: 10.1093/nar/gkt1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lopez-Farfan D, Bart JM, Rojas-Barros DI, Navarro M. 2014. SUMOylation by the E3 ligase TbSIZ1/PIAS1 positively regulates VSG expression in Trypanosoma brucei. PLoS Pathog 10:e1004545. doi: 10.1371/journal.ppat.1004545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang X, Figueiredo LM, Espinal A, Okubo E, Li B. 2009. RAP1 is essential for silencing telomeric variant surface glycoprotein genes in Trypanosoma brucei. Cell 137:99–109. doi: 10.1016/j.cell.2009.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pandya UM, Sandhu R, Li B. 2013. Silencing subtelomeric VSGs by Trypanosoma brucei RAP1 at the insect stage involves chromatin structure changes. Nucleic Acids Res 41:7673–7682. doi: 10.1093/nar/gkt562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gunzl A, Kirkham JK, Nguyen TN, Badjatia N, Park SH. 2015. Mono-allelic VSG expression by RNA polymerase I in Trypanosoma brucei: expression site control from both ends? Gene 556:68–73. doi: 10.1016/j.gene.2014.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horn D. 2014. Antigenic variation in African trypanosomes. Mol Biochem Parasitol 195:123–129. doi: 10.1016/j.molbiopara.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Horn D, Cross GAM. 1997. Analysis of Trypanosoma brucei vsg expression site switching in vitro. Mol Biochem Parasitol 84:189–201. doi: 10.1016/S0166-6851(96)02794-6. [DOI] [PubMed] [Google Scholar]
- 43.Cross M, Taylor MC, Borst P. 1998. Frequent loss of the active site during variant surface glycoprotein expression site switching in vitro in Trypanosoma brucei. Mol Cell Biol 18:198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aitcheson N, Talbot S, Shapiro J, Hughes K, Adkin C, Butt T, Sheader K, Rudenko G. 2005. VSG switching in Trypanosoma brucei: antigenic variation analysed using RNAi in the absence of immune selection. Mol Microbiol 57:1608–1622. doi: 10.1111/j.1365-2958.2005.04795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benmerzouga I, Concepcion-Acevedo J, Kim HS, Vandoros AV, Cross GAM, Klingbeil MM, Li B. 2013. Trypanosoma brucei Orc1 is essential for nuclear DNA replication and affects both VSG silencing and VSG switching. Mol Microbiol 87:196–210. doi: 10.1111/mmi.12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McCulloch R, Barry JD. 1999. A role for RAD51 and homologous recombination in Trypanosoma brucei antigenic variation. Genes Dev 13:2875–2888. doi: 10.1101/gad.13.21.2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marcello L, Barry JD. 2007. Analysis of the VSG gene silent archive in Trypanosoma brucei reveals that mosaic gene expression is prominent in antigenic variation and is favored by archive substructure. Genome Res 17:1344–1352. doi: 10.1101/gr.6421207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morrison LJ, Marcello L, McCulloch R. 2009. Antigenic variation in the African trypanosome: molecular mechanisms and phenotypic complexity. Cell Microbiol 11:1724–1734. doi: 10.1111/j.1462-5822.2009.01383.x. [DOI] [PubMed] [Google Scholar]
- 49.Pays E, van Assel S, Laurent M, Dero B, Michiels F, Kronenberger P, Matthyssens G, van Meirvenne N, LeRay D, Steinert M. 1983. At least two transposed sequences are associated in the expression site of a surface antigen gene in different trypanosome clones. Cell 34:359–369. doi: 10.1016/0092-8674(83)90370-7. [DOI] [PubMed] [Google Scholar]
- 50.Kim HS, Cross GAM. 2010. TOPO3alpha influences antigenic variation by monitoring expression-site-associated VSG switching in Trypanosoma brucei. PLoS Pathog 6:e1000992. doi: 10.1371/journal.ppat.1000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim HS, Cross GAM. 2011. Identification of Trypanosoma brucei RMI1/BLAP75 homologue and its roles in antigenic variation. PLoS One 6:e25313. doi: 10.1371/journal.pone.0025313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jehi SE, Wu F, Li B. 2014. Trypanosoma brucei TIF2 suppresses VSG switching by maintaining subtelomere integrity. Cell Res 24:870–885. doi: 10.1038/cr.2014.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jehi SE, Li X, Sandhu R, Ye F, Benmerzouga I, Zhang M, Zhao Y, Li B. 2014. Suppression of subtelomeric VSG switching by Trypanosoma brucei TRF requires its TTAGGG repeat-binding activity. Nucleic Acids Res 42:12899–12911. doi: 10.1093/nar/gku942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bernards A, van der Ploeg LHT, Frasch ACC, Borst P, Boothroyd JC, Coleman S, Cross GAM. 1981. Activation of trypanosome surface glycoprotein genes involves a gene duplication-transposition leading to an altered 3′ end. Cell 27:497–505. doi: 10.1016/0092-8674(81)90391-3. [DOI] [PubMed] [Google Scholar]
- 55.Robinson NP, Burman N, Melville SE, Barry JD. 1999. Predominance of duplicative VSG gene conversion in antigenic variation in African trypanosomes. Mol Cell Biol 19:5839–5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dreesen O, Cross GAM. 2006. Consequences of telomere shortening at an active VSG expression site in telomerase-deficient Trypanosoma brucei. Eukaryot Cell 5:2114–2119. doi: 10.1128/EC.00059-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hovel-Miner GA, Boothroyd CE, Mugnier M, Dreesen O, Cross GAM, Papavasiliou FN. 2012. Telomere length affects the frequency and mechanism of antigenic variation in Trypanosoma brucei. PLoS Pathog 8:e1002900. doi: 10.1371/journal.ppat.1002900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thon G, Baltz T, Giroud C, Eisen H. 1990. Trypanosome variable surface glycoproteins: composite genes and order of expression. Genes Dev 4:1374–1383. doi: 10.1101/gad.4.8.1374. [DOI] [PubMed] [Google Scholar]
- 59.Morrison LJ, Majiwa P, Read AF, Barry JD. 2005. Probabilistic order in antigenic variation of Trypanosoma brucei. Int J Parasitol 35:961–972. doi: 10.1016/j.ijpara.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 60.Glover L, Jun J, Horn D. 2011. Microhomology-mediated deletion and gene conversion in African trypanosomes. Nucleic Acids Res 39:1372–1380. doi: 10.1093/nar/gkq981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jasin M, Rothstein R. 2013. Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol 5:a012740. doi: 10.1101/cshperspect.a012740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goodarzi AA, Jeggo PA. 2013. The repair and signaling responses to DNA double-strand breaks. Adv Genet 82:1–45. doi: 10.1016/B978-0-12-407676-1.00001-9. [DOI] [PubMed] [Google Scholar]
- 63.Rothkamm K, Kruger I, Thompson LH, Lobrich M. 2003. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol 23:5706–5715. doi: 10.1128/MCB.23.16.5706-5715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Conway C, Proudfoot C, Burton P, Barry JD, McCulloch R. 2002. Two pathways of homologous recombination in Trypanosoma brucei. Mol Microbiol 45:1687–1700. doi: 10.1046/j.1365-2958.2002.03122.x. [DOI] [PubMed] [Google Scholar]
- 65.Burton P, McBride DJ, Wilkes JM, Barry JD, McCulloch R. 2007. Ku heterodimer-independent end joining in Trypanosoma brucei cell extracts relies upon sequence microhomology. Eukaryot Cell 6:1773–1781. doi: 10.1128/EC.00212-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Glover L, McCulloch R, Horn D. 2008. Sequence homology and microhomology dominate chromosomal double-strand break repair in African trypanosomes. Nucleic Acids Res 36:2608–2618. doi: 10.1093/nar/gkn104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barnes RL, McCulloch R. 2007. Trypanosoma brucei homologous recombination is dependent on substrate length and homology, though displays a differential dependence on mismatch repair as substrate length decreases. Nucleic Acids Res 35:3478–3493. doi: 10.1093/nar/gkm249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Baudat F, Imai Y, de Massy B. 2013. Meiotic recombination in mammals: localization and regulation. Nat Rev Genet 14:794–806. doi: 10.1038/nrg3573. [DOI] [PubMed] [Google Scholar]
- 69.Glover L, Alsford S, Horn D. 2013. DNA break site at fragile subtelomeres determines probability and mechanism of antigenic variation in African trypanosomes. PLoS Pathog 9:e1003260. doi: 10.1371/journal.ppat.1003260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sung S, Li F, Park YB, Kim JS, Kim AK, Song OK, Kim J, Che J, Lee SE, Cho Y. 2014. DNA end recognition by the Mre11 nuclease dimer: insights into resection and repair of damaged DNA. EMBO J 33:2422–2435. doi: 10.15252/embj.201488299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, Maity R, S van Rossum-Fikkert Kertokalio A, Romoli F, Ismail A, Ismalaj E, Petricci E, Neale MJ, Bristow RG, Masson JY, Wyman C, Jeggo PA, Tainer JA. 2014. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell 53:7–18. doi: 10.1016/j.molcel.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, Modrich P, Kowalczykowski SC. 2011. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev 25:350–362. doi: 10.1101/gad.2003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mimitou EP, Symington LS. 2008. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455:770–774. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. 2008. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134:981–994. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. 2007. Human CtIP promotes DNA end resection. Nature 450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sugiyama T, Zaitseva EM, Kowalczykowski SC. 1997. A single-stranded DNA-binding protein is needed for efficient presynaptic complex formation by the Saccharomyces cerevisiae Rad51 protein. J Biol Chem 272:7940–7945. doi: 10.1074/jbc.272.12.7940. [DOI] [PubMed] [Google Scholar]
- 77.San Filippo J, Sung P, Klein H. 2008. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- 78.San Filippo J, Chi P, Sehorn MG, Etchin J, Krejci L, Sung P. 2006. Recombination mediator and Rad51 targeting activities of a human BRCA2 polypeptide. J Biol Chem 281:11649–11657. doi: 10.1074/jbc.M601249200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang H, Li Q, Fan J, Holloman WK, Pavletich NP. 2005. The BRCA2 homologue Brh2 nucleates RAD51 filament formation at a dsDNA-ssDNA junction. Nature 433:653–657. doi: 10.1038/nature03234. [DOI] [PubMed] [Google Scholar]
- 80.Symington LS. 2002. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol Mol Biol Rev 66:630–670. doi: 10.1128/MMBR.66.4.630-670.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sung P, Robberson DL. 1995. DNA strand exchange mediated by a RAD51-ssDNA nucleoprotein filament with polarity opposite to that of RecA. Cell 82:453–461. doi: 10.1016/0092-8674(95)90434-4. [DOI] [PubMed] [Google Scholar]
- 82.Lin FL, Sperle K, Sternberg N. 1984. Model for homologous recombination during transfer of DNA into mouse L cells: role for DNA ends in the recombination process. Mol Cell Biol 4:1020–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nassif N, Penney J, Pal S, Engels WR, Gloor GB. 1994. Efficient copying of nonhomologous sequences from ectopic sites via P-element-induced gap repair. Mol Cell Biol 14:1613–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ferguson DO, Holloman WK. 1996. Recombinational repair of gaps in DNA is asymmetric in Ustilago maydis and can be explained by a migrating D-loop model. Proc Natl Acad Sci U S A 93:5419–5424. doi: 10.1073/pnas.93.11.5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW. 1983. The double-strand break repair model for recombination. Cell 33:25–35. doi: 10.1016/0092-8674(83)90331-8. [DOI] [PubMed] [Google Scholar]
- 86.Ho CK, Mazon G, Lam AF, Symington LS. 2010. Mus81 and Yen1 promote reciprocal exchange during mitotic recombination to maintain genome integrity in budding yeast. Mol Cell 40:988–1000. doi: 10.1016/j.molcel.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wechsler T, Newman S, West SC. 2011. Aberrant chromosome morphology in human cells defective for Holliday junction resolution. Nature 471:642–646. doi: 10.1038/nature09790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.De Muyt A, Jessop L, Kolar E, Sourirajan A, Chen J, Dayani Y, Lichten M. 2012. BLM helicase ortholog Sgs1 is a central regulator of meiotic recombination intermediate metabolism. Mol Cell 46:43–53. doi: 10.1016/j.molcel.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zakharyevich K, Tang S, Ma Y, Hunter N. 2012. Delineation of joint molecule resolution pathways in meiosis identifies a crossover-specific resolvase. Cell 149:334–347. doi: 10.1016/j.cell.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wu L, Hickson ID. 2003. The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 426:870–874. doi: 10.1038/nature02253. [DOI] [PubMed] [Google Scholar]
- 91.Michels PAM, Bernards A, van der Ploeg L, Borst P. 1982. Characterization of the expression-linked gene copies of variant surface glycoprotein 118 in two independently isolated clones of Trypanosoma brucei. Nucleic Acids Res 10:2353–2366. doi: 10.1093/nar/10.7.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pays E, Delauw MF, Van Assel S, Laurent M, Vervoort T, Van Meirvenne N, Steinert M. 1983. Modifications of a Trypanosoma b. brucei antigen gene repertoire by different DNA recombinational mechanisms. Cell 35:721–731. doi: 10.1016/0092-8674(83)90105-8. [DOI] [PubMed] [Google Scholar]
- 93.Bernards A, De Lange T, Michels PA, Liu AY, Huisman MJ, Borst P. 1984. Two modes of activation of a single surface antigen gene of Trypanosoma brucei. Cell 36:163–170. doi: 10.1016/0092-8674(84)90085-0. [DOI] [PubMed] [Google Scholar]
- 94.Pays E. 1985. Gene conversion in Trypanosoma antigenic variation. Prog NAR Mol Biol 32:1–27. [DOI] [PubMed] [Google Scholar]
- 95.Gaud A, Carrington M, Deshusses J, Schaller DRG. 1997. Polymerase chain reaction-based gene disruption in Trypanosoma brucei. Mol Biochem Parasitol 87:113–115. doi: 10.1016/S0166-6851(97)00048-0. [DOI] [PubMed] [Google Scholar]
- 96.Suwaki N, Klare K, Tarsounas M. 2011. RAD51 paralogs: roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin Cell Dev Biol 22:898–905. doi: 10.1016/j.semcdb.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 97.French CA, Tambini CE, Thacker J. 2003. Identification of functional domains in the RAD51L2 (RAD51C) protein and its requirement for gene conversion. J Biol Chem 278:45445–45450. doi: 10.1074/jbc.M308621200. [DOI] [PubMed] [Google Scholar]
- 98.Deriano L, Roth DB. 2013. Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annu Rev Genet 47:433–455. doi: 10.1146/annurev-genet-110711-155540. [DOI] [PubMed] [Google Scholar]
- 99.Decottignies A. 2013. Alternative end-joining mechanisms: a historical perspective. Front Genet 4:48. doi: 10.3389/fgene.2013.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang Y, Gostissa M, Hildebrand DG, Becker MS, Boboila C, Chiarle R, Lewis S, Alt FW. 2010. The role of mechanistic factors in promoting chromosomal translocations found in lymphoid and other cancers. Adv Immunol 106:93–133. doi: 10.1016/S0065-2776(10)06004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Deriano L, Stracker TH, Baker A, Petrini JH, Roth DB. 2009. Roles for NBS1 in alternative nonhomologous end-joining of V(D)J recombination intermediates. Mol Cell 34:13–25. doi: 10.1016/j.molcel.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lee-Theilen M, Matthews AJ, Kelly D, Zheng S, Chaudhuri J. 2011. CtIP promotes microhomology-mediated alternative end joining during class-switch recombination. Nat Struct Mol Biol 18:75–79. doi: 10.1038/nsmb.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rass E, Grabarz A, Plo I, Gautier J, Bertrand P, Lopez BS. 2009. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat Struct Mol Biol 16:819–824. doi: 10.1038/nsmb.1641. [DOI] [PubMed] [Google Scholar]
- 104.Xie A, Kwok A, Scully R. 2009. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat Struct Mol Biol 16:814–818. doi: 10.1038/nsmb.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang Y, Jasin M. 2011. An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat Struct Mol Biol 18:80–84. doi: 10.1038/nsmb.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sugawara N, Ira G, Haber JE. 2000. DNA length dependence of the single-strand annealing pathway and the role of Saccharomyces cerevisiae RAD59 in double-strand break repair. Mol Cell Biol 20:5300–5309. doi: 10.1128/MCB.20.14.5300-5309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kramer KM, Brock JA, Bloom K, Moore JK, Haber JE. 1994. Two different types of double-strand breaks in Saccharomyces cerevisiae are repaired by similar RAD52-independent, nonhomologous recombination events. Mol Cell Biol 14:1293–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Boboila C, Alt FW, Schwer B. 2012. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv Immunol 116:1–49. doi: 10.1016/B978-0-12-394300-2.00001-6. [DOI] [PubMed] [Google Scholar]
- 109.Boboila C, Oksenych V, Gostissa M, Wang JH, Zha S, Zhang Y, Chai H, Lee CS, Jankovic M, Saez LM, Nussenzweig MC, McKinnon PJ, Alt FW, Schwer B. 2012. Robust chromosomal DNA repair via alternative end-joining in the absence of X-ray repair cross-complementing protein 1 (XRCC1). Proc Natl Acad Sci U S A 109:2473–2478. doi: 10.1073/pnas.1121470109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Simsek D, Brunet E, Wong SY, Katyal S, Gao Y, McKinnon PJ, Lou J, Zhang L, Li J, Rebar EJ, Gregory PD, Holmes MC, Jasin M. 2011. DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation. PLoS Genet 7:e1002080. doi: 10.1371/journal.pgen.1002080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Conway C, McCulloch R, Ginger ML, Robinson NP, Browitt A, Barry JD. 2002. Ku is important for telomere maintenance, but not for differential expression of telomeric VSG genes, in African trypanosomes. J Biol Chem 277:21269–21277. doi: 10.1074/jbc.M200550200. [DOI] [PubMed] [Google Scholar]
- 112.Janzen CJ, Lander F, Dreesen O, Cross GAM. 2004. Telomere length regulation and transcriptional silencing in KU80-deficient Trypanosoma brucei. Nucleic Acids Res 32:6575–6584. doi: 10.1093/nar/gkh991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Proudfoot C, McCulloch R. 2005. Distinct roles for two RAD51-related genes in Trypanosoma brucei antigenic variation. Nucleic Acids Res 33:6906–6919. doi: 10.1093/nar/gki996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hartley CL, McCulloch R. 2008. Trypanosoma brucei BRCA2 acts in antigenic variation and has undergone a recent expansion in BRC repeat number that is important during homologous recombination. Mol Microbiol 68:1237–1251. doi: 10.1111/j.1365-2958.2008.06230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chen Q, Ijpma A, Greider CW. 2001. Two survivor pathways that allow growth in the absence of telomerase are generated by distinct telomere recombination events. Mol Cell Biol 21:1819–1827. doi: 10.1128/MCB.21.5.1819-1827.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Takata M, Sasaki MS, Tachiiri S, Fukushima T, Sonoda E, Schild D, Thompson LH, Takeda S. 2001. Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol Cell Biol 21:2858–2866. doi: 10.1128/MCB.21.8.2858-2866.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ira G, Malkova A, Liberi G, Foiani M, Haber JE. 2003. Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell 115:401–411. doi: 10.1016/S0092-8674(03)00886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mullen JR, Nallaseth FS, Lan YQ, Slagle CE, Brill SJ. 2005. Yeast Rmi1/Nce4 controls genome stability as a subunit of the Sgs1-Top3 complex. Mol Cell Biol 25:4476–4487. doi: 10.1128/MCB.25.11.4476-4487.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hartung F, Suer S, Knoll A, Wurz-Wildersinn R, Puchta H. 2008. Topoisomerase 3alpha and RMI1 suppress somatic crossovers and are essential for resolution of meiotic recombination intermediates in Arabidopsis thaliana. PLoS Genet 4:e1000285. doi: 10.1371/journal.pgen.1000285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bizard AH, Hickson ID. 2014. The dissolution of double Holliday junctions. Cold Spring Harb Perspect Biol 6:a016477. doi: 10.1101/cshperspect.a016477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Robinson NP, McCulloch R, Conway C, Browitt A, Barry JD. 2002. Inactivation of Mre11 does not affect VSG gene duplication mediated by homologous recombination in Trypanosoma brucei. J Biol Chem 277:26185–26193. doi: 10.1074/jbc.M203205200. [DOI] [PubMed] [Google Scholar]
- 122.Borst P, Rudenko G, Taylor MC, Blundell PA, van Leeuwen F, Bitter W, Cross M, McCulloch R. 1996. Antigenic variation in trypanosomes. Arch Med Res 27:379–388. [PubMed] [Google Scholar]
- 123.Barry JD. 1997. The relative significance of mechanisms of antigenic variation in African trypanosomes. Parasitol Today 13:212–218. doi: 10.1016/S0169-4758(97)01039-9. [DOI] [PubMed] [Google Scholar]
- 124.Stewart JA, Chaiken MF, Wang F, Price CM. 2012. Maintaining the end: roles of telomere proteins in end-protection, telomere replication and length regulation. Mutat Res 730:12–19. doi: 10.1016/j.mrfmmm.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T. 2009. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138:90–103. doi: 10.1016/j.cell.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ye J, Lenain C, Bauwens S, Rizzo A, Saint-Leger A, Poulet A, Benarroch D, Magdinier F, Morere J, Amiard S, Verhoeyen E, Britton S, Calsou P, Salles B, Bizard A, Nadal M, Salvati E, Sabatier L, Wu Y, Biroccio A, Londono-Vallejo A, Giraud-Panis MJ, Gilson E. 2010. TRF2 and apollo cooperate with topoisomerase 2alpha to protect human telomeres from replicative damage. Cell 142:230–242. doi: 10.1016/j.cell.2010.05.032. [DOI] [PubMed] [Google Scholar]
- 127.Miller KM, Rog O, Cooper JP. 2006. Semi-conservative DNA replication through telomeres requires Taz1. Nature 440:824–828. doi: 10.1038/nature04638. [DOI] [PubMed] [Google Scholar]
- 128.Muraki K, Nabetani A, Nishiyama A, Ishikawa F. 2011. Essential roles of Xenopus TRF2 in telomere end protection and replication. Genes Cells 16:728–739. doi: 10.1111/j.1365-2443.2011.01520.x. [DOI] [PubMed] [Google Scholar]
- 129.Canudas S, Houghtaling BR, Kim JY, Dynek JN, Chang WG, Smith S. 2007. Protein requirements for sister telomere association in human cells. EMBO J 26:4867–4878. doi: 10.1038/sj.emboj.7601903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Canudas S, Smith S. 2009. Differential regulation of telomere and centromere cohesion by the Scc3 homologues SA1 and SA2, respectively, in human cells. J Cell Biol 187:165–173. doi: 10.1083/jcb.200903096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.de Lange T. 2005. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 132.Celli GB, de Lange T. 2005. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat Cell Biol 7:712–718. doi: 10.1038/ncb1275. [DOI] [PubMed] [Google Scholar]
- 133.Murnane JP, Sabatier L. 2004. Chromosome rearrangements resulting from telomere dysfunction and their role in cancer. Bioessays 26:1164–1174. doi: 10.1002/bies.20125. [DOI] [PubMed] [Google Scholar]
- 134.Wada M, Nakamura Y. 1996. Unique telomeric expression site of major-surface-glycoprotein genes of Pneumocystis carinii. DNA Res 3:55–64. doi: 10.1093/dnares/3.2.55. [DOI] [PubMed] [Google Scholar]
- 135.Keely SP, Stringer JR. 2009. Complexity of the MSG gene family of Pneumocystis carinii. BMC Genomics 10:367. doi: 10.1186/1471-2164-10-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhang JR, Norris SJ. 1998. Genetic variation of the Borrelia burgdorferi gene vlsE involves cassette-specific, segmental gene conversion. Infect Immun 66:3698–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhang JR, Norris SJ. 1998. Kinetics and in vivo induction of genetic variation of vlsE in Borrelia burgdorferi. Infect Immun 66:3689–3697. [DOI] [PMC free article] [PubMed] [Google Scholar]