

Abstract
Because of resetting, a role for baroreflexes in long-term control of arterial pressure has been commonly dismissed in the past. However, in recent years, this perspective has changed. Novel approaches for determining chronic neurohormonal and cardiovascular responses to natural variations in baroreceptor activity and to electrical stimulation of the carotid baroreflex indicate incomplete resetting and sustained responses that lead to long-term alterations in sympathetic activity and arterial pressure.
If everyone is thinking alike, then no one is thinking.–Benjamin Franklin
Recent clinical trials reporting impressive anti-hypertensive responses to electrical stimulation of the carotid baroreflex in patients with resistant hypertension have renewed interest in the role of the sympathetic nervous system in the pathogenesis of hypertension and other cardiovascular disorders associated with autonomic dysregulation (2, 5, 26, 30, 40, 59). Electrical baroreflex activation (BA) provides global inhibition of sympathetic activity, and suppression of renal sympathetic nerve activity (RSNA) is one major mechanism by which chronic lowering of arterial pressure is achieved. Endovascular radio-frequency renal nerve ablation, another recent device-based therapy for resistant hypertension, directly reduces RSNA and also lowers arterial pressure chronically (16, 64). However, experimental studies indicate that there may be additional mechanisms whereby BA chronically lowers arterial pressure and impacts organ function. In this regard, the focus of this article will not be on past and current clinical trials using these two medical devices for the treatment of resistant hypertension. Rather, the focus of this review will be on experimental studies that have provided a conceptual understanding of the potential role of arterial baroreflexes in long-term control of arterial pressure and the relevance of these studies to understanding the mechanisms that account for chronic lowering of arterial pressure during BA. In turn, the recent development of a safe medical device for chronically activating the carotid baroreflex has provided a unique tool for exploring the efferent mechanisms that lead to long-term blood pressure lowering during sustained baroreflex-mediated suppression of central sympathetic outflow. Some of the responses to chronic BA are consistent with established concepts; others have been quite surprising and need further resolution. Because of space limitations, this article will emphasize only long-term blood pressure control and will exclude discussion of other important actions of BA, such as those on renal hemodynamics (1, 28, 41, 44) and cardiac function (6, 7, 21, 22, 29, 30, 41, 43, 56, 67, 68). Finally, we apologize for including so much personal data, but studies aiming at elucidating the chronic relationships between baroreflex function and arterial pressure are otherwise scarce due to inherent technical challenges.
20th Century Dogma: Resetting Prevents Baroreflexes From Chronically Regulating Arterial Pressure
Although it is well established that the baroreflex plays a critical role in the acute regulation of arterial pressure, the dogma before the turn of the 21st century was that arterial baroreflexes are not important in the long-term control of arterial pressure because they reset in the direction of the prevailing level of arterial pressure (10, 35, 52, 65). Most of the evidence in support of this notion was based on baroreceptor afferent nerve recordings in various acute preparations designed to mimic physiological blood pressure-baroreceptor interactions or short-term recordings in anesthetized animals with chronic hypertension. These studies demonstrated a substantial degree of baroreceptor resetting. Because of experimental limitations in chronically recording nerve activity, these findings were commonly extrapolated to the chronic state to indicate the unimportance of baroreflexes in the long-term regulation of sympathetic activity and arterial pressure. However, the very nature of these acute studies makes them incapable of revealing the precise time course and degree of baroreceptor resetting that is relevant for normal daily activity. The other key argument often made to discount the importance of the baroreflex in long-term control of sympathetic activity and arterial pressure was based on studies following sinoaortic denervation (SAD). These studies demonstrated that the rise in arterial pressure after severing baroreceptor afferent signals to the brain is largely transient and that little or no hypertension is sustained chronically, presumably because initial increases in sympathetic activity return to control levels. Therefore, the presumption has been that if complete loss of baroreceptor inhibition fails to produce appreciable chronic increases in arterial pressure, then it is unlikely that naturally occurring alterations in baroreceptor activity, even if sustained, could have long-term effects on sympathetic activity and arterial pressure. In contrast to the acute studies indicated above, the strength of the SAD studies is that they are based on chronic observations and are not dependent on extrapolating findings from the acute to the chronic state. However, a caveat in the interpretation of these studies is that the temporal effects of SAD on sympathetic activity and arterial pressure are associated with plasticity in the central nervous system structures involved in the baroreceptor circuitry, resulting in alterations in the ability of these neurons to influence sympathetic activity and arterial pressure (9, 31, 57, 58, 63). Although inconsistent with some observations (33, 63), an early study in dogs indicated that central input from cardiopulmonary receptors precluded sustained hypertension after SAD (55). Despite the uncertainties regarding central compensations following SAD, a key implication from these studies is that resetting of the baroreflex may occur centrally, as well as at the level of the baroreceptors. That is, SAD studies indicate that central adaptations or “central resetting” may chronically counteract the magnitude of central sympathetic outflow induced by initial changes in central baroreceptor input.
Challenges to the Dogma at the Turn of the 21st Century: Incomplete Resetting and Long-Term Activation of Baroreflexes in Hypertension
At the turn of the 21st century, a number of observations from studies using novel experimental approaches in chronically instrumented animals challenged the premise that there is complete resetting of baroreflexes in hypertension and that baroreflex-mediated suppression of sympathetic activity is not a sustained physiological response in chronic hypertension. With increasing evidence that increased RSNA plays an important role in mediating some forms of experimental and clinical hypertension (13, 15, 35, 52, 53), a most surprising early finding in chronically instrumented dogs was that renal norepinephrine spillover, an indirect index of RSNA, was actually suppressed throughout the acute and chronic phases of hypertension produced by an infusion of ANG II yielding physiologically relevant increases in plasma levels of the hormone (8). This was a particularly unexpected finding in light of a number of acute studies indicating that ANG II may act centrally to increase sympathetic activity (52). Thus, rather than supporting the contention that activation of the sympathetic nervous system contributes to ANG II hypertension, this finding suggested that reductions in RSNA may actually serve as a compensatory mechanism to attenuate the severity of the hypertension. At this time, the mechanism for renal sympatho-inhibition was puzzling, and, in light of the prevailing dogma regarding baroreflex resetting, it would have been heresy to suggest that sustained suppression of RSNA in hypertension was mediated by chronic activation of the arterial baroreflex. However, this possibility was convincingly established several years later. In elegant studies conducted in chronically instrumented rabbits, continuous electrical recordings of RSNA showed sustained reductions in RSNA throughout both the acute and the chronic phases of ANG II hypertension (4). Furthermore, because the inhibition of RSNA was abolished in rabbits with prior SAD (3), this observation provided direct evidence that the sustained inhibition of RSNA during ANG II hypertension was mediated by chronic activation of arterial baroreflexes.
Further support for the concept that the baroreflex is chronically activated in hypertension comes from neuroanatomical studies in canines using Fos-like (Fos-Li) protein immunohistochemistry to determine the sites of neuronal activation in the central baroreflex pathway. Compared with controls, in dogs with chronic ANG II hypertension, there were significant increases in Fos-Li staining in the nucleus tractus solitarius (NTS), the brain stem site of termination of baroreceptor afferents, and the caudal ventrolateral medulla (CVLM), but no increase in staining in the rostral ventrolateral medulla (RVLM) neurons (49), which project to preganglionic neurons in the spinal cord and account for basal sympathetic outflow to the periphery (FIGURE 1). Since baroreflex suppression of sympathoexcitatory neurons in the RVLM is mediated by stimulation of NTS and CVLM neurons, this study provided further support for the hypothesis that the baroreflex is chronically activated in ANG II hypertension. Furthermore, this same pattern of neuronal activation in the NTS and CVLM was found in canines after 6 wk of obesity hypertension and during chronic electrical activation of the carotid baroreflex (35, 39, 42, 51), supporting the concept that sustained activation of the baroreflex may be a general compensatory response in hypertension. However, a notable difference in the dogs with obesity was that hypertension increased staining in RVLM neurons, which contribute importantly to increased sympathetic activity and arterial pressure in obesity (62). Thus increased staining in RVLM neurons is consistent with this form of hypertension being neurally mediated (17, 24, 41, 44, 53). Collectively, these findings suggest that, in obesity, the sympathoexcitatory projections into the RVLM, which have not been clearly defined, predominate over inhibitory input from baroreceptors, with the resultant response being a net increase in central sympathetic outflow. Furthermore, since baroreflex inhibition of sympathetic activity is progressively impaired during the evolution of obesity hypertension (23), baroreflex dysfunction may make an increasingly important contribution to sympathetic activation and hypertension as obesity extends from weeks to months to years. However, this reasoning assumes that this impairment in the ability of the baroreflex to evoke acute changes in postganglionic efferent sympathetic nerve activity to muscle, measured by microneurography, also reflects long-term alterations in baroreflex control of RSNA.
FIGURE 1.

A schematic representation of the sympathetic carotid baroreflex arc including key medullary brain regions for cardiovascular control
Pressure-induced stretch of carotid sinus baroreceptors is transduced into an electrical signal in afferent fibers in the carotid sinus nerve (Hering's nerve), a branch of the glossopharyngeal nerve (cranial nerve IX), and impulses then travel to the NTS, the initial central site of termination of baroreceptor afferents. In turn, NTS neurons activate inhibitory neurons in the CVLM that project to the RVLM. Projections from RVLM neurons to preganglionic neurons in the intermediolateral cell column of the spinal cord account for basal sympathetic outflow to the periphery. Thus pressure-induced baroreceptor activation of the NTS and CVLM neurons suppresses central sympathetic outflow by inhibiting the RVLM. This medullary brain circuit is also activated by electrical stimulation of the carotid sinus leading to substantial long-term reductions in sympathetic activity and arterial pressure. With the current Barostem neo system, a miniaturized stimulating electrode is sutured to the surface of the carotid sinus and is connected to an internally implantable pulse generator that is externally programmable by radiofrequency control. NTS, nucleus tractus solitarius; CVLM, caudal ventolateral medulla; RVLM, rostral ventrolateral medulla.
With accumulating evidence that the baroreflex is chronically activated in several models of hypertension, it was important to demonstrate that baroreflex-mediated inhibition of RSNA has functional consequences that may lead to long-term reductions in arterial pressure. Given that increases in renal excretory function are essential for permitting chronic reductions in arterial pressure, we focused on whether baroreflex-mediated suppression of RSNA has sustained effects to promote sodium excretion. To obviate the confounding effects of nonneurally mediated mechanisms that may counteract and obscure the direct natriuretic effects of decreased RSNA, we determined temporal changes in sodium excretion during the evolution of both ANG II and norepinephrine-induced hypertension in dogs with unilateral renal denervation and surgical division of the urinary bladder into hemibladders to allow separate 24-h urine collection from denervated and innervated kidneys (47, 48, 50). This is a powerful approach for exposing a functional role of the renal nerves on renal excretory function because both kidneys are exposed to the same perfusion pressure and hormonal influences. Thus any differences in sodium excretion between the kidneys can be attributed to the effects of the renal nerves on renal excretory function. Sodium excretory responses in both forms of hypertension were similar in dogs with intact baroreflexes, and, for the sake of brevity, only those during ANG II hypertension are described below.
With sodium intake held constant, sodium excretory responses in ANG II hypertension were determined before and after SAD (47). During the control period, sodium excretion in the two kidneys was approximately equal (FIGURE 2A). As expected, during ANG II infusion, total sodium excretion (from both kidneys) decreased for 1–2 days before daily sodium balance was subsequently achieved at an elevated arterial pressure. Most notably, throughout the progression of the hypertension, the innervated kidneys excreted more sodium than the denervated kidneys when the baroreflexes were intact (FIGURE 2B). This is consistent with the findings discussed above, indicating baroreflex-mediated inhibition of RSNA in dogs and rabbits with chronic ANG II hypertension. Several weeks after recovery from SAD, the infusion of ANG II was repeated. In marked contrast, sodium excretion in innervated kidneys actually decreased below levels in denervated kidneys (FIGURE 2C), a response diametrically opposite to that observed when arterial baroreflexes were intact. This latter response is consistent with reports that ANG II acts centrally to increase sympathetic activity and highlights the importance of baroreflex activation in abolishing sympathetically mediated sodium retention in ANG II hypertension. Taken together, these findings do not support the hypothesis that increased RSNA impairs sodium excretion and contributes to ANG II hypertension. Rather, these findings indicate just the opposite, that natural baroreflex-mediated suppression of RSNA is a chronic compensatory response that may actually attenuate the antinatriuretic and hypertensive effects of ANG II.
FIGURE 2.

Daily sodium/fluid excretion from innervated and denervated kidneys
Daily sodium/fluid excretion from innervated and denervated kidneys under control conditions (A), during ANG II hypertension (B), and during ANG II hypertension (C) after sinoaortic denervation (SAD). Sodium intake was constant, and daily sodium balance was achieved under all conditions. During ANG II hypertension, there was a relatively greater rate of sodium excretion from innervated than denervated kidneys before but not after SAD, consistent with sustained baroreflex-mediated inhibition of renal sympathetic nerve activity. In the absence of the baroreflex (C), the lower rate of sodium excretion from innervated vs. denervated kidneys may reflect central actions of ANG II to increase renal sympathetic nerve activity.
Parenthetically, the greater rate of sodium excretion in innervated kidneys with reduced RSNA compared with kidneys completely devoid of innervation (FIGURE 2B) presents a conundrum. This suggests adaptive changes in chronically denervated kidneys that actually favor sodium retention. Although the nature of these adaptive changes has not been defined, this abnormality in the ability to excrete sodium is not a consequence of renal denervation supersensitivity (50). Since chronic bilateral renal denervation is a common technique for assessing the physiological and pathophysiological effects of the renal nerves, this may have important interpretive implications.
Notwithstanding the importance of the above studies in demonstrating that baroreflexes do not totally reset in hypertension and do have sustained effects to suppress RSNA and promote sodium excretion, they provide little insight into the quantitative importance of baroreflex activation in attenuating the severity of hypertension. For example, despite the studies discussed above showing sustained baroreflex-mediated suppression of RSNA and attendant increases in neurally mediated sodium excretion in ANG II hypertension, Cowley et al. reported that SAD does not exacerbate the severity of this form of hypertension in canines (11). Does this imply that the natriuresis attributed to baroreflex-mediated suppression of RSNA is relatively insignificant in increasing renal excretory function in the presence of the potent antinatriuretic effects of ANG II? In 2004, the opportunity to provide quantitative insight into the blood pressure-lowering capability of baroreflex activation was provided by CVRx (Minneapolis, MN), a company with an interest in establishing a device-based approach for the treatment of selective cardiovascular disorders including resistant hypertension, which cannot be treated adequately by pharmacological therapy. Their goal was to develop a safe medical device capable of producing controllable and reliable suppression of sympathetic activity and, in the case of resistant hypertension, lowering arterial pressure by chronic electrical activation of the carotid baroreflex.
Device for Chronic Electrical Activation of the Carotid Baroreflex
Chronic electrical stimulation of the carotid sinus provides a novel approach for evaluating quantitatively the time dependency and underlying mechanisms of blood pressure lowering during BA (40). A unique aspect of this technology is that field stimulation of the carotid sinuses bypasses mechanotransduction at the level of the baroreceptors, where most baroreflex resetting occurs, and allows for precise control of baroreceptor afferent input into the central nervous system. With the prototype Rheos system, BA is achieved by implanting stimulating electrodes in the perivascular space around each carotid sinus with the lead bodies connected to a pulse generator. The internally implantable pulse generator is completely programmable by radio-frequency control using an external programming system. This allows for controlled current delivery throughout the day (intensity, frequency, pulse duration), including the option of providing customized patterns of baroreflex activation. However, for experimental and clinical studies conducted to date, the pulse generator has been programmed to deliver continuous impulses throughout the cardiac cycle. Most recently, a second-generation miniaturized electrode has been developed to minimize the invasiveness of the implant procedure. The new electrode (Barostem neo) is sutured to the surface of the carotid sinus and is implanted unilaterally (FIGURE 1).
Chronic Activation of the Carotid Baroreflex Produces Sustained Reductions in Sympathetic Activity and Arterial Pressure
As indicated above, following SAD, which abolishes baroreceptor afferent input into the brain, there are slowly developing central adaptations that greatly diminish, if not completely abolish, the initial sympathetic activation and attendant hypertension. In addition, studies in hypertensive rats suggest that inhibitory adaptations do occur at the level of the NTS to offset sustained baroreceptor input into the brain stem (54). Thus, if central resetting is a powerful mechanism that normally diminishes the sympatho-inhibition induced by increased baroreceptor afferent input, then BA would not be expected to lead to appreciable long-term reductions in sympathetic activity and arterial pressure. Therefore, a critical preclinical test of the Rheos system for chronically lowering arterial pressure was determination of the time-dependent changes in sympathetic activity and arterial pressure during BA. With the pulse generator programmed to achieve a decrease in mean arterial pressure (MAP) of ∼20 mmHg during the first 24 h of BA and with no further adjustments in stimulation parameters thereafter, a most impressive response was that reductions in MAP and plasma norepinephrine concentration (∼ 35%) were sustained throughout the entire 7 days of BA without any trend for adaptation of these responses (45). Heart rate also decreased in parallel with MAP, a response primarily mediated by parasympathetic activation. Despite these exciting findings, we were cognizant of an earlier report by Thrasher indicating that the hypertension in dogs following SAD persisted for 8 days before central adaptations were sufficiently robust to lead to normalization of arterial pressure (66). For that reason, we considered the possibility that 1 wk of BA may have been insufficient time to demonstrate a true sustained blood pressure-lowering response to BA. Therefore, we compared the 3-wk time course of change in MAP in dogs subjected to SAD and BA (43). Results of this study are depicted in FIGURE 3. In confirmation of Thrasher's findings, the initial hypertension after SAD did not persist beyond 1 wk. However, most notably, with stimulation parameters unchanged after days 1–2 of carotid sinus stimulation, the initial reduction in MAP of ∼20 mmHg was sustained throughout the entire 3 wk of BA. Furthermore, in parallel with the fall in MAP, there were sustained reductions in whole body norepinephrine spillover (∼45%), an indirect index of central sympathetic outflow, and heart rate (∼15 beats/min) during the entire duration of BA without any trend for adaptation. Thus, in contrast to the perspective provided from studies using SAD, this study indicates that central resetting is not an important compensatory mechanism that diminishes the suppression of sympathetic activity and attendant lowering of blood pressure, or the bradycardia, in response to chronic increases in baroreceptor afferent input into the central nervous system.
FIGURE 3.
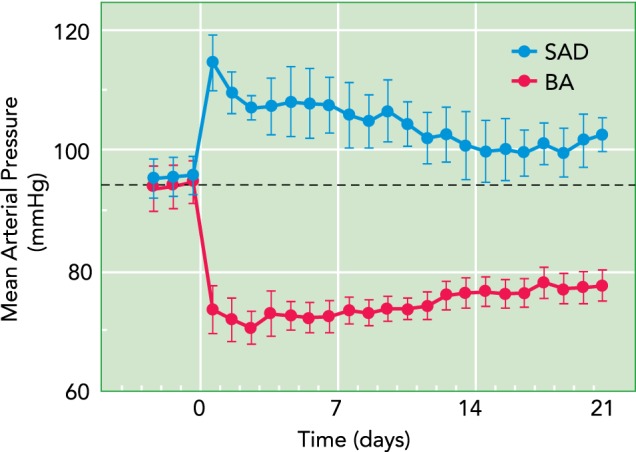
Chronic changes in mean arterial pressure following sinoaortic SAD and electrical baroreflex activation
Substantial reductions in mean arterial pressure were sustained during baroreflex activation (BA), whereas increases in mean arterial pressure were transient following SAD, as expected.
Baroreflex Activation Inhibits the Renin-Angiotensin-Aldosterone System
Activation of the renin-angiotensin system has a powerful role in restoring arterial pressure to control levels following reductions in renal perfusion pressure, and this hormonal system plays an important role in both short-term and long-term control of arterial pressure. Accordingly, compensatory increases in renin secretion have the potential to greatly attenuate the magnitude of blood pressure lowering during BA. However, despite exceeding reductions in arterial pressure (∼15 mmHg) that normally lead to sharp increases in renin secretion (14, 19), it is remarkable that plasma renin activity (PRA) does not increase when arterial pressure is lowered as much as 25 mmHg during BA (36, 45). Thus BA must have an inhibitory effect on renin secretion. Since the renal nerves tonically stimulate renin secretion (13), suppression of RSNA and attendant inhibition of renin secretion may contribute importantly to the lowering of arterial pressure during BA.
The hypothesis that inhibition of renin secretion plays an important role in permitting substantial reductions in MAP during baroreflex activation is supported by the arterial pressure response to equivalent degrees of BA in dogs before and during chronic infusion of ANG II (36), as illustrated in FIGURE 4. Under control conditions, MAP was reduced ∼21 mmHg after 7 days of BA, with no increase in PRA. After recovery from BA, ANG II was chronically infused at a rate calculated to achieve approximately a three- to fivefold increase in the plasma level of the hormone or a level that might normally be expected during a reduction in renal perfusion pressure to ∼25 mmHg. Chronic infusion of ANG II at this rate also increased plasma aldosterone concentration approximately threefold and produced an ∼35 mmHg increase in MAP. Subsequently, acute reductions in arterial pressure during the first hour of BA were pronounced and roughly comparable with those achieved under control conditions. However, in marked contrast, this pronounced acute fall in arterial pressure was not sustained chronically (FIGURE 4). This striking dichotomy between the acute and chronic effects of BA on arterial pressure indicates that global inhibition of sympathetic activity during BA has a powerful effect to counteract the peripheral vasoconstrictor effects of ANG II (7, 25) but only weak actions to offset the antinatriuretic effects of the renin-angiotensin-aldosterone system that lead to increased arterial pressure. This interpretation is consistent with reports that SAD has little or no anti-hypertensive effects in dogs and rabbits with ANG II hypertension (3, 11), despite the sustained suppression of RSNA when the baroreflex is intact. Similarly, in dogs with hypertension induced by chronic aldosterone infusion, the long-term anti-hypertensive effects of BA are diminished in the presence of the high circulating levels of this potent sodium-retaining steroid (46). Thus, although baroreflex-mediated suppression of RSNA would be expected to inhibit the direct stimulatory effects of the renal nerves on sodium reabsorption, these studies suggest that the natriuretic response to BA, and the concomitant blood pressure lowering, is limited when there is activation of the renin-angiotensin-aldosterone system and an inability of this system to respond to neural inhibition.
FIGURE 4.
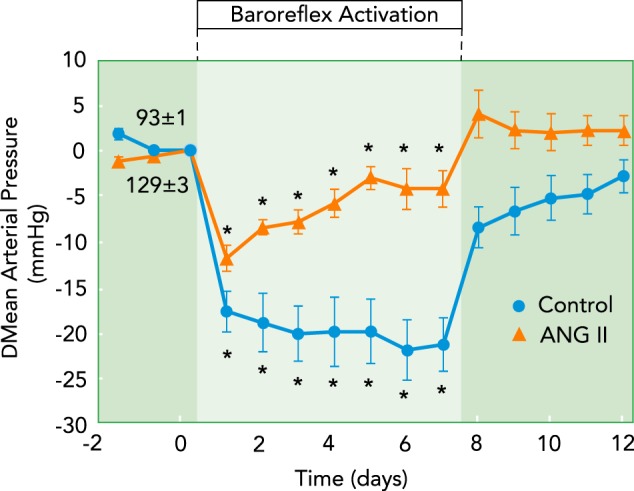
Chronic changes in mean arterial pressure during prolonged BA before and after induction of hypertension by chronic infusion of ANG II
Acute reductions in arterial pressure (not shown) in response to BA were substantial and equivalent under both conditions. In contrast, whereas robust pressure reductions were sustained during BA under control conditions, they were markedly reduced in ANG II hypertension.
A corollary from these studies is that acute responses to inhibition of the sympathetic nervous system do not necessarily reveal the importance of neural mechanisms in the chronic regulation of arterial pressure and, more specifically, their role in mediating hypertension. Acute responses to neural blockade are dominated by changes in peripheral resistance and cardiac output, whereas chronic responses to inhibition of neural activity reflect the impact of the nervous system on the more sluggish renal mechanisms for control of body fluid volume. Most importantly, as discussed above, these studies suggest that BA-mediated inhibition of RSNA has minimal effects to increase renal excretory function and chronically lower arterial pressure in the presence of high circulating levels of the potent sodium retaining hormones ANG II and aldosterone.
Baroreflex Activation Abolishes Obesity-Induced Hypertension
In contrast to the ANG II model of hypertension, obesity-related hypertension has an important neurogenic component that includes activation of the renin-angiotensin system (17, 24, 41, 44, 52). Of particular significance, obesity hypertension, unlike ANG II hypertension, is associated with increased, not decreased, RSNA, and in obesity hypertension circulating levels of ANG II are not fixed by infusion but are responsive to neurally mediated changes in renin secretion. In addition, the canine model of obesity hypertension is highly clinically relevant, since it mimics many of the hemodynamic, neurohormonal, renal, and metabolic changes present in human obesity, and obesity is commonly present in resistant hypertension, a clinical target of BA. Thus it was reasonable to consider whether BA might attenuate or even abolish the hypertension associated with weight gain in dogs because suppression of RSNA would be expected to diminish the direct effects of the renal nerves on sodium reabsorption as well as the indirect effects mediated by stimulation of renin secretion (44).
The development of obesity hypertension and the effects of BA and surgical renal denervation on global and renal-specific inhibition of sympathetic activity, respectively, in the established phase of obesity hypertension are illustrated in FIGURE 5. After control measurements, the regular diet was supplemented with sufficient cooked beef fat to increase body weight to 150% of control over a 4-wk period. Subsequently, fat supplementation was reduced substantially to maintain a constant body weight throughout the remainer of the study. During the 4 wk of fat feeding, there was a progressive increase in arterial pressure and substantial tachycardia. More to the point, in marked contrast to the weak anti-hypertensive response in dogs with ANG II levels fixed modestly above control levels by infusion (FIGURE 4), BA abolished the hypertension and greatly attenuated the tachycardia associated with weight gain. This anti-hypertensive response to BA was associated with suppression of both plasma norepinephrine concentration and PRA, reflecting the actions of BA to suppress the increased sympathetic overactivity and the concomitant neural stimulation of renin secretion.
FIGURE 5.

Development of hypertension with weight gain and the effects of global and renal specific sympathetic inhibition by BA and surgical renal denervation
Development of hypertension with weight gain and the effects of global and renal-specific sympathetic inhibition by BA and surgical renal denervation (RDX), respectively, in the established phase of obesity. Abolition of the hypertension by RDX alone emphasizes the critical importance of increased renal sympathetic nerve activity in the pathogenesis of obesity hypertension and the critical role of renal-specific suppression of sympathetic activity in mediating the anti-hypertensive effects of BA.
After terminating BA and allowing sufficient time for the return of hemodynamics and neurohormones to levels measured before activation of the baroreflex, the same dogs were subjected to bilateral renal denervation to determine the importance of renal-specific sympathetic activation in mediating the hypertension (FIGURE 5). In light of global suppression of sympathetic activity by BA, an impressive response was that abrogating sympathetic drive to the kidneys alone was sufficient to abolish the hypertension. Furthermore, as with BA, the anti-hypertensive response to renal denervation occurred concurrently with suppression of PRA, emphasizing once again the important link between the renal nerves and the renin-angiotensin system in long-term control of arterial pressure and the importance of neurally mediated renin secretion in mediating the hypertension. However, a notable difference between these two approaches for lowering arterial pressure was that there were no changes in plasma norepinephrine concentration or heart rate following renal denervation. This fails to support the possibility that putative sensory afferent signals from the kidneys to the brain contribute to the sustained sympathoexcitation that mediates the hypertension associated with weight gain. Thus this study emphasizes the importance of increased efferent RSNA in the pathogenesis of obesity hypertension and the critical role of renal-specific suppression of efferent sympathetic nerve activity in mediating the anti-hypertensive effects of BA.
Baroreflex Activation Potentiates Lowering of Arterial Pressure by Anti-Hypertensive Medication
Some anti-hypertensive drugs commonly used in the treatment of resistant hypertension, including diuretics, adrenergic blockers, and calcium channel blockers, chronically activate the sympathetic nervous system while lowering arterial pressure (12, 20, 34), a response most likely mediated by chronic unloading of the arterial baroreceptors. This compensatory sympathetic activation would be expected to attenuate the anti-hypertensive effects of these agents and contribute to the failure of pharmacological treatment to lower arterial pressure to acceptable target levels in subjects with resistant hypertension. Indeed, reductions in arterial pressure in dogs chronically administered adrenergic receptor antagonists and calcium channel blockers are associated with substantial increases in plasma norepinephrine concentration and PRA, reflecting increased central sympathetic outflow (28, 38). Most significantly, along with abolishing this neurohormonal activation, concurrent BA lowers arterial pressure further. Thus, by counteracting increased central sympathetic outflow and neurally mediated renin secretion, BA has impressive sustained effects to lower arterial pressure even in the presence of some anti-hypertensive drugs.
Unexpected Findings
Taken together, the studies presented above strongly support the hypothesis that the renal nerves provide an important link between baroreflex-mediated suppression of sympathetic outflow and increased renal excretory function that leads to chronic blood pressure lowering. To directly test this hypothesis, dogs were subjected to BA before and after bilateral renal denervation (37). Much to our surprise, we found that the presence of the renal nerves is not an obligate requirement for chronic lowering of arterial pressure during BA. Similarly, impressive reductions in arterial pressure in response to BA have been reported in subjects with resistant hypertension and prior renal nerve ablation that was unsuccessful in controlling their hypertension (26). The explanation for these finding is not clear but may relate to excessive fluid retention in the absence of the natriuretic effects of the renal nerves (27, 37, 42). This leads to greater activation of redundant natriuretic mechanisms, thus enhancing their contribution to blood pressure lowering. The natriuretic mechanisms identified include increased atrial natriuretic peptide secretion, increased renal interstitial fluid pressure, and reduced plasma protein concentration.
Another unexpected finding was that BA lowered arterial pressure substantially even during complete blockade of adrenergic receptors with well established roles in mediating the long-term effects of the sympathetic nervous system on arterial pressure (38). In dogs chronically administered α1- and β1,2-adrenergic receptor antagonists, MAP decreased ∼20 mmHg in association with an approximately threefold increase in plasma norepinephrine concentration, a response most likely attributed to unloading of arterial baroreceptors. Subsequently, during simultaneous BA, MAP decreased an additional 10 mmHg in parallel with suppression of plasma norepinephrine concentration to control levels. However, whether the added sustained fall in arterial pressure with BA is due to decreased release of norepinephrine (and, for example, diminished activation of postjunctional vasoconstrictor α2-adrenergic receptors) (38) and/or cotransmitters (such as neuropeptide Y and ATP) from adrenergic nerve terminals has not been established. Another possibility not tested is that this added lowering of arterial pressure during BA in the presence of adrenergic blockade is hormonally mediated, analogous to the inhibition of ADH secretion during acute activation of baroreceptors or the more chronic stimulation of atrial natriuretic peptide secretion as discussed above.
Summary, Conclusions, and Perspectives
Novel approaches in chronically instrumented animals have provided strong evidence in support of the contention that arterial baroreflexes may contribute significantly to long-term control of sympathetic activity and arterial pressure. These studies indicate incomplete resetting during natural activation of the baroreflex in hypertension, although the quantitative importance of the resultant suppression of sympathetic activity in attenuating the severity of hypertension is unclear and likely dependent on the etiology of the hypertension. Since a prerequisite for chronic lowering of arterial pressure is increased renal excretory function, studies during the natural activation of the baroreflex suggest that the anti-hypertensive effects of baroreflex activation are linked to chronic inhibition of RSNA. The technology for chronic electrical stimulation of the carotid baroreflex has provided a novel approach for further evaluating this and other concepts related to the role of the baroreflex in long-term control of arterial pressure.
Chronic electrical stimulation of the carotid sinus exploits the sympathoinhibitory potential of the baroreflex by providing controlled and unvarying increases in baroreceptor input into the brain. Studies to date indicate that this leads to sustained suppression of central sympathetic outflow that is not diminished by central adaptations. Consequently, BA has provided novel insight into the effector mechanisms that are causal in mediating chronic blood pressure lowering during activation of the baroreflex afferents. Because BA lowers arterial pressure substantially in kidneys devoid of renal nerves (26, 37), one fundamental concept not confirmed by studies using this technology is the dominance of baroreflex inhibition of RSNA in mediating increases in renal excretory function and attendant long-term reductions in blood pressure. Based on limited data, we hypothesized that other hormonal and hemodynamic mechanisms contribute to increased renal excretory function during BA and that the importance of these redundant mechanisms to this response intensifies at the expense of excessive fluid retention when the primary mechanism for fluid excretion (suppressed RSNA) is rendered nonfunctional by renal denervation. However, along with studies designed to identify and quantify the importance of these redundant mechanisms, corroboration of this hypothesis will require temporal measurements of RSNA (renal norepinephrine spillover) to confirm that suppression of sympathetic activity during BA does, in fact, include the sympathetic outflow to the kidneys. To date, this key measurement has not been made.
Electrical stimulation of the carotid sinus bypasses the mechanosensory complex at the level of the baroreceptors and permits controlled electrical activation of baroreceptor afferents. Therefore, by having the capacity to chronically control stimulation patterns and parameters, BA technology provides the unique opportunity to study the central processing and function of subtypes of baroreceptors. However, to date, this aspect of the technology has been underutilized. As discussed above, measurements of whole body norepinephrine spillover and arterial pressure during 3 wk of continuous, invariant baroreceptor input into the central nervous system indicate that central baroreflex resetting does not play an important role in offsetting the sympathoinhibitory and blood pressure-lowering effects of baroreceptor afferent signals into the brain (43). Accordingly, a logical follow up to these observations would be to determine whether a continuous pulsatile pattern closely mimicking the natural input into the central nervous system during the cardiac cycle would lead to similar or even enhanced long-term reflex responses to BA. In a related issue, by controlling stimulation parameters (voltage, frequency), BA technology may be used to explore whether there are chronic functional differences attributable to the central processing of input from different baroreceptor subtypes. Based on functional responses to changes in pressure, acute studies in the dog have identified two types of carotid baroreceptors with properties that differ substantially (60, 61). Type I baroreceptors (with mostly A-fiber myelinated axons) undergo acute resetting, whereas type II baroreceptors (C-fiber unmyelinated and small A-fiber myelinated axons) have higher threshold pressures and do not reset. This has led to the hypothesis that type I baroreceptors are important in the stabilization of arterial pressure, whereas those with type II receptors are involved in signaling the absolute level of arterial pressure. Additionally, it has been reported that the tonic input from baroreceptors with unmyelinated baroreceptor afferents fibers is increased in hypertension (32). Because published studies during BA have used stimulation parameters that most likely activate both myelinated and unmyelinated baroreceptor afferents, functional differences in the central processing of these two types of baroreceptors have not been established. To this end, by manipulating stimulation parameters during BA to achieve relatively selective activation of myelinated and nonmyelinated baroreceptor afferents (18), it may be possible to determine whether more sustained functional responses are achieved with central integration of type II vs. type I baroreceptor inputs, supporting the hypothesis that activation of the former may have sustained effects to attenuate the severity of hypertension.
The mechanistic insight from the recent studies discussed in this review lends credence to the contention that arterial baroreflexes play a role in long-term control of arterial pressure. At the same time, questions remain, and resolution of these uncertainties is needed before the baroreflex can be placed in the hierarchy of neurohormonal mechanisms that contribute importantly to long-term blood pressure regulation. The necessity for additional information notwithstanding, the current knowledge from these studies would be expected to be helpful in predicting which patients will likely benefit the most in current clinical trials designed to evaluate the efficacy of BA therapy in the treatment of resistant hypertension and heart failure, conditions in which activation of the sympathetic nervous system plays an important role in the pathogenesis of these disorders.
Footnotes
The author's studies cited in this report were funded by National Heart, Lung, and Blood Institute Grant HL-51971.
Thomas E. Lohmeier has received consulting fees from CVRx, Inc., and has served on the Scientific Advisory Board, CVRx., Inc.
Author contributions: T.E.L. and R.I. conception and design of research; T.E.L. and R.I. performed experiments; T.E.L. and R.I. analyzed data; T.E.L. and R.I. interpreted results of experiments; T.E.L. and R.I. prepared figures; T.E.L. and R.I. drafted manuscript; T.E.L. and R.I. edited and revised manuscript; T.E.L. and R.I. approved final version of manuscript.
References
- 1.Alnima T, de Leeuw PW, Tan ES, Kroon AA. Renal responses to long-term carotid baroreflex activation therapy in patients with drug-resistant hypertension. Hypertension 61: 1334–1339, 2013. [DOI] [PubMed] [Google Scholar]
- 2.Bakris GL, Nadim MK, Haller H, Lovett EG, Schafer JE, Bisognano JD. Baroreflex activation therapy provides durable benefit in patients with resistant hypertension: results of long-term follow-up in the Rheos Pivotal Trial. J Am Soc Hypertens 6: 152–158, 2012. [DOI] [PubMed] [Google Scholar]
- 3.Barrett CJ, Guild SJ, Ramchandra R, Malpas SC. Baroreceptor denervation prevents sympathoinhibition during angiotensin II-induced hypertension. Hypertension 46: 1–5, 2005. [DOI] [PubMed] [Google Scholar]
- 4.Barrett CJ, Ramchandra R, Guild SJ, Lala A, Budgett DM, Malpas SC. What sets the long-term level of renal sympathetic nerve activity: a role for angiotensin II and baroreflexes? Circ Res 92: 1330–1336, 2003. [DOI] [PubMed] [Google Scholar]
- 5.Bisognano JD, Bakris G, Nadim MK, Sanchez L, Kroon AA, Schafer J, de Leeuw PW, Sica DA. Baroreflex activation therapy lowers blood pressure in patients with resistant hypertension: results from the double-blind, randomized, placebo-controlled Rheos Pivotal trial. J Am Coll Cardiol 58: 765–773, 2011. [DOI] [PubMed] [Google Scholar]
- 6.Bisognano JD, Kaufmann CL, Bach DS. Improved cardiac structure and function with chronic treatment using an implantable device in resistant hypertension; results from European and United States trials of the Rheos system. J Am Coll Cardiol 17: 1787–1791, 2011. [DOI] [PubMed] [Google Scholar]
- 7.Burgoyne S, Georgakopoulos D, Belenkie I, Tyberg JV. Systemic vascular effects of acute electrical baroreflex stimulation. Am J Physiol Heart Circ Physiol 307: H236–H241, 2014. [DOI] [PubMed] [Google Scholar]
- 8.Carroll RG, Lohmeier TE, Brown AJ. Chronic angiotensin II infusion decreases renal norepinephrine overflow in conscious dogs. Hypertension 6: 675–681, 1984. [DOI] [PubMed] [Google Scholar]
- 9.Cavalleri MT, Burgi K, Cruz JC, Jordao MT, Ceroni A, Michelini LC. Afferent signaling drives oxytocinergic preautonomic neurons and mediates training-induced plasticity. Am J Physiol Regul Integr Comp Physiol 301: R958–R966, 2011. [DOI] [PubMed] [Google Scholar]
- 10.Cowley AW., Jr Long-term control of arterial pressure. Physiol Rev 72: 231–300, 1992. [DOI] [PubMed] [Google Scholar]
- 11.Cowley AW Jr, DeClue JW. Quantification of baroreceptor influence on arterial pressure changes seen in primary angiotensin-induced hypertension in dogs. Circ Res 39: 779–787, 1976. [DOI] [PubMed] [Google Scholar]
- 12.De Champlain J, Karas M, Nguyen P, Cartier P, Wistaff R, Toal CB, Nadeau R, Larochelle P. Different effects of nifedipine and amlodipine on circulating catecholamine levels in essential hypertensive patients. J Hypertens 16: 1357–1369, 1998. [PubMed] [Google Scholar]
- 13.DiBona GF, Kopp UC. Neural control of renal function. Physiol Rev 77: 75–197, 1997. [DOI] [PubMed] [Google Scholar]
- 14.Ehmke H, Persson P, Fisher S, Hackenthal E, Kirchheim H. Resettimg of pressure-dependent renin release by intrarenal alpha-1-adrenoceptors in conscious dogs. Pflügers Arch 413: 261–266, 1989. [DOI] [PubMed] [Google Scholar]
- 15.Esler M, Jennings G, Korner P, Willett I, Dudley F, Hasking G, Anderson W, Lambert G. Assessment of human sympathetic nerve activity from measurements of norepinephrine turnover. Hypertension 11: 3–20, 1988. [DOI] [PubMed] [Google Scholar]
- 16.Esler MD, Krum H, Schlaich M, Schmieder RE, Bohm M, Sobotka PA. Renal sympathetic denervation for treatment of drug-resistant hypertension. One-year results from the Simplicity HTN-2 randomize, controlled trial. Circulation 126: 2976–2982, 2012. [DOI] [PubMed] [Google Scholar]
- 17.Esler M, Straznicky N, Eikelis N, Masuo K, Lambert G, Lambert E. Mechanisms of sympathetic activation in obesity-related hypertension. Hypertension 48: 787–796, 2006. [DOI] [PubMed] [Google Scholar]
- 18.Fan W, Andresen MC. Differential frequency-dependent reflex regulation of myelinated and nonmyelinated rat aortic baroreceptors. Am J Physiol Heart Circ Physiol 275: H632–H640, 1998. [DOI] [PubMed] [Google Scholar]
- 19.Finke R, Gross R, Hackenthal E, Huber J, Kirchheim HR. Threshold pressure for pressure-dependent renin release in the autoregulating kidney of conscious dogs. Pflügers Arch 399: 102–110, 1983. [DOI] [PubMed] [Google Scholar]
- 20.Fu Q, Witkowski S, Arbab-Zadeh A, Prasad A, Okazaki K, Levine BD. Persistent sympathetic activation during chronic antihypertensive therapy. A potential mechanism for long-term morbidity. Hypertension 45: 513–521, 2005. [DOI] [PubMed] [Google Scholar]
- 21.Georgakopoulos D, Little WC, Abraham WT, Weaver FA, Zile MR. Chronic baroreflex activation: a potential therapeutic approach to heart failure with preserved ejection fraction. J Card Fail 17: 167–178, 2011. [DOI] [PubMed] [Google Scholar]
- 22.Georgakopoulos D, Wagner D, Cates AW, Irwin E, Lovett EG. Effects of electrical stimulation of the carotid sinus baroreflex using the Rheos device on ventricular-vascular coupling and myocardial efficiency assessed by pressure-volume relations in non-vagotomized anesthesized dogs. Conf Proc IEEE Med Biol Soc 2009: 2025–2029, 2009. [DOI] [PubMed] [Google Scholar]
- 23.Grassi G, Seravalle G, Dell'Oro R, Turri C, Bolla GB, Mancia G. Adrenergic and reflex abnormalities in obesity-related hypertension. Hypertension 36: 538–542, 2000. [DOI] [PubMed] [Google Scholar]
- 24.Hall JE, da Silva AA, Brandon E, Stec DE, Ying Z, Jones DW. Pathophysiology of obesity-induced hypertension and target organ damage. In: Comprehensive Hypertension, edited by Lip GYH, Hall JE. Philadelphia, PA: Elsevier, 2007, p. 447–468. [Google Scholar]
- 25.Heusser K, Engeli S, Diedrich A, Menne J, Eckert S, Peters T, Sweep FCGJ, Haller H, Pichlmaier AM, Luft FC, Jordan J. Carotid baroreceptor stimulatio, sympathetic activity, baroreflex function, and blood pressure in hypertensive patients. Hypertension 55: 619–626, 2010. [DOI] [PubMed] [Google Scholar]
- 26.Hoppe UC, Brandt MC, Wachter R, Beige J, Rump LC, Kroon AA, Cates AW, Lovett EG, Haller H. Minimally invasive system for baroreflex activation therapy chronically lowers blood pressure with pacemaker-like safety profile: results from the Barostim neo trial. J Am Soc Hypertens 6: 270–276, 2012. [DOI] [PubMed] [Google Scholar]
- 27.Iliescu R, Lohmeier TE. Lowering of blood pressure during chronic suppression of central sympathetic outflow: insight from computer simulations. Clin Exptl Pharmacol Physiol 37: 24–33, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iliescu R, Irwin ED, Georgakopoulos D, Lohmeier TE. Renal responses to chronic suppression of central sympathetic outflow. Hypertension 60: 749–756, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iliescu R, Tudorancea I, Irwin ED, Lohmeier TE. Chronic baroreflex activation restores spontaneous baroreflex control and variability of heart rate in obesity-induced hypertension. Am J Physiol Heart Circ Physiol 305: H1080–H1088, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iliescu R, Tudorancea I, Lohmeier TE. Baroreflex activation: from mechanisms to therapy for cardiovascular disease. Curr Hypertens Rep 16: 453–461, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ito S, Sved AF. Influence of GABA in the nucleus of the solitary tract on blood pressure in baroreceptor-denervated rats. Am J Physiol Regul Integr Comp Physiol 273: R1657–R1662, 1997. [DOI] [PubMed] [Google Scholar]
- 32.Jones JV, Thoren P. Characteristics of aortic baroreceptors with non-myelinated afferents arising from the aortic arch of rabbits with chronic renovascular hypertension. Acta Physiol Scand 101: 286–293, 1977. [DOI] [PubMed] [Google Scholar]
- 33.Just A, Wittmann U, Nafz B, Wagner CD, Ehmke H, Kirchheim HR, Persson PB. The blood pressure buffering capacity of nitric oxide by comparison to the baroreceptor reflex. Am J Physiol Heart Circ Physiol 267: H521–H527, 1994. [DOI] [PubMed] [Google Scholar]
- 34.Ligtenberg G, Blankestijin PJ, Oey PL, Klein IH, Dijkhorst-Oei LT, Boomsma F, Wieneke GH, van Huffelen AC, Koomans HA. Reduction in sympathetic hyperactivity by enalapril in patients with chronic renal failure. N Engl J Med 340: 1321–1328, 1999. [DOI] [PubMed] [Google Scholar]
- 35.Lohmeier TE, Drummond HA. The baroreflex in the pathogenesis of hypertension. In: Comprehensive Hypertension, edited by Lip GYH, Hall JE. Philadelphia, PA: Elsevier, 2007, p. 265–279. [Google Scholar]
- 36.Lohmeier TE, Dwyer TM, Hildebrandt DA, Irwin ED, Rossing MA, Sedar DJ, Kieval RS. Influence of prolonged baroreflex activation on arterial pressure in angiotensin hypertension. Hypertension 46: 1194–1200, 2005. [DOI] [PubMed] [Google Scholar]
- 37.Lohmeier TE, Hildebrandt DA, Dwyer TM, Barrett AM, Irwin ED, Rossing MA, Kieval RS. Renal denervation does not abolish sustained baroreflex-mediated reductions in arterial pressure. Hypertension 49: 373–379, 2007. [DOI] [PubMed] [Google Scholar]
- 38.Lohmeier TE, Hildebrandt DA, Dwyer TM, Iliescu R, Irwin ED, Cates AW, Rossing MA. Prolonged activation of the baroreflex decreases arterial pressure even during chronic adrenergic blockade. Hypertension 53: 833–838, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lohmeier TE, Hildebrandt DA, Warren S, may P, Cunningham JT. Recent insights into the interactions between the baroreflex and the kidneys in hypertension. Am J Physiol Regul Integr Comp Physiol 288: R828–R836, 2005. [DOI] [PubMed] [Google Scholar]
- 40.Lohmeier TE, Iliescu R. Chronic lowering of blood pressure by carotid baroreflex activation. Mechanisms and potential for hypertension therapy. Hypertension 57: 880–886, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lohmeier TE, Iliescu R. The sympathetic nervous system in obesity hypertension. Curr Hypertens Rep 15: 409–416, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lohmeier TE, Iliescu R. Lowering of blood pressure by chronic suppression of central sympathetic outflow. J Appl Physiol 113: 1652–1658, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lohmeier TE, Iliescu R, Dwyer TM, Irwin ED, Cates AW, Rossing MA. Sustained suppression of sympathetic activity and arterial pressure during chronic activation of the carotid baroreflex. Am J Physiol Heart Circ Physiol 299: H402–H409, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lohmeier TE, Iliescu R, Liu B, Henegar JR, Maric-Bilkan C, Irwin ED. Systemic and renal-specific sympathoinhibition in obesity hypertension. Hypertension 59: 331–338, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lohmeier TE, Irwin ED, Rossing MA, Sedar DJ, Kieval RS. Prolonged activation of the baroreflex produces sustained hypotension. Hypertension 43: 306–311, 2004. [DOI] [PubMed] [Google Scholar]
- 46.Lohmeier TE, Liu B, Georgakopoulos D, Cates A, Irwin E. Cardiovascular responses to chronic baroreflex activation in aldosterone hypertension. Hypertension 62: A354, 2013. [Google Scholar]
- 47.Lohmeier TE, Lohmeier JR, Haque A, Hildebrandt DA. Baroreflexes prevent neurally-induced sodium retention in angiotensin hypertension. Am J Physiol Regul Integr Comp Physiol 279: R1437–R1448, 2000. [DOI] [PubMed] [Google Scholar]
- 48.Lohmeier TE, Lohmeier JR, Reckelhoff JF, Hildebrandt DA. Sustained influence of the renal nerves to attenuate sodium retention in angiotensin hypertension. Am J Physiol Regul Integr Comp Physiol 281: R434–R443, 2001. [DOI] [PubMed] [Google Scholar]
- 49.Lohmeier TE, Lohmeier TE, Warren S, May PJ, Cunningham JT. Sustained activation of the central baroreflex pathway in angiotensin hypertension. Hypertension 39: 550–556, 2002. [DOI] [PubMed] [Google Scholar]
- 50.Lohmeier TE, Reinhart GA, Mizelle HL, Han M, Dean MM. Renal denervation supersensitivity revisited. Am J Physiol Regul Integr Comp Physiol 275: R1239–R1246, 1998. [DOI] [PubMed] [Google Scholar]
- 51.Lohmeier TE, Warren S, Cunningham JT. Sustained activation of the central baroreceptor pathway in obesity hypertension. Hypertension 42: 96–102, 2003. [DOI] [PubMed] [Google Scholar]
- 52.Malpas SC. Sympathetic nervous system overactivity and its role in the development of cardiovascular diseases. Physiol Rev 90: 513–557, 2010. [DOI] [PubMed] [Google Scholar]
- 53.Mancia G, Grassi G. The autonomic nervous system and hypertension. Cir Res 114: 1804–1814, 1014. [DOI] [PubMed] [Google Scholar]
- 54.Mifflin SW. What does the brain know about blood pressure? News Physiol Sci 16: 266–271, 2001. [DOI] [PubMed] [Google Scholar]
- 55.Persson PB, Ehmke H, Kirchheim H, Seller H. Effect of sino-aortic denervation in comparison to cardiopulmonary deafferentation on long-term blood pressure in conscious dogs. Pflügers Arch 411: 160–166, 1988. [DOI] [PubMed] [Google Scholar]
- 56.Sabbah HN, Gupta RC, Imai M, Irwin ED, Rastogi S, Rossing MA, Kieval RS. Chronic electrical stimulation of the carotid sinus baroreflex improves left ventricular function and promotes reversal of ventricular remodeling in dogs with advanced heart failure. Cir Heart Fail 4: 65–70, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schreihofer AM, Ito S, Sved AF. Nucleus tractus solitarius and control of blood pressure in chronic sinoaortic denervated rats. Am J Physiol Regul Integr Comp Physiol 263: R258–R266, 1992. [DOI] [PubMed] [Google Scholar]
- 58.Schreihofer AM, Ito S, Sved AF. Brain stem control of arterial pressure in chronic arterial baroreceptor-denervated rats. Am J Physiol Regul Integr Comp Physiol 289: R1746–R1755, 2005. [DOI] [PubMed] [Google Scholar]
- 59.Scheffers IJM, Kroon AA, Schmidli J, Jordan J, Tordoir JJM, Mohaupt MG, Luft FC, Haller H, Menne J, Engeli S, Ceral J, Eckert S, Erglis A, Narkiewicz K, Philipp T, de Leeuw PW. Novel baroreflex activation therapy in resistant hypertension. J Am Coll Cardiol 56: 1254–1258, 2010. [DOI] [PubMed] [Google Scholar]
- 60.Seagard JL, Gallenberg LA, Hopp FA, Dean C. Acute resetting in two functionally different types of carotid baroreceptors. Circ Res 70: 559–565, 1972. [DOI] [PubMed] [Google Scholar]
- 61.Seagard JL, Hopp FA, Drummond HA, Van Wynsberghe DM. Selective contribution of two types of carotid sinus baroreceptors to the control of blood pressure. Circ Res 72: 1011–1022, 1993. [DOI] [PubMed] [Google Scholar]
- 62.Stocker SD, Meador R, Adams JM. Neurons of the rostral ventrolateral medulla contribute to obesity-induced hypertension in rats. Hypertension 49: 640–646, 2007. [DOI] [PubMed] [Google Scholar]
- 63.Sved AF, Schreihofer AM, Kost CK Jr. Blood pressure regulation in baroreceptor-denervated rats. Clin Exp Pharmacol Physiol 24: 77–82, 1997. [DOI] [PubMed] [Google Scholar]
- 64.Symplicity HTN2 Investigators. Renal sympathetic denervation in patients with treatment-resistant hypertension (The Symplicity HTN-2 Trial). Lancet 376: 1903–1909, 2011. [DOI] [PubMed] [Google Scholar]
- 65.Thrasher TN. Baroreceptors and long-term control of blood pressure. Exp Physiol 89: 331–341, 2004. [DOI] [PubMed] [Google Scholar]
- 66.Thrasher TN. Effects of chronic baroreceptor unloading on blood pressure in the dog. Am J Physiol Regul Integr Comp Physiol 288: R863–R871, 2005. [DOI] [PubMed] [Google Scholar]
- 67.Wustmann K, Kucera JP, Scheffers I, Mohaupt M, Kroon AA, de Leeuw PW, Schmidli J, Allemann Y, Delacrétaz E. Effects of chronic baroreflex stimulation on the autonomic cardiovascular regulation in patients with drug-resistant arterial hypertension. Hypertension 54: 530–536, 2009. [DOI] [PubMed] [Google Scholar]
- 68.Zucker IH, Hackley JF, Cornish KG, Hiser BA, Anderson NR, Kieval R, Irwin ED, Serdar DJ, Peuler JD, Rossing MA. Chronic baroreceptor activation enhances survival in dogs with pacing-induced heart failure. Hypertension 50: 904–910, 2007. [DOI] [PubMed] [Google Scholar]