

Abstract
The role(s) of the epidermal growth factor receptor (EGFR) in hepatocytes is unknown. We generated a murine hepatocyte specific-EGFR knockout (KO) model to evaluate how loss of hepatocellular EGFR expression affects processes such as EGF clearance, circulating EGF concentrations, and liver regeneration following 70% resection or CCl4-induced centrilobular injury. We were able to disrupt EGFR expression effectively in hepatocytes and showed that the ability of EGF and heregulin (HRG) to phosphorylate EGFR and ERBB3, respectively, required EGFR. Loss of hepatocellular EGFR impaired clearance of exogenous EGF from the portal circulation but paradoxically resulted in reduced circulating levels of endogenous EGF. This was associated with decreased submandibular salivary gland production of EGF. EGFR disruption did not result in increased expression of other ERBB proteins or Met, except in neonatal mice. Liver regeneration following 70% hepatectomy revealed a mild phenotype, with no change in cyclin D1 expression and slight differences in cyclin A expression compared with controls. Peak 5-bromo-2′-deoxyuridine labeling was shifted from 36 to 48 h. Centrilobular damage and regenerative response induced by carbon tetrachloride (CCl4) were identical in the KO and wild-type mice. In contrast, loss of Met increased CCl4-induced necrosis and delayed regeneration. Although loss of hepatocellular EGFR alone did not have an effect in this model, EGFR-Met double KOs displayed enhanced necrosis and delayed liver regeneration compared with Met KOs alone. This suggests that EGFR and Met may partially compensate for the loss of the other, although other compensatory mechanisms can be envisioned.
Keywords: liver, submandibular gland, EGFR, Met, hepatectomy, carbon tetrachloride
multiple hormone and growth factor receptors and their ligands have been implicated in the regulation of hepatocyte proliferation, survival, and motility. Two important and well-studied receptor tyrosine kinases (RTK) in the liver are the epidermal growth factor receptor (EGFR) and Met, which are the receptors for EGF-like ligands and hepatocyte growth factor (HGF), respectively. Both receptors have extracellular ligand binding, transmembrane, and intracellular tyrosine kinase domains. Ligand binding to the extracellular domain alters the configuration of the protein, activating the kinase domain, which initiates intracellular signaling pathways and eventually gene transcription. Gene targeting experiments have shown that Met and its ligand, HGF, but not EGFR or EGF are required for normal liver development during embryogenesis. Global knockout (KO) mice lacking Met or HGF die during midgestation with multiple abnormalities, including liver dysgenesis (2, 47). Global KO mice lacking EGFR also die during embryogenesis or shortly after birth, but the specific cause of death is variable and mouse strain dependent (53). The embryonic or neonatal lethality associated with the loss of either receptor precludes the study of Met or EGFR gene disruption in global KO adult mice. However, hepatocyte-specific (HS) Met (3, 18) and EGFR (27) KO mice do survive as adults. Studies with these mice have suggested that both receptors are required for efficient liver regeneration after partial hepatectomy (PH).
EGFR is a member of the ERBB family of RTKs, which also includes ERBB2, ERBB3, and ERBB4. These RTKs form homo- or heterodimers with each other, which are the active signaling units. EGFR, ERBB2, and ERBB3, but not ERBB4, are expressed in mouse liver, but the expression of ERBB2 plummets after weaning (7). The expression of ERBB3, the receptor for heregulin (HRG), persists in adult mice. This receptor was previously thought to lack intrinsic kinase activity; however, it is now known to have weak kinase activity, turned on by the interaction with other ERBB molecules (51). In contrast to other ERBB kinases, which can be activated by ligand binding alone within a homo- or heterodimeric kinase signaling unit, activation of the ERBB3 tyrosine kinase requires a transient physical interaction with its dimeric ERBB binding partner. ERBB3 monomers, once activated, can dissociate from the initial heterodimeric pairings and subsequently form HRG-activated ERBB3 homodimers.
Radioligand binding studies indicate that each hepatocyte of the adult male rodent liver expresses ∼600,000 EGFR (1) but only 20,000 ERBB3 receptors (6). Relatively little is known about the histological localization of these RTK in the liver or whether ERBB3 can signal with a kinase other than EGFR. An EGFR monomer can form active signaling homodimers with other EGFR molecules or active signaling heterodimers with other ERBB family members, including ERBB3. The signaling outcomes of an EGFR-EGFR homodimer compared with an EGFR-ERBB3 heterodimer are unique, in part because of the multiple PI3-kinase binding sites in the intracellular regulatory domain of ERBB3 (24). Moreover, ERBB3 can be activated not only by EGFR and other ERBB proteins, but also under some circumstances by other RTKs, such as Met (11). Along the same line, some HGF-mediated Met actions in cultured hepatocytes can be blocked by inhibition of the EGFR kinase (41).
We generated a hepatocyte specific-EGFR conditional model (HS-EGFRKO) by deleting exon 3 of the EGFR gene in postnatal hepatocytes. We crossed EGFRf/f mice with albumin-Cre transgenic mice. Deletion of exon 3 introduces a frameshift resulting in two stop codons in exon 4 and early termination of translation in hepatocytes, which uniquely synthesize albumin (23). This transgene includes only albumin regulatory elements. It lacks the α-fetoprotein enhancers present in the α-fetoprotein-Cre transgene, used in an earlier liver regeneration study to disrupt EGFR expression in parenchymal cells (including bile duct cells) (27). We have used this model to localize EGFR and ERBB3 in the liver and to analyze some of the potential roles played by EGFR in hepatocytes. In this article, we evaluated the role of EGFR in ERBB3 signaling, in exogenous EGF ligand clearance, and in EGF production by the submandibular salivary gland. We also evaluated the loss of hepatocyte EGFR on liver regeneration in surgical and chemical models of hepatocellular loss. Because the liver regenerates following surgical resection, which removes parenchymal as well as nonparenchymal cells, we assessed the importance of EGFR in liver regeneration following 70% hepatectomy (25, 26). We found a weaker effect of EGFR gene disruption on liver regeneration following hepatectomy than in a previous study (27).
We also examined how loss of hepatocyte EGFR affected regeneration following chemical injury caused by carbon tetrachloride (CCl4), a centrilobular hepatotoxin. We compared the loss of hepatocellular EGFR with that of a single gene hepatocyte specific-Met conditional model (HS-MetKO) generated by crossing Metf/f mice with albumin-Cre transgenic mice. In addition, we generated a hepatocyte specific-EGFR-Met double KO (HS-EGFR/Met DKO). Compared with wild-type mice or single gene KOs, dual disruption of EGFR and Met yielded a more severe phenotype in CCl4-injected mice. These mice had smaller livers, increased liver necrosis, and delayed regeneration. Surprisingly, in contrast to the hepatectomy results, the loss of EGFR alone in this chemical injury model had no effect, either on necrosis or subsequent liver regeneration. These results indicate that hepatocytes can support liver regeneration following centrilobular necrosis even in the absence of EGFR.
EXPERIMENTAL PROCEDURES
Mice and genetic crosses.
Transgenic mouse models Egfrf/f (23), Metf/f (18), and Alb-Cre+/− (The Jackson Laboratory) mouse strains were generated as previously described. Egfrf/f has two loxP sites flanking exon3 of the EGFR. Deletion of exon 3 caused a frameshift that resulted in two stop codons in exon 4 and early termination of translation (23). To generate hepatocyte-specific EGFR KO, Alb-Cre mice were crossed with Egfrf/f to generate Alb-Cre Egfrf/f or their siblings Egfrf/f. A similar approach was used to establish Alb-Cre/Metf/f. To minimize the influence of sex differences in the response of mice to hepatectomy or CCl4, we mainly studied male mice but did have female subgroups at select times. Since deletion of floxed DNA in hepatocytes is age dependent, we used mice that were at least 8 wk old in regeneration experiments. Mice were fed Purina Mills Lab Diet and water ad libitum under specific pathogen-free conditions in an American Association for the Accreditation of Lab Animal Care-approved facility. Mice were raised under conditions of regulated lighting (lights on 0600–1800), temperature, and humidity. The Vanderbilt Institutional Animal Care and Use Committee approved all experiments.
Genotyping.
Met DNA was extracted from ear punches or tail biopsies for genotyping by incubating at 95°C in 100 μl of 25 mM NaOH/0.2 mM EDTA for 20 min and then neutralizing with 100 μl of 40 mM Tris·HCl, pH 5.0. For the subsequent genotyping reactions, 1 μl of lysed tissue sample was used per reaction. PCR conditions were 35 cycles at 94°C for 45 s, 62°C for 60 s, and 72°C for 120 s. The EGFR allele was amplified by PCR with the following primers: EGFR, 5′-CTTTGGAGAACCTGCAGATC-3′ and EGFR, 5′-CTGCTACTGGCTCAAGTTTC-3′. PCR products were run on a 3% agarose gel to separate a 320-bp product corresponding to wild-type EGFR and a 370-bp product corresponding to the KO EGFR allele. The Met allele was amplified by PCR with the following primers: Met-L, 5′- GCAACTGTCTTTTGATCCCTGC-3′ and Met-R, 5′-TGTCCAGCAAAGTCCCATGATAG-3′. PCR products were run on a 3% agarose gel to separate a 500-bp product corresponding to wild-type and a 580-bp set of product corresponding to the KO Metf/f allele.
Partial hepatectomy.
PH (70%) was performed on male mice under isoflurane anesthesia by ligating and removing the left lateral and median lobes, including the gallbladder as previously described (39). There were four to six mice per time point and no perioperative mortality. Warm isotonic saline (0.5 ml) was deposited into the peritoneum before closure.
Collection of livers and other organ samples.
Mice were anesthetized with 3% isoflurane. They were subjected to a thoracotomy and cardiac puncture to obtain blood. Organs were rapidly dissected from each animal and the wet weights were recorded. Portions of the liver were either frozen in liquid nitrogen for protein and RNA analyses or fixed in phosphate-buffered 4% paraformaldehyde for subsequent paraffin embedding and various histological analyses. The weights of the regenerating liver and the mouse at the time of euthanasia were used to calculate a liver-to-body weight per cent (liver/body wt ratio × 100%).
CCl4 model of liver injury and regeneration.
CCl4 (0.8 μl/g body wt) in olive oil (200 μl) was injected into the intraperitoneal (ip) cavity and the mice were then euthanized at various times after injections. All injections were performed during the middle of the light phase to minimize circadian differences in toxicity. There were five or more mice per time point and no postinjection mortality, even in mice with delayed liver regeneration.
Serum chemistry.
Plasma was obtained at the time of mouse euthanasia and tested for serum alanine aminotransferase (ALT) (TecoDiagnostics, Anaheim, CA).
BrdU labeling and immunohistochemistry.
Mice that underwent PH were injected with 100 μg/g of 5-bromo-2′-deoxyuridine (BrdU; Boehringer Mannheim, Indianapolis, IN) 1 h before euthanasia. Mice from the CCl4 experiments were not injected with BrdU. Livers were fixed in PBS-buffered 4% paraformaldehyde. After overnight fixation, liver samples were transferred to 70% ethanol, dehydrated in a graded series of ethanols and xylenes, and embedded in paraffin, and 5-μm sections were cut by use of a Leica Biocut 2030 microtome. Sections were deparaffinized and rehydrated in a graded series of ethanols. They were immunostained with anti-BrdU (Ab-2, Oncogene Research Products, La Jolla, CA); anti-Ki67 (Clone SP6, Lab Vision, Fremont, CA); anti-cyclin A (sc-596, Santa Cruz Biotechnology, Santa Cruz, CA), anti-EGFR (sc-03, Santa Cruz Biotechnology); anti-PY 1068 EGFR (CS3777, Cell Signaling Technology, Beverly, MA); or anti-PY 1197 ERBB3 (CS4561, Cell Signaling Technology). For BrdU immunostaining, tissue sections were pretreated with 1 N HCl for 8 min at 60°C to denature the DNA. Histological images were photographed on an Olympus Vanex AHBT3 microscope by using a NIKON E5000 connected by a PTEM 257009 camera to an objective adaptor. In the CCl4 experiments, sections were stained with periodic acid-Schiff to highlight the necrotic areas. These images were photographed and uploaded to a computer, and the percent necrotic area was determined by use of the Image J program. For immunohistochemistry, sections were processed by the manufacturer's protocol (Vectastain Elite ABC kit; Vector Laboratories, Burlingame, CA). Endogenous peroxidase activity was quenched by placing the slides in 0.3% H2O2 in methanol for 30 min. After blocking the slides in a 3.0% goat serum solution for 1 h, the tissue sections were incubated with the antibodies for 90 min in a humidified chamber. The sections were then washed, incubated with biotinylated secondary antibody, rinsed in PBS, and incubated with the Vectastain Elite ABC reagent. Diaminobenzidine was used as the peroxidase substrate.
Western blotting.
Pieces of liver (∼100 mg) were weighed and then homogenized on ice using a 2 ml Wheaton glass tissue homogenizer in TGH buffer (20 mM HEPES, 1% Triton X-100, 10% glycerol, 50 mM NaCl). This buffer included protease inhibitors (1 mM PMSF, 1 mM sodium orthovanadate, 10 μg/ml aprotinin, and 1 μg/ml leupeptin) as well as phosphatase inhibitors (10 mM sodium molybdate and 10 mM β-glycerol phosphate). Lysates were immunoblotted as previously described (42). We used the following affinity-purified antibodies from Santa Cruz Biotechnology (Santa Cruz, CA): sc-03 for EGFR; sc-284 for ERBB2; sc-285 for ERBB3; sc-283 for ERBB4; sc-162 for Met; sc-8312 for Akt 1,2,3; sc-33827 for Akt 1,2; sc-13 for Erk 1,2. Antibodies from Cell Signaling Technology (Beverly, MA) included anti-phosphotyrosine [Y1068]-EGFR (CS3777), [Y1197]-ERBB3 (CS4561), and phospho-AKT (CS927). The ppErk 42,44 antibody anti-ACTIVE MAPK pTEpY (V8031) was from Promega (Fitchburg, WI). The lanes in each immunoblot contained equal amounts of protein, as determined by the Bio-Rad DC Protein Assay (Bio-Rad Laboratories, Hercules, CA). After each transfer, we confirmed equal protein loading and transfer by Ponceau S staining of immunoblots, scanning the image for future reference. All sample cohorts were analyzed on a single blot to ensure reliable comparison. Immunoreactive signal was detected by the enhanced chemiluminescent method using either the SuperSignal West Pico Chemiluminescent Substrate or the SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific, Rockford, IL). The decision to use one or the other depended on the quality of the antibody as well as the abundance of the protein. We performed densitometry using an Epson scanner and the Image J program (48).
Statistical analysis.
Data are expressed as means ± SE. Statistical analysis was performed by an unpaired, two-tailed Student's t-test assuming equal variances between compared groups. A P value of < 0.05 was determined to be statistically significant.
RESULTS
EGFR expression and functionality is effectively reduced in the HS-EGFRKO mice.
Figure 1A shows that there was no difference in the liver weights in the wild-type and HS-EGFRKO mice. To document successful disruption of EGFR in hepatocytes, liver homogenates were prepared from the livers of male and female mice and immunoblotted for EGFR or ERBB3. As shown in Fig. 1B, these results confirmed a sexual dimorphism in EGFR in male compared with female livers (1), with the male livers expressing over eight times as much EGFR protein. In the HS-EGFRKO mice, no EGFR was detected until the chemiluminescent exposure was much longer (shown later in Fig. 1D). To confirm that this resulted in reduced EGF binding, we carried out a 125I-EGF binding analysis of liver membranes from male and female mice (9, 40) (Fig. 1C). Specific ligand binding was lower in female compared with male liver membranes and markedly reduced in membranes from male and female HS-EGFRKO mice. Figure 1D shows that expression of EGFR persisted in other tissues known to harbor EGFR. The immunoblot was overexposed to show the abundance of EGFR in the liver relative to the other tissues. This blot reveals several smaller discrete EGFR fragments in the wild-type mouse liver, which may arise from in vivo or postmortem proteolysis. Evidence for alternatively processed RNA transcripts has been reported previously, but these transcripts encode for the extracellular domain and the related proteins would not be detected by our COOH-terminal antibody (32). Some EGFR was detected upon long-term exposure in the livers of HS-EGFRKO mice, presumably derived from nonparenchymal cells or from hepatocytes that failed to undergo recombination. Figure 1E shows the phosphorylated isoforms of ERBB3 and EGFR induced by exogenous HRG or EGF. To induce tyrosine phosphorylation of hepatic EGFR or ERBB3, we injected into the portal veins of anesthetized adult male mice either recombinant murine EGF (1.5 μg in 100 μl PBS), recombinant human HRG-β1 (1.875 μg in 100 μl PBS), both peptides together, or PBS (100 μl) (the peptides were from PeproTech, Rocky Hill, NJ). Mice were euthanized 3 min after injection. No phosphorylation of ERBB3 or EGFR was detected in the HS-EGFRKO livers at this chemiluminescent film exposure. EGF strongly stimulated the tyrosine phosphorylation of EGFR whereas HRG strongly stimulated the tyrosine phosphorylation of ERBB3. Although HRG stimulated the phosphorylation of ERBB3 more strongly than EGFR, this phosphorylation was entirely dependent on the presence of EGFR given the absence of phosphorylation in the HS-EGFRKO homogenates. This finding validates that the vast majority of ERBB3 in the liver resides in hepatocytes as opposed to the nonparenchymal cells and that the EGFR-ERBB3 heterodimer is the initial signaling dimer employed by HRG.
Fig. 1.

Gene disruption of EGFR effectively blocks EGFR protein expression in the liver. A: we weighed male and female HS-EGFRKO mice and their wild-type counterparts and found no significant differences in body weight between them over a 15-wk period. B: immunoblots revealed the absence of EGFR in HS-EGFRKO mice and a sex difference in the expression of EGFR. ERBB3 expression was not altered in the livers of HS-EGFRKO mice. C: EGF binding to liver membranes was measured by radioligand binding studies. Specific binding was virtually extinguished in HS-EGFRKO liver membranes. D: we analyzed by immunoblot the expression of EGFR in different organs, using an antibody that recognizes the COOH-terminus. This blot shows that EGFR expression persisted in HS-EGFRKO mice in the lung and colon. The chemiluminescent images were from the same blot exposed for equal periods of time to show the relative abundance of EGFR in the liver compared with the other organs. Note that a number of EGFR fragments or cleavage products are evident in the liver blot; however, it is not known whether they represents in vivo or ex vivo degradation products or alternatively spliced forms of EGFR. E: mice were injected with heregulin (HRG), EGF, or EGF/HRG into the portal vein and then killed 5 min later. We carried out immunoblots on liver lysates using antibodies that recognize phospho-specific forms of ERBB3 or EGFR. This blot shows that the injection HRG results mainly in phosphorylation of ERBB3 and EGF results mainly in phosphorylation of EGFR. HRG-induced phosphorylation of ERBB3 requires EGFR. *P < 0.003; **P < 0.0001.
Immunohistochemical localization of EGFR and ERBB3 expression in HS-EGFRKO and wild-type mice.
We immunostained liver sections from wild-type or HS-EGFRKO adult male mice to localize EGFR. The HS-EGFRKO microscopic sections proved to be a useful way to validate the specificity of our EGFR and phospho-EGFR antibodies. We evaluated EGFR expression early in the light and dark periods because the number and affinity of the EGFR varies in a circadian manner based on radioligand binding studies (43). We confirmed that binding was higher early in the light period than early in the dark period (Fig. 2A). The binding was entirely localized to the cell surface, and we did not detect nuclear staining. The staining for total EGFR was uniform between the portal to central vein. We did not see a portal-to-central vein gradient in EGFR protein, which has been previously described in tissue autoradiography studies using radiolabeled EGF (4, 5, 50). As suggested by the immunoblots of total protein, only isolated hepatocytes and nonparenchymal cells, including bile duct cells, showed EGFR staining in the HS-EGFRKO sections. Disruption of EGFR expression occurred uniformly across the entire acinus.
Fig. 2.

Immunohistochemical localization of EGFR and phospho-EGFR and phospho-ERBB3. A: we evaluated the immunohistochemical localization of EGFR in the liver in wild-type (left) and HS-EGFRKO mice (right) 2 h after lights on (AM) and 2 h after lights off (PM). Only a few hepatocytes expressed EGFR in the HS-EFGRKO mice. Nonparenchymal cells continued to express EGFR in the HS-EGFRKO livers. The expression in the early light phase was stronger than that in the early dark phase. Hepatocytes at the beginning of the light phase are larger because of glycogen synthesis during the nighttime feeding phase. B: we injected EGF or HRG into the portal vein (PV) and then used immunohistochemistry to identify the localization of the phosphorylated forms of EGFR (top 2 panels) or ERBB3 (bottom 2 panels). Clear differences in zonal distribution of the 2 receptor tyrosine kinases were observed. The phosphorylated form of EGFR had a striking periportal localization (in contrast to the relatively uniform distribution of “total” EGFR as shown in Fig. 2A) whereas phosphorylated ERBB3 tended to be stronger around the central vein (CV).
We next analyzed the localization of EGFR and ERBB3 phosphorylation in liver sections of wild-type mice using phosphotyrosine-specific antibodies (Fig. 2B). We injected recombinant murine EGF (1.5 μg in 100 μl PBS), recombinant human HRG-β1 (1.875 μg in 100 μl PBS), or PBS (100 μl) into the portal vein to enhance EGFR or ERBB3 tyrosine phosphorylation in wild-type or KO mice. Mice were euthanized 3 min after injection. The microscopic sections came from the same mice used to obtain liver lysates for the immunoblot in Fig. 1E above. Control tissue sections, which were obtained from mice euthanized in the middle of the light phase 5 min after the injection of PBS (the carrier), showed little or no phosphorylation, probably because circulating levels of EGF in mice peak in the late dark phase (22). As expected, liver sections of HS-EGFRKO mice injected with either EGF or HRG showed no phosphorylation (data not shown). Consistent with the aforementioned immunoblot results, HRG had a much greater effect on ERBB3 phosphorylation, and EGF on EGFR phosphorylation. The histological patterns of phospho-EGFR and phospho-ERBB3 differed. For EGFR, there was a well-defined acinar gradient, with the level of phosphorylation being greater near the periportal tract (4, 5, 50) (Fig. 2B). This was in contrast to the uniform staining for “total” EGFR (Fig. 2A). In contrast, phospho-ERBB3 was greater near the central vein. In contrast to EGFR, we saw specific phospho-EGFR signal in the nuclei of some hepatocytes, even though most of the staining was on the cell surfaces.
EGFR gene disruption alters the ontological patterns of expression of EGFR, ERBB2, ERBB3, and Met.
We analyzed the expression patterns of EGFR in the livers of mice from different ages after birth by carrying out immunoblots and performing densitometry. Figure 3 shows the results for EGFR, ERBB2, ERBB3, and Met. We previously had shown that EGFR expression is completely extinguished as early as 6 wk of age in our HS-EGFRKO. We observed that EGFR expression in the wild-type mice is always lower than in the HS-EGFRKO livers as early as 1 day of age (Fig. 1A). Interestingly, the EGFR expression increased at weaning (21 days) and remained elevated during the first year of life. For ErbB2, we confirmed in mice our previous finding in rats that the expression of ERBB2 drops at the time of weaning and remains low during the first year of life (7, 46) (Fig. 2B). In contrast to EGFR and ERBB2, ERBB3 expression increased by day 7 and remained elevated. In contrast to the ERBB proteins, Met expression appeared to be less developmentally regulated. We noted in preweaned animals that ERBB2, ERBB3, and Met were consistently higher in the HS-EGFRKO mouse liver compared with the wild-type liver, raising the possibility that other RTKs can compensate for lack of EGFR through increased expression during the neonatal period. Other possibilities for the enhanced expression of ERBB2 and ERBB3 in the HS-EGFRKO mouse liver include decreased EGFR-driven degradation of ERBB2 and ERBB3 or a negative influence of EGFR on the expression of these proteins in normal mice.
Fig. 3.
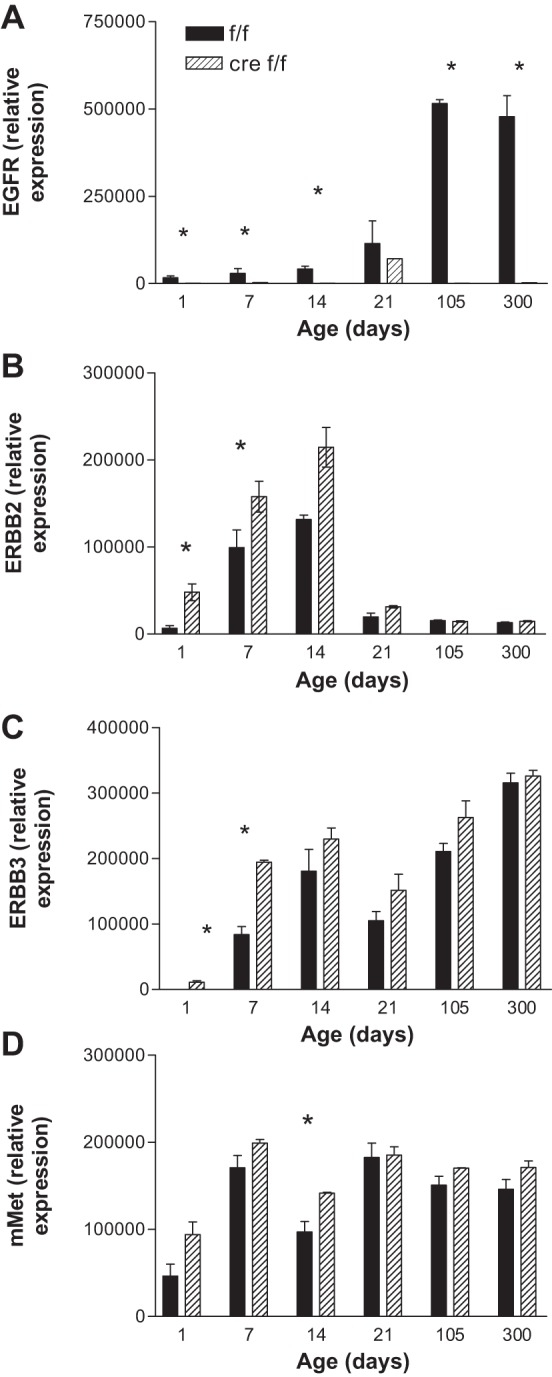
Developmental expression of EGFR, ERBB2, ERBB3, and Met in HS-EGFRKO and wild-type mice. We performed immunoblots and then analyzed by densitometry the relative expression of EGFR (A), ERBB2 (B), ERBB3 (C), and Met (D) in HS-EGFRKO and wild-type mice from 1 to 300 days of age. This figure shows decreased expression of EGFR even before weaning in the HS-EGFRKO mice. During early development there are statistically significant increases in ERBB2, ERBB3, and Met. No such increases are observed after weaning. *P < 0.05.
EGFR gene disruption decreases the clearance of portal vein-injected EGF, with signaling and proliferating consequences for peripheral organs.
We injected recombinant murine EGF (5 μg in 200 μl PBS) into the portal vein of wild-type and HS-EGFRKO mice and analyzed the effect on the clearance of EGF 5 min after injection. Figure 4A shows the abundance of phosphoproteins p-EGFR, p-AKT, and pp-ERK1,2 in the liver and colon of wild-type and HS-EGFRKO mice. These phospho-proteins were barely detected in PBS-injected mice (data not shown), and all samples shown were from EGF-injected mice. This figure shows that the wild-type mouse has enhanced EGFR, AKT, and ERK 1,2 phosphorylation in response to EGF, as would be expected. It also shows that these proteins are more heavily phosphorylated in the colons of the HS-EGFRKO mouse than the wild-type mouse. This is consistent with the hypothesis that EGFR in the liver may serve as a clearance receptor that diverts portal EGF to the bile duct system and eventually the intestinal lumen. To show that the circulating levels of EGF were higher in the HS-EGFRKO mice than in the wild-type mice, we measured the EGF levels in cardiac plasma at the time of euthanasia, using a sensitive and specific radioimmunoassay (RIA) (36). We found that plasma EGF levels were about fourfold higher in the KO mice (Fig. 4B). We also used this RIA to measure in the same mice the EGF levels in the membranes as well as the cytosolic fraction of cells. We boiled tissue membranes, pelleted them, and assayed the EGF in the resultant supernatants and the tissue lysate cytosols. This study, as shown in Fig. 4C, also demonstrated an increased localization of EGF in peripheral tissues compared with the liver in the EGF-injected HS-EGFRKO mice.
Fig. 4.

HS-EGFRKO mice have reduced EGF clearance. A: we injected EGF intraperitoneally (ip) (20 μg in 200 μl ip) and then analyzed by immunoblot the phosphorylation of EGFR, AKT, and ERK 1,2 in the liver and colon 5 min after injection. Note that all 3 proteins are more heavily phosphorylated in the colon of the HS-EGFRKO mice, implying that the loss of EGFR in hepatocytes decreases the clearance of the ip injected EGF. B: we measured the EGF levels in the plasma in mice injected with EGF ip and found that the levels were severalfold higher in the HS-EGFRKO mice, again indicative of reduced clearance of exogenous EGF in these mice. C: we measured EGF in the membranes (left) and cytosol (right) in the liver, pancreas, and colon in mice that had been injected with EGF ip, noting that the levels were higher in the pancreas and colon of the HS-EGFRKO mice. D: we injected EGF at different concentrations into mice ip and then monitored DNA synthesis 8 h later in the basal layer of the tongue, an EGF target organ. Note that the effect of EGF is more pronounced in the HS-EGFRKO mice for the dosages tested. *P < 0.02; **P < 0.001.
Finally, in a separate experiment, we injected EGF ip and examined whether it would alter the cell proliferation of a target organ in the HS-EGFRKO mice. We have previously reported that EGF stimulates DNA synthesis in organs of the intestinal tract as early as 4–12 h after ip injection (44, 45). One of these organs is the tongue. We injected five wild and five HS-EGFRKO mice with varying dosages of EGF and euthanized the mice 8 h later. We then histochemically evaluated DNA synthesis in cross sections of tongue, as judged by nuclear cyclin-A labeling in the basal layer of this stratified epithelium (Fig. 4D). The tongue shows a pronounced circadian rhythm of DNA synthesis and these mice were euthanized late in the light phase, when DNA synthesis is at its lowest point. We observed that EGF, at all dosages tested, increased DNA synthesis in the HS-EGFRKO mouse tongue compared with that in the wild-type mice, again pointing to a clearance role for hepatocellular EGFR.
HS-EGFRKO mice have paradoxically lower plasma levels of EGF.
The results of the previous experiments led us to hypothesize that EGFR may serve as a clearance receptor and that the circulating levels of endogenous EGF would be higher in the HS-EGFRKO mouse plasma. By RIA analysis, we discovered that plasma obtained the hearts of HS-EGFRKO mice actually had lower circulating EGF levels than wild-type mice. Figure 5A shows that ratio of wild-type plasma EGF to HS-EGFRKO plasma EGF increases with age after weaning. Figure 5B shows that plasma EGF remains constant during a 24 h fast in both wild-type and HS-EGFRKO mice. In contrast, insulin decreases by 80% in the plasma of fasted mice over this same period (data not shown). We also measured by RIA the circulating levels of transforming growth factor-α (TGF-α), a distinct EGFR ligand, in wild-type and HS-EGFRKO mice and observed no differences (data not shown).
Fig. 5.
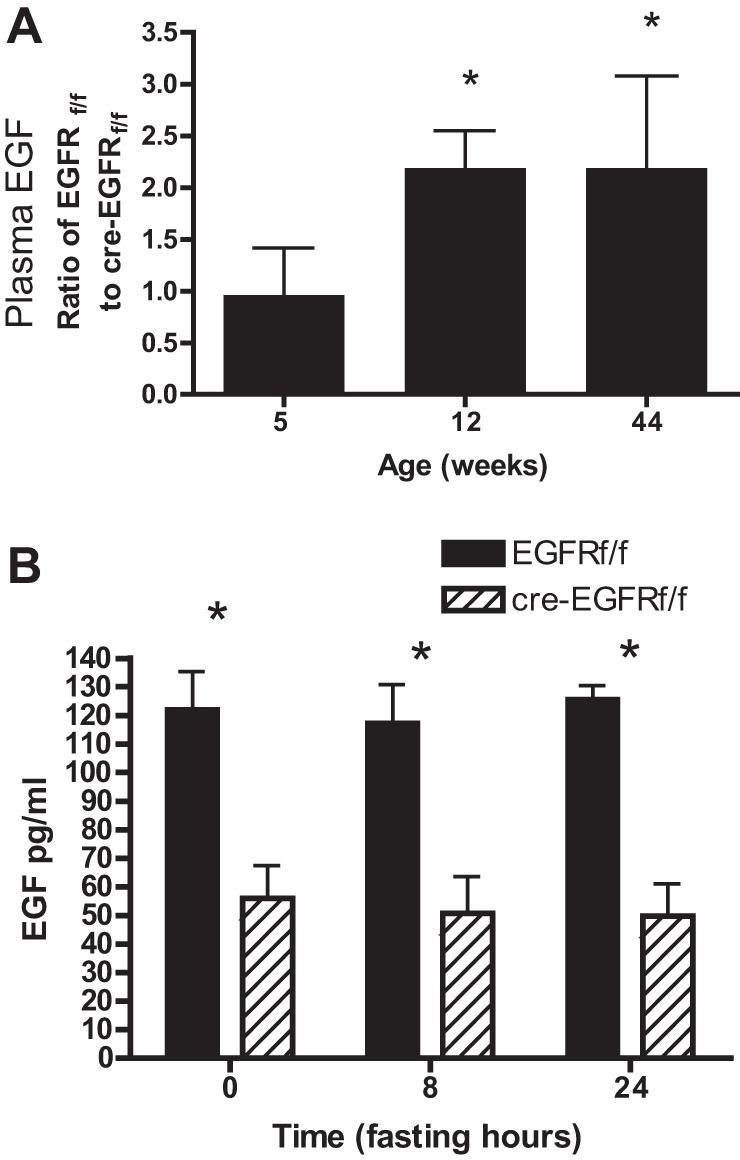
HS-EGFRKO mice have paradoxically lower plasma levels of EGF. A: we monitored mid-light phase plasma EGF levels in HS-EGFRKO and wild-type mice at different ages and observed that the relative amount of EGF in the wild-type mice increased relative to HS-EGFRKO mice. There was twice as much circulating EGF in the wild-type mice compared with knockout (KO) mice at 12 and 44 wk of age. B: we measured mid-light phase plasma EGF in fasted mice and observed that in contrast to insulin, which decreased markedly (data not shown) the EGF levels remained constant and were always less in the HS-EGFRKO mice compared with the wild-type counterparts. *P < 0.02.
We next examined the submandibular salivary gland and its EGF content, since this is the main source of circulating EGF (22). We observed that the submandibular glands of 11-mo-old male mice were smaller in the HS-EGFRKO mice. In contrast, the livers had a normal weight, although the kidneys in the HS-EGFRKO mice were larger (Fig. 6, A and B). Indeed, when we measured the amount of EGF in the submandibular gland, we found decreased EGF levels (Fig. 6C). We immunostained the salivary gland of male 11-mo-old mice for EGF and analyzed the staining pattern. EGF is produced by the granular convoluted cells (16), which are easily distinguished from the serous-producing acinar cells. This organ normally shows a sexual dimorphism after weaning, with female mice having fewer granular convoluted tubular cells and more acinar cells. We observed that HS-EGFRKO male mice had a reduced number of tubules (Fig. 6, D and E) relative to the acinar cells. Thus the organ of the male HS-EGFRKO appeared to have become feminized over the first year of life.
Fig. 6.

HS-EGFRKO mice show reductions in the weight and EGF production by the submandibular salivary gland (SMG). A: we weighed various organs in 11-mo-old old HS-EGFRKO mice and compared the weight to those in wild-type mice. The kidneys (Kid.) were larger whereas the submandibular glands were smaller. There was no change in the size of the liver (Liv.) itself. Tes., testes; Blad., bladder; Spl., spleen; Hrt., heart. B: we looked at the size of the submandibular glands as a function of age and noted that there was a progressive decrease in size that was first apparent at 2 mo. C: we used an RIA to measure the amount of EGF in the submandibular gland and found the EGF glandular content was reduced by ∼30% at 11 mo of age. D: we calculated the area occupied by the granular convoluted duct cells in 11-mo-old old mice and found that it was significantly reduced in the HS-EGFRKO compared with the controls. These results indicate that the reduction in circulating EGF observed in the HS-EGFRKO mice (Fig. 5) is caused by decreased production of EGF by atrophic granular convoluted tubular cells, which synthesize nearly all of the EGF in circulation. E: sections of the submandibular salivary gland were immunostained with a rabbit anti-polyclonal against mouse EGF to identify the granular convoluted tubular cells (GCT). Darkness in the cytoplasm of the tubular cells correlates with EGF levels in this grayscale image. The acini (Ac) expressed no EGF. Note that the tubules are more numerous and robust in the wild-type mouse (left) compared with the HS-EGFRKO mouse sections (right). The EGF content was also higher in the GCT cells, consistent with the RIA results. These sections are representative, but there was some variation within each submandibular gland. *P < 0.01; **P < 0.001.
Liver regeneration following 70% hepatectomy is delayed in HS-EGFRKO mice.
Considerable evidence suggests that EGFR plays a stimulatory role in liver regeneration. It has been reported that in another model of EGFR gene disruption that peak DNA synthesis is reduced by 80–90% at 36 and 48 h after liver resection (27). To confirm this finding, we carried out 70% hepatectomies in wild-type and HS-EGFRKO mice and then harvested regenerating livers at 30, 36, 48, and 72 h after resection. We found that the weights of the male but not the female livers were slightly larger in the wild-type mice than the KO mice after hepatectomy (Fig. 7A; P < 0.04). To evaluate DNA synthesis, we analyzed cyclin A, a broad S-phase marker, by immunohistochemistry (Fig. 7B) and by immunoblot analysis of total liver homogenates (Fig. 7C). Surprisingly, the increase in cyclin A in the HS-EGFRKO livers paralleled that in the wild-type mice by immunohistochemical or immunoblot analysis. When we evaluated cyclin D by immunoblot, we found no difference in induction or levels of cyclin D (data not shown). However, when we analyzed BrdU labeling, we did note a shift in the peak from 36 to 48 h even though the initial timing of BrdU labeling in the HS-EGFRKO livers paralleled that of the wild-type livers (Fig. 7, D and E). In addition, we saw higher Ki67 nuclear labeling in wild-type compared with HS-EGFRKO livers at 30 h (Table 1). Ki67 is a broad marker of proliferation competent cells that identifies not only cells in the S phase, but also cells in the G1-S and M phases (10). We did not rigorously examine whether there was a G2-M1 lag in the HS-EGFRKO mice as has been reported for HS-MetKO mice after PH (12); however, mitotic figures were more numerous in the HS-EGFRKO mice at 72 h compared with control mice, again suggesting a delay in regeneration. Overall, the effect of EGFR gene disruption was very modest compared with the previous study (27). In addition, we saw no effect on Ki67 nuclear labeling in female HS-EGFRKO mice at 36 and 48 h (Table 1).
Fig. 7.

HS-EGFRKO mice show delayed 5-bromo-2′-deoxyuridine (BrdU) labeling in mice subjected to 70% hepatectomy. A: livers were removed from animals that underwent 70% hepatectomy and expressed as a percent of total body weight. Statistically significant differences were found in weight at 36 and 72 h after partial hepatectomy (PH), with the livers from the male HS-EGFRKO mice being significantly smaller than those of the wild-type mice at 36 h. No such differences were seen for female mice. B: we monitored nuclear cyclin A, a specific S-phase marker, by immunohistochemistry and observed that peak labeling occurred at 48 h in both the HS-EGFRKO and wild-type mice. Labeling at this time was comparable in both groups. C: we monitored cyclin A expression by immunoblotting and observed that the appearance of cyclin A in the livers of HS-EGFRKO mice after PH paralleled that in the wild-type mice. The level at 48 h was slightly higher in the wild-type mice. D: we monitored BrdU nuclear labeling by immunohistochemistry. BrdU defines cells actively synthesizing DNA during the instant it is injected into the mice. Although the kinetic of BrdU labeling was similar between KO and wild-type mice, peak labeling in KO mice lagged behind that in wild-type mice, suggestive of delayed cell proliferation. E: photomicrograph shows nuclei that are positive for BrdU incorporation into DNA. *P < 0.03; **P < 0.006.
Table 1.
Ki67 nuclear labeling after 70% hepatectomy
| Hours | 30 | 36 | 48 | 72 | 36 (F) | 48 (F) |
|---|---|---|---|---|---|---|
| f/f | 12.1 ± 1.9 | 41.5 ± 2.6 | 48.2 ± 5.6 | 33.6 ± 4.5 | 15.6 ± 1.5 | 44.1 ± 3.4 |
| Cre f/f | 5.3 ± 1.3 | 30.7 ± 4.6 | 59.7 ± 3.4 | 31.5 ± 2.2 | 13.5 ± 2.5 | 47.0 ± 5.9 |
| Ratio | 2.28 | 1.35 | 0.81 | 0.93 | 1.16 | 0.94 |
Liver sections from groups of hepatocyte-specific EGFR wild-type (f/f) or knockout (Cre f/f) (N = 4–6) were analyzed for nuclear Ki67 staining by immunohistochemistry (% of total hepatocyte nuclei). Means and standard errors were calculated for sections obtained from mice euthanized at different times after hepatectomy, including female (F) mice. The only statistically significant difference between was for the livers of male mice at 30 h (P < 0.005). The 36 and 48 h means of male mice approached statistical significance (P = 0.1140 and P = 0.08, respectively). The ratios of the means (ff/cre ff) are shown in the bottom row.
Liver regeneration following CCl4 is impaired in HS-MetKO mice but not HS-EGFRKO mice.
When the liver is injured by a chemical agent, the injured cells die, later replaced by proliferating hepatocytes that migrate into the wounded area. This requires not only cell proliferation but also cell migration and inflammatory removal of the dead cells. We injected CCl4 into wild-type and HS-EGFRKO mice to determine whether loss of EGFR affected liver injury or cell proliferation in this model. We compared the effects of EGFR loss by carrying out similar experiments in a HS-MetKO and a HS-cMet-EGFR double KO (HS-Met-EGFRDKO). We harvested livers from mice euthanized at 24, 36, 48, 72, and 120 h after CCl4 administration. We found that EGFR gene disruption had no effect on plasma ALT levels, a marker of liver damage (Fig. 8). We found that loss of hepatocyte EGFR loss had no effect on the liver weight in this model (Fig. 9A), whereas loss of hepatocyte Met caused a consistent decrease in liver weight. Dual hepatocyte loss of EGFR and Met led to an even greater reduction in liver weight at most time points. When we analyzed the effect of EGFR gene disruption on the area of centrilobular necrosis over time, we found that loss of EGFR had no effect on necrosis (Fig. 9B). The histological evidence confirmed the biochemical finding with respect to plasma ALT (Fig. 8). In contrast, loss of Met led to more necrosis at 72 h, but we did not see a difference at 48 or 120 h (Fig. 9B). In addition, loss of both EGFR and Met resulted in striking and highly significant differences at the time points studied (Fig. 9B). Finally, we quantified Ki67 nuclear labeling to evaluate cell proliferation (Fig. 9C). We found no difference in nuclear labeling between wild-type and HS-EGFRKO, but striking differences between wild-type mice and their HS-MetKO and HS-Met-EGFRDKO counterparts. The nuclear labeling was low in the latter two groups at 48 h but did rebound at 120 h.
Fig. 8.
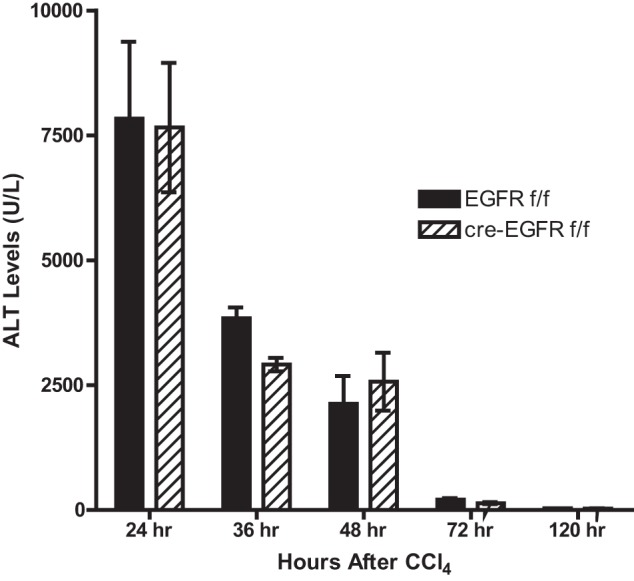
Serum alanine aminotransferase (ALT) levels in HS-EGFRKO mice after CCl4 are similar to those in control mice. This figure shows that the levels and kinetic of plasma ALT, a marker of liver damage, were similar for HS-EGFRKO and wild-type mice over a 5-day period after CCl4 injection. This was consistent with the similar histological appearance of the livers in these mice (see Fig. 9B, left).
Fig. 9.

Loss of Met and the loss of both Met and EGFR have a more detrimental effect on CCl4-induced liver necrosis and regeneration than the loss of EGFR alone. A: livers were removed from mice and weighed at various times after a CCl4 ip injection, and liver weight was expressed as a function of body weight. Note that the greatest loss of weight occurred for the animals that lost hepatocellular expression of both EGFR and Met. Loss of EGFR alone had no effect on liver weight. B: we monitored the necrotic area of the liver. Note that the greatest necrosis occurred for the animals that lost expression of both EGFR and Met. Loss of EGFR alone had no effect on liver necrosis. C: we evaluated cell proliferation by monitoring nuclear expression of Ki67 by immunohistochemistry at 48 and 120 h after CCl4 injection. Note that the greatest delay in Ki67 synthesis occurred for the animals that lost expression of both EGFR and Met. Loss of EGFR alone had no significant effect on cell proliferation as judged by this marker. *P ≤ 0.002; **P ≤ 0.001.
DISCUSSION
In this article we developed an HS-EGFRKO model to evaluate how loss of hepatocellular EGFR expression affects varied processes such as EGF clearance, circulating EGF concentrations, and liver regeneration following 70% resection or CCl4-induced centrilobular injury. We showed that the EGFR expression was effectively disrupted in the livers of male and female mice by immunoblot (Fig. 1, A and C), radioligand binding (Fig. 1B), and EGFR kinase activation studies (Fig. 1D). By injecting EGF or HRG into the portal vein and evaluating by immunoblot the phosphorylation of EGFR and ERBB3 at 5 min after injection, we demonstrated that each ligand induced phosphorylation primarily of its own receptor (Fig. 1D). Since ERBB3 is a weak kinase and no ERBB3 phosphorylation was observed in the HS-EGFRKO mice, a sufficient number of activated EGFR molecules must exist at the time of injection to phosphorylate ERBB3, even in the absence of exogenous EGF. This supports our prior hypothesis that EGFR and ERBB3 are the major heterodimeric ERBB signaling partners in hepatocytes of mature adult mice, which is based on low ERBB2 (Fig. 3B) and no ERBB4 expression (7). When we examined the immunohistochemical phosphorylation patterns of EGFR and ERBB3 in the same animal liver (Fig. 2B), we noted that EGFR was preferentially phosphorylated in hepatocytes near the portal vein whereas phospho-ERBB3 was more prominent in hepatocytes near the central vein. This has implications for understanding the signaling outcomes of these diverse ligands. The actions of EGF or HRG signaling may lead to distinct signaling outcomes not only because differential signal transduction launched by an EGFR homodimer as opposed to an EGFR-ERBB3 heterodimer (or even an ERB3 homodimer), but also because they occupy different zones in the hepatic acinus. This is important because liver parenchyma display metabolic zonation (19). For example, oxidative energy metabolism, glucose release, amino acid usage, cholesterol synthesis, ammonia detoxification, glutathione peroxidation and conjugation, and bile formation are predominately localized to the periportal zone, whereas glucose uptake, ketogenesis, glutamine formation, and xenobiotic metabolism are localized to the perivenous zone.
The strong periportal localization of EGF is consistent with prior work showing a similar pattern using autoradiographic localization of portal vein-injected iodinated EGF. Early researchers showed that the liver effectively removed EGF from the portal circulation much in the way it clears and regulates the circulating levels of insulin (4, 5, 50). Nevertheless, EGFR itself is uniformly distributed across the liver acinus when localized by immunohistochemistry using an antibody that recognizes nonphosphorylated as well as phosphorylated EGFR (Fig. 2A). In the HS-EGFRKO mice, portal vein-injected EGF resulted in higher concentration of EGF in plasma (Fig. 4B) and in peripheral tissues (Fig. 4C), leading to a greater tyrosine phosphorylation of EGFR in these tissues (Fig. 4C). Intraperitoneally injected EGF also stimulated cell proliferation to a greater extent in EGF-target cell populations, such as the basal layer of the tongue epithelium (Fig. 4D). Clearance of EGF from the blood may be a general mechanism to tamp down the proliferative effects of EGF target tissues. Since ∼10% of the internalized EGF crosses the hepatocyte to be delivered into the bile ducts (4), EGF clearance may also be a mechanism to provide the biliary tract with EGF, a well-established epithelial protectant. Numerous articles have shown that luminal EGF above the apical surface membrane of a polarized epithelial cell, such as an enterocyte, is normally sequestered from its basolateral receptor (40) but can gain access during conditions of mucosal disruption, initiating repair through migration and even replication of tight-junction disrupted cells (8, 54).
On the basis of studies showing that EGFR can be effectively cleared, we hypothesized that the serum levels of EGF would be higher in the HS-EGFRKO mice than their wild-type counterparts. However, when we measured the circulating levels by RIA, we discovered that they were consistently lower in the former compared with the latter (Fig. 5, A and B). This reduction occurred shortly after weaning when EGF salivary gland levels rise, particularly in male mice (15). When we compared organs weights of 11-mo-old mice, we found that HS-EGFKO had statistically significant increases in the average kidney weight and decreases in the submandibular gland (Fig. 6, A and B), with no changes in the testes, bladder, spleen, or heart or in the liver itself (Fig. 6A). When we measured the amount of EGF in the submandibular gland, we found significant reductions in the HS-EGFRKO mice (Fig. 6C), consistent with the decreased plasma levels. Microscopic analysis of the area in the submandibular gland occupied by the granulated convoluted cells ducts revealed a reduction in the total area in the HS-EGFKO mice consistent with emasculation of this organ (Fig. 6, D and E). Testosterone as well as triiodothyronine (T3) are responsible for the sustenance of these tubules and decreased testosterone or T3 production or increased sex hormone binding protein in HS-EGFRKO mouse could lead to this outcome (13, 14). Chronic insulin or dexamethasone injections also enlarge the submandibular gland and increase its EGF content (17, 39). The reason for the increase in kidney size is also a matter of speculation, but cannot be ascribed to decreased free testosterone since this hormone positively regulates renal size. Hepatocytes do synthesize IGF-1, which also positively regulates remnant kidney size following unilateral nephrectomy. If EGF negatively regulates IGF-1 production in hepatocytes (38), then the loss of EGFR signaling therein could increase IGF-1 leading to renal enlargement.
EGFR has been implicated in the positive regulation of hepatic regeneration after PH. Removal of the submandibular salivary gland, the major source of circulating and salivary EGF, retards liver regeneration in mice 2 wk after sialoadenectomy (28). However, the submandibular salivary gland synthesizes many biologically active peptides, including NGF and renin (35). In addition, the liver itself increases local expression of TGF-α, another important EGF-like ligand, during regeneration; however, targeted disruption of this growth factor had no effect on regeneration following PH (37).
Perinatal deletion of EGFR in hepatocytes expressing the Alfp-cre or Mx transgene showed strikingly impaired cell proliferation and cell cycle progression, with a pronounced reduction and delay in cyclin D1 expression, even though these mice did not show altered activation of Erk or Akt within the first few hours after PH (27). In these studies, DNA synthesis was barely detectable at 36 and 48 h after 70% hepatectomy. In contrast, we found a relatively mild phenotype. For example, cyclin A expression displayed similar kinetics in the livers of HS-EGFRKO and their wild-type counterparts (Fig. 7, B and C). We found no differences between wild-type and HS-EGFRKO with respect to cyclin D1 expression either (data not shown). We did see a slight lag in BrdU labeling in the livers HS-EGFRKO mice, with the peak occurring at 48 h rather than 36 h, but it was more modest than that of the earlier study (Fig. 7, D and E).
The reasons for the differences between the two studies may be related to experimental design. The Alfp-cre or Mx driven Cre recombinase tissue distributions disrupt EGFR expression in cells other than hepatocytes. The Mx promoter is active in hepatocytes, bile duct cells, and nonparenchymal cells of the liver as well as in other organs (21). The Alfp-cre transgene includes not only albumin regulatory elements but also α-fetoprotein enhancers that are not present in our Alb-cre transgenic mice (31). It drives Cre-recombinase in bile duct cells as well as hepatocytes (20). The Alfp-cre EGFRKO mice also show a reduced body weight, which began as early as the third postnatal week and persisted through adulthood. In contrast, our HS-EGFRKO and wild-type mice had a similar body weight regardless of sex (Fig. 1A). In the earlier study, a more complicated mechanism involving EGFR loss in some other cell type besides hepatocytes, such as bile duct cells, may have interfered with liver regeneration after hepatectomy.
Several other factors may be involved. In the previous study, the mice were the C57BL/6 strain, whereas our mice were a mixed 129SV-C57BL/6 strain. There may be genetic modifiers in the 129SV mouse that attenuate the loss of EGFR in hepatocytes. Strain has been shown to alter the effect of EGFR deletion in other models (52). For example, global KOs die during embryogenesis or the neonatal period of distinct strain-specific causes. In agreement with the previous study, we did not see compensatory upregulation of Met or the other ERBB proteins during in adult mice; however, increased levels of ERBB2, ERBB3, and Met were seen prior to weaning, perhaps contributing to the normalization of early liver development in the absence of EGFR (Fig. 3). Finally, even the wild-type PH mice in the previous study showed significant mortality as early as 48 h after PH. We had no mortality. This raises a question as to whether there may have been an additional stress-associated factor that accentuated the role of EGFR previously. The sex of the mice used in the prior study was not reported; however, we carried out a preliminary PH study with female mice and found no difference in the expression of Ki67, the nuclear cell proliferation marker, at 36 and 48 h after resection (Table 1).
Having seen a relatively minimal effect of EGFR gene disruption on regeneration after 70% hepatectomy, we examined the response of the liver to acute CCl4 injury, which is metabolized by cytochrome P450 2E1. The CCl4 metabolites cause centrilobular necrosis and apoptosis, provoking over a 5- to 7-day period an inflammatory clearing of dead hepatocytes as well as an ingrowth of proliferating hepatocytes. This restores the “wounded” area with functional parenchyma. When we measured the serum ALT levels in the HS-EGFRKO plasmas, we found no differences in the timing or levels of this marker of liver damage (Fig. 8). We then looked at liver weight, necrotic damage, and cell proliferation in the livers of not only EGFR, but also for Met single gene KO mice and EGFR-cMet double KO mice (Fig. 9). Again, loss of EGFR seemed to have little or no effect in this model (Fig. 9A). In contrast, Met did show a reduction in liver weight at specific times, particularly when combined with loss of EGFR in a HS-EGFR-cMet “double” KO. We found no difference in cytochrome P450 2E1 expression in the various wild-type strains and in the HS-EGFRKO mice, but preliminary studies revealed a small but statistically significant increase in the HS-MetKO (15%) and EGFR-Met double KO mice (10%; data not shown). This increase, which requires further study, is insufficient to account for the increased injury in either KO stain or the even greater injury in the EGFR-Met double KO mice. Collectively, these results suggest to us that EGFR and to a greater extent Met can compensate for one another, although a signaling mechanism downstream from these cell surface receptors may also be at work.
Met may be more important than EGFR in the regulation of liver regeneration, particularly in the CCl4 chemical injury model. Met has been shown to play a more important role than EGFR in liver development (18). In earlier studies, it has also been difficult to show that EGFR is phosphorylated immediately after PH when the circulating levels of EGF increase by 30%, although there is evidence that the receptor is internalized and ubiquitinated (49). Still, the receptor does undergo enhanced tyrosine phosphorylation relative to sham-operated mice in protein tyrosine phosphatase 1B (34) and Mig-6 (33) KO mice, both of which normally negatively regulate EGFR tyrosine phosphorylation. These mice do show accelerated liver regeneration after PH. However, Mig-6 also negatively regulates Met tyrosine phosphorylation (29). An alternative explanation to account of the lack of effect of EGFR may be the existence of redundant mechanisms to compensate for EGFR loss. Given the several-week delay between EGFR deletion and PH, the hepatocyte might acquire or upregulate redundant signaling mechanisms that allow it to overcome this loss. This might account for the acute effectiveness of EGFR siRNA in the rat to retard liver regeneration after PH (30). The latter finding could also be interpreted that EGFR plays a more important role in the rat than the mouse liver, for which numerous differences exist, including the earlier timing of peak regeneration in the rat (24 h) compared with the mouse (anywhere from 36 to 48 h in different studies). Yet others have reported that AEE788, an immediate pharmacological inhibitor of EGFR, had no effect on regeneration in the rat after 70 or 90% PH (9).
Much has changed since EGF and its receptor were discovered almost 50 years ago. The key-lock model used to describe ligand-receptor interactions clearly misjudged the complexity and redundancy built into ligand-receptor signaling networks. This complexity is now well appreciated even if the understanding is still limited. Each RTK may have multiple ligands or distinct tyrosine kinase signaling partners. Moreover, for every kinase there is a phosphatase or direct kinase inhibitor that can negatively regulate downstream effects. There is also built-in signaling redundancy that enables cells to compensate for the loss of a specific protein. Unfortunately, such issues create difficulties in the use of KO studies to analyze the role of specific RTKs in normal events, such as liver regeneration. Similar problems are faced by oncologists treating cancers with specific inhibitors of tyrosine kinase receptors. Cancer cells can reprogram themselves and achieve alternative proliferative and metastatic fates by using a panoply of signaling molecules. Presumably, positive or negative modulation of various hierarchical signaling mechanisms involved in hepatocyte cell cycle progression enable the livers of double KO EGFR and Met mice to mount a regenerative response even though they lack two kinases, such as EGFR and Met, once thought to be crucial for regeneration. This avoids the need to employ a slower hepatic oval cell-mediated liver regeneration, relied on when hepatocyte proliferation is terminally blocked.
GRANTS
This work was supported by R01DK53804 and R21CA149708 (to W. E. Russell) and R01CA092479 (to D. W. Threadgill).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
L.A.S., D.W.T., and W.E.R. conception and design of research; L.A.S., X.Z., and M.C.S. performed experiments; L.A.S., X.Z., and M.C.S. analyzed data; L.A.S., X.Z., and M.C.S. interpreted results of experiments; L.A.S. and M.C.S. prepared figures; L.A.S. drafted manuscript; L.A.S., D.W.T., and W.E.R. edited and revised manuscript; L.A.S., D.W.T., and W.E.R. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Drs. Snorri S. Thorgeirsson and Pat Levitt for providing the floxed Met mice used to create the HS-MetKO mice. The authors thank Dr. Shyamal K. Roy for providing rabbit anti mouse-EGF antibody used in the EGF RIA.
REFERENCES
- 1.Benveniste R, Danoff TM, Ilekis J, Craig HR. Epidermal growth factor receptor numbers in male and female mouse primary hepatocyte cultures. Cell Biochem Funct 6: 231–235, 1988. [DOI] [PubMed] [Google Scholar]
- 2.Bladt F, Riethmacher D, Isenmann S, Aguzzi A, Birchmeier C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature 376: 768–771, 1995. [DOI] [PubMed] [Google Scholar]
- 3.Borowiak M, Garratt AN, Wustefeld T, Strehle M, Trautwein C, Birchmeier C. Met provides essential signals for liver regeneration. Proc Natl Acad Sci USA 101: 10608–10613, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burwen SJ, Barker ME, Goldman IS, Hradek GT, Raper SE, Jones AL. Transport of epidermal growth factor by rat liver: evidence for a nonlysosomal pathway. J Cell Biol 99: 1259–1265, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burwen SJ, Barker ME, Goldman IS, Jones AL. The effect of concentration on hepatic transport of exogenous epidermal growth factor. Hepatology 5: 211–214, 1985. [DOI] [PubMed] [Google Scholar]
- 6.Carver RS, Sliwkowski MX, Sitaric S, Russell WE. Insulin regulates heregulin binding and ErbB3 expression in rat hepatocytes. J Biol Chem 271: 13491–13496, 1996. [DOI] [PubMed] [Google Scholar]
- 7.Carver RS, Stevenson MC, Scheving LA, Russell WE. Diverse expression of ErbB receptor proteins during rat liver development and regeneration. Gastroenterology 123: 2017–2027, 2002. [DOI] [PubMed] [Google Scholar]
- 8.Casaletto JB, McClatchey AI. Spatial regulation of receptor tyrosine kinases in development and cancer. Nat Rev Cancer 12: 387–400, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deng M, Huang H, Jin H, Dirsch O, Dahmen U. The anti-proliferative side effects of AEE788, a tyrosine kinase inhibitor blocking both EGF- and VEGF-receptor, are liver-size-dependent after partial hepatectomy in rats. Invest New Drugs 29: 593–606, 2011. [DOI] [PubMed] [Google Scholar]
- 10.Endl E, Gerdes J. The Ki-67 protein: fascinating forms and an unknown function. Exp Cell Res 257: 231–237, 2000. [DOI] [PubMed] [Google Scholar]
- 11.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Janne PA. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316: 1039–1043, 2007. [DOI] [PubMed] [Google Scholar]
- 12.Factor VM, Seo D, Ishikawa T, Kaposi-Novak P, Marquardt JU, Andersen JB, Conner EA, Thorgeirsson SS. Loss of c-Met disrupts gene expression program required for G2/M progression during liver regeneration in mice. PLoS One 5: e12739, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fang J, Yamamoto R, Takatsuka D, Tsuji M, Terada N. Effects of pretreatment with androgen or thyroid-hormone on androgen-induced proliferation of granular convoluted tubular cells in mouse submandibular glands. Anat Rec 236: 679–684, 1993. [DOI] [PubMed] [Google Scholar]
- 14.Fujieda M, Murata Y, Hayashi H, Kambe F, Matsui N, Seo H. Effect of thyroid-hormone on epidermal growth-factor gene-expression in mouse submandibular-gland. Endocrinology 132: 121–125, 1993. [DOI] [PubMed] [Google Scholar]
- 15.Gattone VH, Sherman DA, Hinton DA, Niu FW, Topham RT, Klein RM. Epidermal growth-factor in the neonatal mouse salivary-gland and kidney. Biol Neonate 61: 54–67, 1992. [DOI] [PubMed] [Google Scholar]
- 16.Gresik EW. The granular convoluted tubule (Gct) cell of rodent submandibular glands. Microsc Res Tech 27: 1–24, 1994. [DOI] [PubMed] [Google Scholar]
- 17.Hosoi K, Maruyama S, Ueha T, Sato S, Gresik EW. Additive and or synergistic effects of 5-alpha-dihydrotestosterone, dexamethasone, and triiodo-l-thyronine on induction of proteinases and epidermal growth-factor in the submandibular-gland of hypophysectomized mice. Endocrinology 130: 1044–1055, 1992. [DOI] [PubMed] [Google Scholar]
- 18.Huh CG, Factor VM, Sanchez A, Uchida K, Conner EA, Thorgeirsson SS. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad Sci USA 101: 4477–4482, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jungermann K, Kietzmann T. Zonation of parenchymal and nonparenchymal metabolism in liver. Annu Rev Nutr 16: 179–203, 1996. [DOI] [PubMed] [Google Scholar]
- 20.Kellendonk C, Opherk C, Anlag K, Schutz G, Tronche F. Hepatocyte-specific expression of Cre recombinase. Genesis 26: 151–153, 2000. [DOI] [PubMed] [Google Scholar]
- 21.Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science 269: 1427–1429, 1995. [DOI] [PubMed] [Google Scholar]
- 22.Kurachi H, Oka T. Changes in epidermal growth factor concentrations of submandibular gland, plasma and urine of normal and sialoadenectomized female mice during various reproductive stages. J Endocrinol 106: 197–202, 1985. [DOI] [PubMed] [Google Scholar]
- 23.Lee TC, Threadgill DW. Generation and validation of mice carrying a conditional allele of the epidermal growth factor receptor. Genesis 47: 85–92, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lemmon MA, Schlessinger J, Ferguson KM. The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harb Perspect Biol 6: a020768, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michalopoulos GK. Advances in liver regeneration. Expert Rev Gastroenterol Hepatol 8: 897–907, 2014. [DOI] [PubMed] [Google Scholar]
- 26.Michalopoulos GK. Principles of liver regeneration and growth homeostasis. Compr Physiol 3: 485–513, 2013. [DOI] [PubMed] [Google Scholar]
- 27.Natarajan A, Wagner B, Sibilia M. The EGF receptor is required for efficient liver regeneration. Proc Natl Acad Sci USA 104: 17081–17086, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noguchi S, Ohba Y, Oka T. Influence of epidermal growth factor on liver regeneration after partial hepatectomy in mice. J Endocrinol 128: 425–431, 1991. [DOI] [PubMed] [Google Scholar]
- 29.Pante G, Thompson J, Lamballe F, Iwata T, Ferby I, Barr FA, Davies AM, Maina F, Klein R. Mitogen-inducible gene 6 is an endogenous inhibitor of HGF/Met-induced cell migration and neurite growth. J Cell Biol 171: 337–348, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paranjpe S, Bowen WC, Tseng GC, Luo JH, Orr A, Michalopoulos GK. RNA interference against hepatic epidermal growth factor receptor has suppressive effects on liver regeneration in rats. Am J Pathol 176: 2669–2681, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem 274: 305–315, 1999. [DOI] [PubMed] [Google Scholar]
- 32.Reiter JL, Threadgill DW, Eley GD, Strunk KE, Danielsen AJ, Sinclair CS, Pearsall RS, Green PJ, Yee D, Lampland AL, Balasubramaniam S, Crossley TD, Magnuson TR, James CD, Maihle NJ. Comparative genomic sequence analysis and isolation of human and mouse alternative EGFR transcripts encoding truncated receptor isoforms. Genomics 71: 1–20, 2001. [DOI] [PubMed] [Google Scholar]
- 33.Reschke M, Ferby I, Stepniak E, Seitzer N, Horst D, Wagner EF, Ullrich A. Mitogen-inducible gene-6 is a negative regulator of epidermal growth factor receptor signaling in hepatocytes and human hepatocellular carcinoma. Hepatology 51: 1383–1390, 2010. [DOI] [PubMed] [Google Scholar]
- 34.Revuelta-Cervantes J, Mayoral R, Miranda S, Gonzalez-Rodriguez A, Fernandez M, Martin-Sanz P, Valverde AM. Protein tyrosine phosphatase 1B (PTP1B) deficiency accelerates hepatic regeneration in mice. Am J Pathol 178: 1591–1604, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rougeot C, Rosinski-Chupin I, Mathison R, Rougeon F. Rodent submandibular gland peptide hormones and other biologically active peptides. Peptides 21: 443–455, 2000. [DOI] [PubMed] [Google Scholar]
- 36.Roy SK, Greenwald GS. Immunohistochemical localization of epidermal growth factor-like activity in the hamster ovary with a polyclonal antibody. Endocrinology 126: 1309–1317, 1990. [DOI] [PubMed] [Google Scholar]
- 37.Russell WE, Kaufmann WK, Sitaric S, Luetteke NC, Lee DC. Liver regeneration and hepatocarcinogenesis in transforming growth factor-alpha-targeted mice. Mol Carcinog 15: 183–189, 1996. [DOI] [PubMed] [Google Scholar]
- 38.Santhanam M, Chia DJ. Hepatic-specific accessibility of Igf1 gene enhancers is independent of growth hormone signaling. Mol Endocrinol 27: 2080–2092, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schaudies RP, Grimes J, Davis D, Koldovsky O. Modulation of immunoreactive epidermal growth-factor levels in the submandibular-gland, pancreas, liver, kidney and gastrointestinal-tract of suckling rats by cortisone and triiodothyronine. J Endocrinol 131: 95–100, 1991. [DOI] [PubMed] [Google Scholar]
- 40.Scheving LA, Shiurba RA, Nguyen TD, Gray GM. Epidermal growth factor receptor of the intestinal enterocyte. Localization to laterobasal but not brush border membrane. J Biol Chem 264: 1735–1741, 1989. [PubMed] [Google Scholar]
- 41.Scheving LA, Stevenson MC, Taylormoore JM, Traxler P, Russell WE. Integral role of the EGF receptor in HGF-mediated hepatocyte proliferation. Biochem Biophys Res Commun 290: 197–203, 2002. [DOI] [PubMed] [Google Scholar]
- 42.Scheving LA, Stevenson MC, Zhang X, Russell WE. Cultured rat hepatocytes upregulate Akt and ERK in an ErbB-2-dependent manner. Am J Physiol Gastrointest Liver Physiol 295: G322–G331, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scheving LA, Tsai TH, Cornett LE, Feuers RJ, Scheving LE. Circadian variation of epidermal growth factor receptor in mouse liver. Anat Rec 224: 459–465, 1989. [DOI] [PubMed] [Google Scholar]
- 44.Scheving LA, Yeh YC, Tsai TH, Scheving LE. Circadian phase-dependent stimulatory effects of epidermal growth factor on deoxyribonucleic acid synthesis in the duodenum, jejunum, ileum, caecum, colon, and rectum of the adult male mouse. Endocrinology 106: 1498–1503, 1980. [DOI] [PubMed] [Google Scholar]
- 45.Scheving LA, Yeh YC, Tsai TH, Scheving LE. Circadian phase-dependent stimulatory effects of epidermal growth factor on deoxyribonucleic acid synthesis in the tongue, esophagus, and stomach of the adult male mouse. Endocrinology 105: 1475–1480, 1979. [DOI] [PubMed] [Google Scholar]
- 46.Scheving LA, Zhang L, Stevenson MC, Kwak ES, Russell WE. The emergence of ErbB2 expression in cultured rat hepatocytes correlates with enhanced and diversified EGF-mediated signaling. Am J Physiol Gastrointest Liver Physiol 291: G16–G25, 2006. [DOI] [PubMed] [Google Scholar]
- 47.Schmidt C, Bladt F, Goedecke S, Brinkmann V, Zschiesche W, Sharpe M, Gherardi E, Birchmeier C. Scatter factor/hepatocyte growth factor is essential for liver development. Nature 373: 699–702, 1995. [DOI] [PubMed] [Google Scholar]
- 48.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Skarpen E, Oksvold MP, Grosvik H, Widnes C, Huitfeldt HS. Altered regulation of EGF receptor signaling following a partial hepatectomy. J Cell Physiol 202: 707–716, 2005. [DOI] [PubMed] [Google Scholar]
- 50.St Hilaire RJ, Hradek GT, Jones AL. Hepatic sequestration and biliary secretion of epidermal growth factor: evidence for a high-capacity uptake system. Proc Natl Acad Sci USA 80: 3797–3801, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Steinkamp MP, Low-Nam ST, Yang S, Lidke KA, Lidke DS, Wilson BS. erbB3 is an active tyrosine kinase capable of homo- and heterointeractions. Mol Cell Biol 34: 965–977, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Strunk KE, Amann V, Threadgill DW. Phenotypic variation resulting from a deficiency of epidermal growth factor receptor in mice is caused by extensive genetic heterogeneity that can be genetically and molecularly partitioned. Genetics 167: 1821–1832, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Threadgill DW, Dlugosz AA, Hansen LA, Tennenbaum T, Lichti U, Yee D, LaMantia C, Mourton T, Herrup K, Harris RC,. Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science 269: 230–234, 1995. [DOI] [PubMed] [Google Scholar]
- 54.Vermeer PD, Einwalter LA, Moninger TO, Rokhlina T, Kern JA, Zabner J, Welsh MJ. Segregation of receptor and ligand regulates activation of epithelial growth factor receptor. Nature 422: 322–326, 2003. [DOI] [PubMed] [Google Scholar]