

Abstract
Elastin plays a pivotal role in lung development. We therefore queried if elastin haploinsufficient newborn mice (Eln+/−) would exhibit abnormal lung structure and function related to modified extracellular matrix (ECM) composition. Because mechanical ventilation (MV) has been linked to dysregulated elastic fiber formation in the newborn lung, we also asked if elastin haploinsufficiency would accentuate lung growth arrest seen after prolonged MV of neonatal mice. We studied 5-day-old wild-type (Eln+/+) and Eln+/− littermates at baseline and after MV with air for 8–24 h. Lungs of unventilated Eln+/− mice contained ∼50% less elastin and ∼100% more collagen-1 and lysyl oxidase compared with Eln+/+ pups. Eln+/− lungs contained fewer capillaries than Eln+/+ lungs, without discernible differences in alveolar structure. In response to MV, lung tropoelastin and elastase activity increased in Eln+/+ neonates, whereas tropoelastin decreased and elastase activity was unchanged in Eln+/− mice. Fibrillin-1 protein increased in lungs of both groups during MV, more in Eln+/− than in Eln+/+ pups. In both groups, MV caused capillary loss, with larger and fewer alveoli compared with unventilated controls. Respiratory system elastance, which was less in unventilated Eln+/− compared with Eln+/+ mice, was similar in both groups after MV. These results suggest that elastin haploinsufficiency adversely impacts pulmonary angiogenesis and that MV dysregulates elastic fiber integrity, with further loss of lung capillaries, lung growth arrest, and impaired respiratory function in both Eln+/+ and Eln+/− mice. Paucity of lung capillaries in Eln+/− newborns might help explain subsequent development of pulmonary hypertension previously reported in adult Eln+/− mice.
Keywords: lung growth and development, elastic fiber formation, extracellular matrix components, collagen, lysyl oxidase and fibrillins, pulmonary capillaries, lung cell apoptosis
elastin plays a prominent role in lung growth and development, providing a formative framework for future alveoli and pulmonary blood vessels. Mutant mice lacking the elastin gene die within a few days of birth from cardiorespiratory failure linked to smooth muscle overgrowth in systemic and pulmonary arteries, incomplete airway branching, and lack of alveolar septation (18, 39). These developmental defects in elastin-null mice can be attributed to the missing network of elastic fibers within the lung that helps to provide structural integrity and distensibility to the conducting airways while enabling expansion and contraction of alveoli, pulsation of blood vessels, and elastic recoil of the surrounding matrix. Mutant mice lacking one elastin allele (Eln+/−) typically survive to adulthood but are more susceptible to injury, in the form of severe emphysema when exposed to cigarette smoke (31), and impaired epithelial cell proliferation and restricted lung growth after pneumonectomy (13, 32).
With respect to the pulmonary circulation, elastin hemizygosity in humans and in mice is associated with vascular defects characterized by a compensatory increase in the number of rings of elastic lamellae and smooth muscle during arterial development, leading to prominent arterial thickening and increased risk of obstructive vascular disease (7, 19). Elastin-deficient mice, compared with wild-type mice, exhibited both systemic and pulmonary hypertension that was ascribed to decreased arterial elastin content and adaptive vascular remodeling during development, with pulmonary arteries that had smaller internal diameters, thinner walls, and increased numbers of elastic lamellae (8, 30, 38).
Extracellular matrix (ECM) remodeling, resulting in abundant, disordered lung elastin, is a prominent pathological feature of neonatal chronic lung disease, commonly known as bronchopulmonary dysplasia, that typically develops in premature infants and animals treated with lengthy assisted ventilation and increased oxygen (1, 5, 12, 22, 28, 36). We previously showed that prolonged mechanical ventilation (MV) of newborn mice with either air or 40% oxygen uncouples synthesis and assembly of lung elastin and leads to increased apoptosis, resulting in impaired alveolar formation and lung growth arrest (3, 25). Because elastin deficiency increases susceptibility to lung injury in adult mice (13, 31), we surmised that lengthy MV of elastin-deficient neonatal mice would accentuate their lung injury, as assessed by measurement of lung structure and function, when compared with lungs of mechanically ventilated elastin-sufficient mice. To test this hypothesis, 5-day-old wild-type (Eln+/+) and elastin haploinsufficient (Eln+/−) mice received MV with air for up to 24 h; spontaneously breathing littermates served as unventilated controls.
This study had two main objectives: first, to define the lung phenotype linked to elastin haploinsufficiency in neonatal mice, with particular attention to differences in ECM protein composition that may impact growth and development of the lung and its vasculature; and second, to assess lung molecular, structural, and functional responses to injury imposed by prolonged cyclic stretch in neonatal Eln+/+ vs. Eln+/− mice. Despite notable differences in lung ECM and vascular endothelial proteins of unventilated wild-type vs. mutant pups, these differences did not translate to discernible structural or functional differences in the lungs of Eln+/− pups when compared with those of Eln+/+ pups after lengthy MV.
MATERIALS AND METHODS
Transgenic Mice
Transgenic mice for breeding were provided by Dr. Dean Y. Li (University of Utah, Salt Lake City, UT). To obtain litters of newborn mice for these studies, we mated female C57BL/6J wild-type mice (Eln+/+) with male C57BL/6J transgenic mice bearing a heterozygous deletion of exon 1 in the elastin gene (Eln+/−) that had been backcrossed for more than five generations into the C57BL/6J background (8). Genotyping was performed on all mouse pups that were used in this study, with confirmation of mutant status by qRT-PCR for tropoelastin mRNA and immunoblot analysis for tropoelastin protein in lung tissue extracts. Eln+/+ littermates from the same crosses were used as controls for the Eln+/− pups in both the baseline (spontaneously breathing air) and intervention (MV with air) components of the study.
Experimental Design
We used full-term 5- to 6-day-old Eln+/+ (n = 23, body wt 4.1 ± 0.7 g) and Eln+/− (n = 20, body wt 4.0 ± 0.6 g) mice that spontaneously breathed air without MV to assess the impact of elastin haploinsufficiency on important molecular and structural features of the lung and its microvasculature. In tandem with these baseline studies, randomly selected 5- to 6-day-old Eln+/+ and Eln+/− littermates received MV with air at 90 breaths/min for 24 h to assess the effects of MV on key molecular and structural features of the lung and its microvasculature in Eln+/+ compared with Eln+/− neonatal mice. Additional 8-h studies were done to assess the effects of MV on lung elastase activity and elastance of the respiratory system in neonatal Eln+/+ compared with Eln+/− neonatal mice.
Mice selected for MV underwent a tracheotomy after intramuscular ketamine (∼60 μg/g body wt) and xylazine (∼12 μg/g body wt), followed by MV applied with a customized small animal respirator (MicroVent 848; Harvard Apparatus, Holliston, MA). Unventilated control pups were sedated with ketamine and xylazine for sham surgery (superficial neck incision) and then spontaneously breathed room air for the duration of the study. Table 1 shows descriptive data for the Eln+/+ and Eln+/− newborn mice and respiratory data from the 24-h MV studies.
Table 1.
Summary of respiratory data from the 24-h MV studies performed with Eln+/+ and Eln+/− newborn mice
| Group | n | Weight, g | TV, μl/g body wt | PIP, cmH2O | MAP, cmH2O | Lung Wt, mg/g body wt | Lung Volume, μl/g body wt |
|---|---|---|---|---|---|---|---|
| Eln+/+ | 13 | 4.0 ± 0.5 | 9.8 ± 0.6 | 20 ± 3 | 6 ± 1 | 16.0 ± 1.0 | 56 ± 10 |
| Eln+/− | 12 | 4.1 ± 0.6 | 9.8 ± 0.7 | 21 ± 3 | 6 ± 1 | 15.6 ± 1.2 | 60 ± 8 |
Values are means ± SD; n, no. of mice.
MV, mechanical ventilation; Eln+/+, wild type; Eln+/−, elastin haploinsufficient; TV, tidal volume; PIP, peak inspiratory pressure; MAP, mean airway pressure.
No statistically significant differences.
During MV, mice were kept in a neutral thermal environment and fed every 2 h via an orally placed gastric tube, initially with 10% glucose-0.25% saline solution, and later with an elemental amino acid-based infant formula (AA Lipil; Mead-Johnson, Evansville, IN), yielding a daily fluid intake of ∼100–120 μl/g body wt. A single intramuscular dose of ampicillin (200 μg/g body wt) and gentamicin (4 μg/g body wt) was given at the start of each study to reduce the risk of infection. Intramuscular injections of ketamine and xylazine (10 and 2 μg/g body wt, respectively) were repeated as needed for sedation during MV. Urine was obtained by suprapubic needle aspiration when the bladder became visibly distended, and 24-h urine collections were frozen for later measurement of desmosine and creatinine concentrations. At the end of each study, pups were killed with an intraperitoneal dose of ∼150 μg/g body wt pentobarbital sodium, and the lungs were excised for various studies as described below.
All surgical and animal care procedures and experimental protocols were reviewed and approved by the Stanford University Institutional Animal Care and Use Committee.
Functional Studies of The Respiratory System
To evaluate mechanical properties of the respiratory system in Eln+/+ compared with Eln+/− newborn mice, we used a modification of a previously described method for measuring lung mechanics in small rodents (24). Each pup had a tracheotomy that was connected to a small animal respirator (MicroVent 848; Harvard Apparatus) that delivered an initial tidal volume of 10 μl/g body wt at a frequency of 120 cycles/min, followed by placement in a whole body plethysmograph (model PLY3111; Buxco Electronics, Wilmington, NC). The respiratory flow signal was measured through a flow transducer connected to the plethysmograph. Tidal volume, determined from electrical integration of the flow signal, and airway pressures, measured from a pressure transducer connected to an airway side port, were monitored continuously to assess dynamic compliance and resistance of the respiratory system in real time. As soon as baseline measurements became stable, quasistatic respiratory system mechanics were assessed by stepwise increases in tidal volume from 10 to 45 μl/g body wt. For each incremental change in volume, a 60-s period elapsed to allow stabilization, after which measurements were recorded for an additional 30 s. A volume-pressure plot was generated from sequential measurements, and respiratory system compliance was calculated from the slope of the plot. Respiratory system elastance was expressed as the reciprocal of compliance (elastanceRS = 1/dynamic complianceRS). Measurements were performed in a set of Eln+/+ and Eln+/− pups for comparison at baseline (no prior MV) and in another set of Eln+/+ and Eln+/− pups immediately after 8 h of MV.
Postmortem Processing of Lungs for Quantitative Histology
The lungs were fixed intratracheally with 4% buffered paraformaldehyde overnight at 20 cmH2O, as described previously (4). On the day following initial fixation, the lungs were excised and their volumes were measured by fluid displacement (29), after which they were embedded in paraffin for isotropic uniform random sectioning, as previously described (25). Alveolar area was measured in randomly selected hematoxylin- and eosin-stained 4-μm sections using the Bioquant image analysis system (R&M Biometrics, Nashville, TN), and radial alveolar counts provided an index of alveolar number (6). Dual assessment of alveolar area and radial alveolar counts serves as a surrogate to judge the impact of MV in yielding lung growth arrest.
Elastin and Collagen-1 Distribution in Lung Parenchyma
The relative amount and distribution of insoluble elastin in lung was assessed by quantitative image analysis of Hart's-stained tissue sections using the Bioquant True Color Windows Image Analysis system (R&M Biometrics), as previously described (3). Localization and semiquantitative assessment of collagen-1 in distal lung was done by immunohistochemical staining of 4-μm-thick lung tissue sections. Antigen retrieval with citrate buffer was performed for 20 min at 95°C, followed by quenching of endogenous peroxidase activity using 0.3% H2O2 for 20 min. Sections were incubated overnight at 4°C with the primary antibody (polyclonal rabbit anti-mouse collagen-1, catalog no. 21286; Abcam, Cambridge, MA) at 1:1,000 dilution. Immune complexes were detected with an anti-rabbit avidin/biotin-3,3′-diaminobenzidine (DAB) detection system (no. PK6101, Vectastain ABC Kit; Vector Laboratories) and liquid DAB+ substrate chromagen system (no. K3467; Dako). Slides were counterstained with hematoxylin (no. H-3404; Vector Laboratories). Quantitative image analysis of stained/total lung parenchyma was performed using the Bioquant imaging system applied to ten ×400 fields in random tissue sections for each set of lungs. In addition, vessel-associated collagen-1 was assessed by determining the ratio of stained/total arterial wall area of arteries >100 μm in the random tissue sections.
The distribution and intensity of elastin deposition in sprouting secondary septa were further evaluated by immunofluorescence using confocal scanning microscopy. For these studies, terminal lung fixation included perfusion of the pulmonary circulation with ice-cold PBS injected through the right ventricle, followed by fixation with 10% formalin (Thermo Scientific, Waltham, MA) via the vasculature and trachea, which was ligated for overnight fixation. Tissue sections from 5-day-old Eln+/+ and Eln+/− mice were deparaffinized, rehydrated, and boiled in EDTA (0.25 mM) for epitope retrieval. Sections were treated with hydrogen peroxide to block endogenous peroxidase and then washed with PBS and immersed in 5% bovine serum albumin, followed by overnight incubation at 4°C with the primary elastin antibody (sc17580, 1:500; Santa Cruz). On the next day, sections were washed in PBS three times and incubated with AlexaFluor-488-tagged secondary antibody (no. A11055; Life Technologies, Carlsbad, CA) for 1 h at room temperature. Sections were mounted with Vectashield mounting media (Vector Laboratories) and viewed with an Olympus IX81 microscope using a FV1000 confocal scanning system to prepare images.
Serine Elastase Activity
Lung tissue was snap-frozen in liquid N2 and stored at 80°C for measurement of serine elastase activity by a previously described method (3, 5) that uses DQ-elastin substrate (no. E12056, EnzChek Elastase Assay Kit; Invitrogen, Camarillo, CA).
Lung Tissue Protein Extraction and Immunoblots
Protein extraction and immunoblot analysis were carried out on lung tissue samples, as previously described (5), to measure lung content of proteins linked to the ECM (tropoelastin, lysyl oxidase, fibrillin-1 and -2, collagen-1) and blood vessels [vascular endothelial (VE)-cadherin, CD-31], and to lung growth [vascular endothelial growth factor (VEGF)-A, platelet-derived growth factor (PDGF)-A, cleaved caspase-3]. With respect to the ECM, we specifically focused on those proteins that are known to play a major role in the synthesis and assembly of elastic fibers in the lung, namely tropoelastin, lysyl oxidase, and the fibrillins (3, 37), as well as collagen-1, the predominant collagen subtype found in the newborn mouse lung (34), and collagen-4, the most abundant collagen subtype in the basement membranes of alveoli and pulmonary blood vessels (27).
The following antibodies were applied for immunoblot protein measurements: tropoelastin, 1:1,000 dilution of a rabbit polyclonal antibody (kindly provided by Dr. Robert Mecham, Washington University, St. Louis, MO); lysyl oxidase, 1:800 dilution of a rabbit polyclonal antibody (no. ab6586; Abcam); fibrillin-1, 1:200 dilution of a rabbit polyclonal antibody (from Dr. Mecham); fibrillin-2, 1:300 dilution of a rabbit polyclonal antibody (from Dr. Mecham); collagen-1, 1:2,000 dilution of a rabbit polyclonal antibody (no. 21286; Abcam); VE-cadherin, 1:300 dilution of a rabbit polyclonal antibody (no. ab33168; Abcam); CD-31, 1:2,500 dilution of a rat anti-mouse monoclonal antibody (no. DIA310; Dianova, New Paltz, NY); VEGF-A, 1:200 dilution of a rat anti-mouse monoclonal antibody (no. MAB493; R&D, Minneapolis, MN); PDGF-A, 1:200 dilution of a mouse monoclonal antibody (no. sc9974, Santa Cruz Biotechnology, Santa Cruz, CA); and cleaved caspase-3, 1:700 dilution of a rabbit monoclonal antibody (no. 9664; Cell Signaling Technology, Danvers, MA).
Membranes with applied antibodies were incubated overnight at 4°C, washed three times in a solution containing 0.1% Tween 20 and Tris-buffered saline, and incubated at room temperature for 1 h with a dilute (1:5,000) solution of a secondary antibody (goat anti-rabbit IgG-horseradish peroxidase, no. sc2301; Santa Cruz) conjugated to horseradish peroxidase, followed by three washes. Membranes were stripped and reprobed with a 1:8,000 dilution of rabbit polyclonal anti-β-actin antibody (no. ab8227; Abcam) and incubated at room temperature for 1 h to provide an internal loading control. Images were detected by chemiluminescence (ECL or ECL+ Detection Kit; Amersham Life Science, Piscataway, NJ) and quantified by densitometry using a Gel Documentation System (Bio-Rad, Hercules, CA). All immunoblot analyses compared two sets of lung tissue protein extracts (Eln+/+ vs. Eln+/− or no MV vs. MV) run simultaneously on the same membrane.
Urinary Desmosine
Cumulative 24-h urine specimens were frozen for later radioimmunoassay measurements of desmosine and colorimetric assay for creatinine, as previously described (33).
Quantification of Pulmonary Arterioles and Capillaries
Lung tissue sections, prepared as described above, were processed for detection of the endothelial cell marker CD-31 by application of the primary antibody, rat anti-mouse CD-31 (1:100; Dianova). Immune complexes were visualized using the Vectastain Kit (Vector Laboratories, Burlingame, CA), with application of the relevant peroxidase-coupled secondary antibody. We used a modification of a previously described immunohistochemical and morphometric technique (2, 25) to assess the number of pulmonary arterioles (20–100 μm diameter) per 100 alveoli counted in ten ×200 microscopic fields of distal lung per animal. To quantify pulmonary microvessels, including capillaries, we used a similar approach to assess the number of CD-31-stained microvessels (<20 μm diameter) per 100 alveoli counted in 10 ×400 microscopic fields of distal lung per animal. For quantification of microvessels, including capillaries, we counted all vessels that displayed a lumen, whether their shape was circular or oval.
Quantification of Medial Smooth Muscle Thickness in Pulmonary Arterioles
Lung tissue sections, prepared as described above, were processed for detection of α-smooth muscle (SM) actin to assess the degree of muscularization of pulmonary arterioles as an index of vascular remodeling typically associated with pulmonary hypertension. Epitope retrieval was performed by boiling the sections in citrate buffer, pH 6.0 for 20 min, followed by blocking endogenous peroxidase with hydrogen peroxide, then washing and stabilization with Sea Block (Thermo Fisher Scientific, Rockford, IL). Sections were incubated with the primary antibody against α-SM actin (no. A2547, 1:500; Sigma-Aldrich, St. Louis, MO) for 20 min at room temperature. After streptavidin-biotin amplification (Dako, Carpenteria, CA), slides were incubated with DAB and counterstained with hematoxylin. All muscularized and partially muscularized (75% surrounding muscle) vessels in a section were assessed for circularity using perpendicular vessel diameters that intersected through the center of the vessel. Arterioles were included in the analysis provided that the difference in the length of the intersecting perpendicular lines was ≤10%. Noncircular vessels were not included in the analysis to avoid oblique planes of section that could introduce bias due to uneven smooth muscle profiles. Circular arterioles were assessed for smooth muscle thickness and vessel diameter at ×400 magnification using the Bioquant image analysis system. Smooth muscle thickness was measured at two opposite points of the vessel wall and was related to the vessel diameter as a percentage according to the formula, 2 × mean smooth muscle thickness/vessel diameter × 100 = %SM thickness. Image analysis was performed on all ×400 fields in two random tissue sections per animal.
Statistical Analysis
Data in the text, Table 1, and Figs. 1–10 are expressed as means ± SD. For comparison of single variable data between Eln+/+ and Eln+/− mice, or between unventilated controls and mechanically ventilated pups (either Eln+/+ or Eln+/−), we used Student's unpaired t-test to assess for significant differences or the nonparametric Mann-Whitney test for data sets bearing a nonnormal distribution. When more than two data sets were compared, we applied two-way ANOVA and the Bonferroni multiple-comparison test. Statistical analysis was done using Prism 4 software (GraphPad, San Diego, CA). Differences between groups were considered statistically significant if the P value was <0.05.
Fig. 1.

Compared with lungs of wild-type (Eln+/+) neonatal mice, lungs of elastin haploinsufficient newborn (Eln+/−) pups display, by immunoblot (A), a ∼50% reduction of tropoelastin protein content, by histochemical analysis of Hart's stained tissue sections (×400; arrows point to elastin at septal tips) (B), and an ∼50% reduction of elastin surface density (C). Summary data, means and SD; n = 5–6/group; *significant difference, P < 0.05. D: confocal microscopy of lung tissue sections (×400) prepared for immunofluorescence imaging of elastin (green) show thinner and less abundant elastic fibers at sites of sprouting secondary septa (arrows) in lungs of Eln+/− compared with Eln+/+ pups.
Fig. 10.

Immunoblots of VE-cadherin protein (A and B) showed a consistent decrease in the lungs of both Eln+/+ and Eln+/− mice after MV for 24 h. The pattern was similar for CD-31 protein (C and D), but the decrease was statistically significant only in lungs of Eln+/− pups. E: quantitative image analysis of lung tissue sections stained for CD-31 showed no difference in the number of arterioles (20–100 μm diameter)/100 alveoli counted in the lungs of Eln+/+ and Eln+/− mice after MV for 24 h. F: quantification of smaller microvessels (<20 μm diameter)/100 alveoli, which were mainly capillaries (expressed as capillary surface density), was less after MV in both Eln+/+ and Eln+/− pups compared with unventilated Eln+/+ and Eln+/− controls, as depicted in Fig. 3C. Summary data, means ± SD; *significant difference, P < 0.05. For E and F, n = 6/group; §significant difference, MV vs. no MV (Fig. 3C), P < 0.05.
RESULTS
Baseline Conditions (No MV)
ECM proteins in lung.
Lungs of Eln+/− neonatal mice, when compared with those of Eln+/+ pups, displayed a 50% reduction in tropoelastin protein, as expected (Fig. 1, A–C). Elastin was especially prominent at sites of sprouting secondary septa in Eln+/+ pups, in marked contrast to the attenuated elastic fibers noted in secondary septa of Eln+/− pups (Fig. 1D). Diminished lung elastin was associated with a twofold increase in collagen-1 (Fig. 2A) and lysyl oxidase (Fig. 2B) proteins. Lung abundance of fibrillin-1 protein was similar in wild-type and mutant mice (Fig. 2C), whereas fibrillin-2 protein was ∼30% less in the Eln+/− pups (Fig. 2D). Collagen-1 was distributed in the walls of distal alveoli and was also prominent around lung blood vessels, especially in the Eln+/− mice (Fig. 2E). Lung content of other ECM proteins, namely collagen-4, lysyl oxidase like-1, fibulin-5, emilin-1, and microfibril-associated glycoprotein, was not significantly different in Eln+/+ compared with Eln+/− pups (data not shown).
Fig. 2.

Immunoblot analysis of extracellular matrix (ECM) proteins in lungs of Eln+/+ and Eln+/− neonatal mice, showing increased abundance of collagen-1 (A) and lysyl oxidase proteins in Eln+/− (B) pups; similar amounts of fibrillin-1 protein in Eln+/+ and Eln+/− pups (C); and less fibrillin-2 protein in Eln+/− pups (D), compared with Eln+/+ pups. Summary data; means and SD; *significant difference, P < 0.05. E: immunohistochemical localization of collagen-1 (arrows, brown stain) in lung sections of Eln+/+ and Eln+/− pups, showing prominent collagen-1 around microvessels, especially notable in the Eln+/− lung (panel on right and inset). Lungs: ×400 magnification; insets: ×680 magnification; scale bar = 50 μm.
Lung microvessels and alveoli.
Immunoblot analysis for VE-cadherin showed significantly less VE-cadherin protein in lungs of Eln+/− compared with Eln+/+ neonatal mice (Fig. 3A). Whereas the number of pulmonary arterioles (20–100 μm diameter)/100 alveoli was similar between Eln+/+ and Eln+/− mice (Fig. 3B), Eln+/− pups had significantly fewer microvessels, including lung capillaries (<20/100 μm)/alveoli than did Eln+/+ pups (Fig. 3, C and D). This prompted us to determine if the modest (<10%) difference in the number of lung microvessels could have initiated vascular remodeling reflective of increased pulmonary arterial pressure in the Eln+/− mice. Medial smooth muscle thickness, however, was less in the pulmonary arterioles of Eln+/− pups than it was in Eln+/+ littermates (Fig. 3E), indicating that pulmonary arterial hypertension (PAH), as reported in adult Eln+/− mice (30), appears to evolve developmentally beyond the newborn period. Despite the apparent differences in lung microvasculature of wild-type vs. mutant mice, morphometric analysis showed no significant structural differences between Eln+/+ and Eln+/− pups with respect to alveolar area (Fig. 4, A and B) and number (Fig. 4C).
Fig. 3.

A–C: immunoblots showing decreased vascular endothelial (VE)-cadherin protein in lungs of Eln+/− compared with Eln+/+ pups (A), quantitative assessment of pulmonary arterioles (20–100 μm diameter) (B) and smaller microvessels (<20 μm diameter) (C), predominantly capillaries (<10 μm diameter), showing less capillary surface density/100 alveoli in lungs of Eln+/− compared with Eln+/+ pups. Summary data, means and SD; n = 6/group; *significant difference, P < 0.05. D: images of Eln+/+ and Eln+/− lung sections (×400; scale bars 20 μm) stained with CD-31 antibody (brown), denoting discernible pulmonary capillaries (arrows). E: Eln+/+ and Eln+/− lung sections stained with α-smooth muscle (SM) actin antibody showed less medial smooth muscle thickness, expressed as a percent of vessel diameter, in pulmonary arterioles (<40 and 40–80 μm diameter) of Eln+/− compared with Eln+/+ newborn mice. Summary data, means and SD; n = 135 for Eln+/+ <40-μm arterioles, n = 17 for Eln+/+ 40- to 80-μm arterioles, n = 136 for Eln+/− <40-μm arterioles, n = 44 for Eln+/− 40- to 80-μm arterioles; *significant difference, P < 0.05.
Fig. 4.

A: representative lung tissue sections (×100; scale bar = 100 μm) showing similar alveolar structure in Eln+/+ and Eln+/− neonatal mice. B: summary data (mean and SD) for alveolar area, assessed by quantitative image analysis of lung tissue sections from Eln+/+ and Eln+/− pups, showing no difference in alveolar size. C: summary data (means and SD) for radial alveolar counts, reflecting similar numbers of alveoli in lungs of Eln+/+ and Eln+/− mice; n = 5/group.
Functional assessment of respiratory system elastance.
Dynamic respiratory system elastance, expressed as Δpressure/Δvolume relative to body weight, was significantly less in Eln+/− (1.14 ± 0.12 cmH2O·μl−1·g body wt−1) than in Eln+/+ (1.44 ± 0.13 cmH2O·μl−1·g body wt−1) mice (Fig. 5). This diminished elastance is the expected functional correlate of the decreased elastic fiber deposition noted in the secondary septa and alveolar walls of the Eln+/− lungs (Fig. 1, B and C).
Fig. 5.
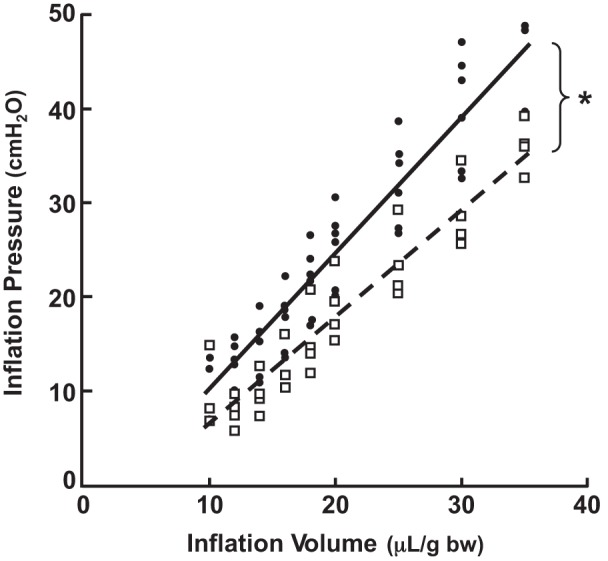
Respiratory system elastance (elastanceRS) is less in Eln+/− than in Eln+/+ neonatal mice. Shown are in vivo measurements of lung inflation pressures in response to serial increases of tidal volume (μl/g body wt) delivered via tracheotomy in 6-day-old Eln+/+ (●) and Eln+/− (□) pups. Oblique lines are linear regression plots of elastanceRS [Δpressure (P)/Δvolume (V) = 1/complianceRS] for Eln+/+ mice (solid line, n = 6, R2 = 0.899) and for Eln+/− mice (broken line, n = 4, R2 = 0.900). ElastanceRS: Eln+/+ 1.44 ± 0.13; Eln+/− 1.14 ± 0.12 cmH2O·μl−1·g body wt−1. *Significant difference, Eln+/+ vs. Eln+/− groups, P < 0.01.
Effects of MV
ECM proteins in lung.
MV for 24 h caused a significant increase of tropoelastin protein in lungs of Eln+/+ mice (Fig. 6A). This increased production of tropoelastin was accompanied by a significant increase of serine elastase activity in the lungs of the wild-type pups (Fig. 7A), which in turn was associated with a significant increase of urinary desmosine (Fig. 7C), a biomarker of elastin degradation. In contrast, MV yielded a 50% reduction of tropoelastin protein in the lungs of Eln+/− pups (Fig. 6B), without a significant change of either lung elastase activity (Fig. 7B) or urinary desmosine excretion (Fig. 7D). Consistent with previous observations (3, 25), MV caused redistribution of elastic fibers from septal tips (Fig. 1B) to alveolar walls (Fig. 7E), with less elastin surface density in the lungs of Eln+/− pups compared with Eln+/+ pups (Fig. 7F).
Fig. 6.
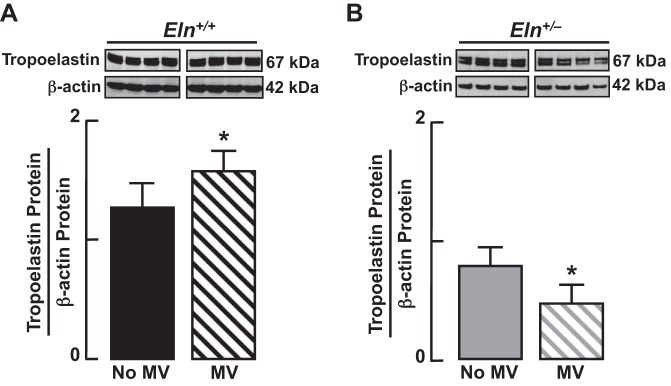
Immunoblots showing increased lung abundance of tropoelastin protein in response to mechanical ventilation (MV) of Eln+/+ mice for 24 h (A) and decreased lung abundance of tropoelastin protein after 24 h of MV of Eln+/− mice (B). Summary data, means ± SD; *significant difference, P < 0.05.
Fig. 7.

Serine elastase activity increased in the lungs of Eln+/+ mice (A), but did not change in Eln+/− mice (B) after 8 h of MV. Urinary desmosine excretion increased in Eln+/+ pups (C) but did not change in Eln+/− pups (D) after MV for 24 h. E: Hart's-stained lung tissue sections (×400; scale bars = 100 μm) showing dispersion of elastic fibers (arrows, brown stain) in alveolar walls of both Eln+/+ and Eln+/− mice after 24 h of MV. F: while the distribution pattern of elastin was similar in the lungs of Eln+/+ and Eln+/− pups after MV, quantitative image analysis of lung tissue sections showed less elastin surface density in Eln+/− mice than in Eln+/+ mice. Summary data, means ± SD; n = 4/group; *significant difference, P < 0.05.
MV for 24 h yielded no significant change of collagen-1 protein abundance in lungs of either Eln+/+ or Eln+/− mice (Fig. 8, A and B); there was a trend toward greater lung content of collagen-1 in Eln+/− than in Eln+/+ mice after MV, but this difference was not statistically significant (P > 0.05). Likewise, there was no significant change in lung content of lysyl oxidase protein in response to MV in either Eln+/+ or Eln+/− pups, although there was a downward trend in both groups (Fig. 8, C and D). Fibrillin-1 protein, however, increased in the lungs of both Eln+/+ and Eln+/− mice after 24 h of MV, with a significantly greater increase observed in the lungs of mutants compared with wild-type pups (Fig. 8, E and F). For fibrillin-2, the patterns of response to MV differed between the two groups: lung content of fibrillin-2 decreased with MV in Eln+/+ mice, whereas it increased in the Eln+/− pups (Fig. 8, G and H).
Fig. 8.

Immunoblots of ECM proteins in lungs of Eln+/+ and Eln+/− mice, comparing unventilated control pups with those that received MV for 24 h. Lung content of collagen-1 (A and B) and lysyl oxidase (C and D) did not change significantly after MV in either Eln+/+ or Eln+/− mice, although both proteins trended down in wild-type pups in response to MV. In contrast, lung content of fibrillin-1 increased significantly with MV in both Eln+/+ (E) and Eln+/− (F) mice, most notably in Eln+/− pups. For fibrillin-2, the response to MV differed between Eln+/+ and Eln+/− mice: lung content of fibrillin-2 decreased in Eln+/+ (G) but increased in Eln+/− mice (H) after MV for 24 h. Summary data, means ± SD; *significant difference vs. no MV, P < 0.05; §significant difference vs. Eln+/+ after MV for 24 h, P < 0.05.
Growth factors related to apoptosis and failed alveolar septation.
Because prolonged MV has been reported to disrupt VEGF and PDGF signaling in lungs of newborn mice, which is linked to increased apoptosis and lung growth arrest (4, 25), we measured lung abundance of VEGF-A (Fig. 9, A and B) and PDGF-A (Fig. 9, C and D) proteins and found that these critical growth factors were decreased in both Eln+/+ and Eln+/− mice after MV for 24 h. Consistent with previous reports, these changes in VEGF-A and PDGF-A protein abundance were associated with a 5- to 10-fold increase of cleaved caspase-3 protein in the lungs of wild-type and mutant pups (Fig. 9, E and F), reflecting increased apoptosis in both groups.
Fig. 9.

Immunoblots of growth factors known to impact formation of alveoli and lung capillaries showed similar effects of MV in lungs of Eln+/+ and Eln+/− mice. Lung content of vascular endothelial growth factor (VEGF, A and B) and platelet-derived growth factor (PDGF, C and D) proteins decreased, compared with unventilated controls, during MV of both Eln+/+ and Eln+/− mice. These changes were associated with a 5- to 10-fold increase of cleaved caspase-3 protein (E and F), indicative of MV-induced lung cell apoptosis. Summary data, means ± SD; *significant difference vs. no MV, P < 0.05.
Lung microvessels and alveoli.
MV reduced lung abundance of the endothelial markers VE-cadherin (Fig. 10, A and B) and CD-31 (Fig. 10, C and D) proteins to a similar degree in Eln+/+ and Eln+/− mice. Quantitative image analysis of pulmonary arterioles (20–100 μm diameter) and microvessels (<20 μm), which were predominantly capillaries, showed no significant differences between Eln+/+ and Eln+/− mice after 24 h of MV (Fig. 10, E and F). For both groups of mice, however, the number of lung microvessels, including capillaries, expressed per 100 alveoli was significantly less after MV than it was in unventilated pups (Fig. 3C), indicating that MV leads to capillary loss in both wild-type and mutant newborn mice. Likewise, MV of both Eln+/+ and Eln+/− pups, compared with their respective unventilated controls, yielded similar lung structural changes (Fig. 11A), shown as increased alveolar size (Fig. 11B) and diminished alveolar number (radial alveolar counts; Fig. 11C).
Fig. 11.

MV inhibits alveolar formation in lungs of both Eln+/+ and Eln+/− mice. A: lung sections from 6-day-old mice showing increased alveolar size and reduced alveolar septation after MV with air for 24 h compared with lung sections obtained from unventilated wild-type and mutant control pups (×100 magnification; scale bars = 100 μm). Measured lung volumes were similar between groups. Summary data (means ± SD) are shown for alveolar size (B) and alveolar number (C), assessed by quantitative image analysis of 12 random fields (×200) in 4 lung sections/animal. After 24 h of MV, lungs of both Eln+/+ and Eln+/− mice showed a 2- to 3-fold increase in alveolar area (B), and a ∼30% reduction in alveolar number (C), as determined by radial alveolar counts, compared with lungs of unventilated control pups; n = 5–6/group; *significant difference, MV vs. no MV, P < 0.05.
Functional assessment of respiratory system elastance.
In response to MV, dynamic respiratory system elastance, expressed as Δpressure/Δvolume relative to body weight, was similar in Eln+/− and Eln+/+ mice (Fig. 12; elastanceRS: Eln+/− 1.40 ± 0.52, Eln+/+ 1.24 ± 0.40 cmH2O·μl−1·g body wt−1). Whereas there was no apparent change of elastanceRS in the wild-type pups after 8 h of MV, the increased elastanceRS observed in mechanically ventilated compared with unventilated Eln+/− pups (Fig. 5) is indicative of increased lung stiffness associated with MV, apparently accentuated by elastin deficiency and elastic fiber redistribution from the septal tips to the alveolar walls.
Fig. 12.
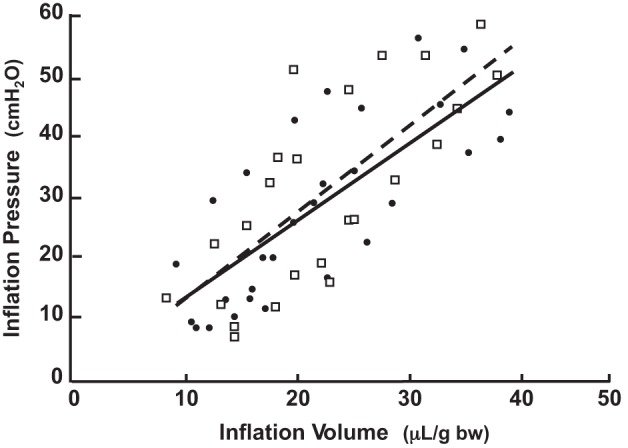
Respiratory system elastance is similar in Eln+/+ and Eln+/− neonatal mice after MV for 8 h. Shown are in vivo measurements of lung inflation pressures in response to serial increases of tidal volume (μl/g body wt) delivered via tracheotomy in 6-day-old Eln+/+(●) and Eln+/− (□) pups that had been exposed to MV. Oblique lines are linear regression plots of respiratory system elastance (ΔP/ΔV = 1/complianceRS) for Eln+/+ mice (solid line, n = 5, R2 = 0.556) and for Eln+/− mice (broken line, n = 4, R2 = 0.576). ElastanceRS, Eln+/+ 1.24 ± 0.40; Eln+/− 1.40 ± 0.52 cmH2O·μl−1·g body wt−1. No significant difference between groups.
DISCUSSION
The first aim of this study was to define the newborn lung phenotype associated with elastin haploinsufficiency, focusing on differences in ECM composition and its impact on lung structure and function, and on the pulmonary microvasculature of neonatal mice. Here we show that mutant pups lacking a single allele of the elastin gene exhibit a twofold increase in lung content of collagen-1 and lysyl oxidase proteins compared with wild-type pups, with no detectable difference in alveolar size or number. As expected, assessment of respiratory system elastance was significantly less in elastin-deficient newborns than it was in the wild-type pups, suggesting that the 50% reduction of lung elastin coupled with the twofold increase in collagen-1 yielded significant impairment of respiratory function in the Eln+/− pups.
Because lung microvessels are considered a central link to alveolar formation and lung growth, we were surprised to find that the number of pulmonary microvessels, including capillaries, was significantly lower in Eln+/− than in Eln+/+ mice, whereas alveolar size and number were similar, irrespective of differences in lung elastin. This raises the interesting notion that differences in the mechanical properties of the lung, related to elastin deficiency, and perhaps to increased collagen-1, adversely impact angiogenesis while sparing alveolar formation in the developing lung. Our finding of similar alveolar structure in Eln+/+ and Eln+/− neonatal mice is consistent with previous observations of Shifren et al. (31), who reported identical alveolar size in 3-mo-old mice with elastin haploinsufficiency compared with wild-type mice of the same age. Although lung content of collagen-1 was not measured in these adult mice, lung content of total collagen, assessed by measurement of hydroxyproline per gram lung weight, was similar in the mutant and wild-type animals. In a subsequent study (30), the same investigators showed that adult mice with elastin deficiency had significantly increased right ventricular pressures and right ventricular hypertrophy, with remodeling of their lung vasculature. Pulmonary arterioles of the Eln+/− mice showed increased medial smooth muscle, and there were fewer 15- to 50-μm arterioles noted in the lungs of the mutants compared with the wild-type adult mice. Interestingly, the impact of reduced numbers of pulmonary arterioles as a possible contributor to PAH was not addressed in that report. Our finding of fewer lung microvessels, including capillaries, in Eln+/− neonatal pups supports speculation that defective angiogenesis linked to reduced elastin may contribute to the development of PAH reported in adult mice with elastin haploinsufficiency (8, 30, 38).
The prominent role of the ECM in regulating formation of alveoli and microvessels in the developing lung is well recognized (14, 23). A number of recent reports have highlighted the importance of ECM protein composition and associated tissue stiffness in regulating postnatal development of the lung and its vasculature (20, 21, 26, 40). Our finding of reduced lung elastance associated with diminished numbers of pulmonary microvessels in neonatal mice bearing a single allele of the elastin gene is consistent with the previous report that mechanical signals conveyed by variations in ECM elasticity can impact VEGF signaling and thereby alter vascular development (20).
The second aim of this study was to assess lung molecular, structural, and functional responses to injury imposed by prolonged cyclic stretch in neonatal Eln+/+ vs. Eln+/− mice. In earlier studies we showed that MV of newborn mice for 24 h uncoupled synthesis and assembly of elastin and increased lung cell apoptosis, resulting in impaired alveolarization and lung growth arrest (3). Based on prior reports that Eln+/− mice, compared with wild-type controls, acquired more severe emphysema when exposed to cigarette smoke (31), and exhibited abnormal lung growth in response to partial pneumonectomy (32), we surmised that MV would inhibit lung growth and impair lung function to a greater degree in Eln+/− neonatal mice than in their Eln+/+ littermates. We therefore were surprised to discover that MV yielded a similar increase of alveolar size and decrease of alveolar number, indicative of lung growth arrest, and similar evidence of respiratory system malfunction in Eln+/+ and Eln+/− pups. What might account for this unexpected outcome?
To address this question, we examined the impact of MV on various components of the ECM and on growth factors that are known to regulate alveolar septation and lung growth during postnatal development. Here we focused particularly on elastin, which together with microfibrils constitutes the major component of mature elastic fibers (37). Because microfibrils play an important role in elastic fiber assembly, we also examined the effects of MV on the amount and distribution of fibrillins, which are the major structural elements of microfibrils.
We first discovered that, in response to MV for 24 h, lung content of tropoelastin, the monomeric secreted form of elastin, increased in Eln+/+ pups but decreased in their Eln+/− littermates. We wondered if this might reflect a difference in lung elastase activity that has been linked to MV of newborn mice (10, 11). We indeed found that MV increased serine elastase activity in the lungs of Eln+/+ mice but not in the Eln+/− mutants. This result might be attributed to more elastin substrate in the Eln+/+ mice, allowing for greater degradation of lung elastin, which in turn can trigger synthesis of new elastin. There was little or no inflammation noted in the lungs of either Eln+/+ or Eln+/− mice (data not shown), which might account for differences in elastase activity elicited by MV. It is possible, however, that inherent or evoked differences of ECM composition in the Eln+/− lungs somehow prevent activation of elastase and resultant elastin degradation, thereby contributing to the increased respiratory system elastance observed in the mutant lungs exposed to MV.
In response to MV, the wild-type pups exhibited increased tropoelastin production without a corresponding increase of lysyl oxidases or fibrillin-2, ECM proteins that play an important role in elastin assembly. The resultant uncoupling of elastin synthesis and assembly during MV leads to the production of poorly formed elastic fibers that are ill-suited to serve as an effective scaffold for normal alveolarization (3, 5), thereby causing lung growth arrest. Eln+/− mice, with accentuated elastin deficiency in response to MV, likewise exhibited impaired lung growth and dysfunction, similar to that seen in Eln+/+ mice.
MV led to increased lung content of both fibrillin-1 and fibrillin-2 in Eln+/− mice such that both proteins were significantly more abundant in the lungs of mutants compared with wild-type pups after 24 h of MV. Fibrillins provide a critical scaffold to which secreted elastin binds to form the structural framework of elastic fibers that enable expansion and contraction of alveoli, pulsation of blood vessels, and elastic recoil of the surrounding matrix. Because fibrillins are the predominant protein constituent of microfibrils, their upregulation in mechanically ventilated Eln+/− mice may help compensate for the reduced abundance of elastin noted in the lungs of mutant pups after MV, and thereby account for the similarities in lung structure and function observed in mechanically ventilated Eln+/+ and Eln+/− pups. Increased abundance of fibrillins, however, without a corresponding increase of tropoelastin and lysyl oxidase in the lungs of Eln+/− pups, might help explain the increased respiratory system elastance (i.e., reduced compliance) seen in response to MV of the mutant pups. Fibrillins normally are coexpressed with elastin to form a competent network of elastic fibers in the lung, a linkage that likely is disrupted by the further reduction of tropoelastin exhibited in Eln+/− pups, but not in Eln+/+ littermates, exposed to lengthy MV.
VEGF-A and PDGF-A are growth factors that play a critical role in regulating alveolar septation and postnatal lung growth. As in previous studies of mechanically ventilated newborn mice (4) and preterm lambs with CLD (5), we found that lung abundance of VEGF-A and PDGF-A was reduced in both wild-type and elastin-deficient pups after MV for 24 h. In the earlier studies, the newborn animals received MV with O2-rich gas, whereas in this study MV was applied with air, suggesting that mechanical force, rather than increased oxygen, is responsible for suppressing these two key growth factors, irrespective of lung elastin content.
Our findings of a >10-fold increase in apoptosis associated with MV is consistent with previous studies showing increased apoptosis of cultured fetal and adult rat lung epithelial cells that were subjected to mechanical stretch for up to 24 h (9, 17). Likewise, previous studies of newborn mice that received MV with air for 24 h displayed a >10-fold increase in apoptosis, sufficient to account for significant inhibition of lung growth and loss of lung microvessels that was attributed to prolonged cyclic stretch of the developing lung, associated with activation of TGF-β (25). A recent study showed that MV of newborn rats for 24 h increased apoptosis of distal lung epithelial cells, which was attributed to activation of the extrinsic FasL/Fas pathway, resulting in impaired alveolar formation (16). Here we show that lengthy MV of newborn mice, irrespective of lung elastin content, also induces apoptosis of endothelial cells, resulting in loss of pulmonary microvessels, a finding that has been linked to defective alveolar septation and resultant lung growth arrest (15, 35).
In contrast to other animal models of neonatal lung injury, this model exhibits little or no inflammatory response to MV, perhaps related to the use of air, rather than extra oxygen, and the relatively modest inflation pressures and tidal volumes that are applied for no more than 24 h. As such, this model offers the advantage of demonstrating the specific effects of moderate cyclic stretch on lung growth and development, without the confounding influence of extreme overinflation, or increased inspired oxygen, or infection, all of which may contribute to inflammation and likely account for much of the apoptosis and lung injury observed in other models, notably those that have applied MV to premature lambs for periods of up to 3 wk (1, 2, 5, 10, 11, 25). This is not intended to be a model of neonatal chronic lung disease, but it provides a useful approach by which to tease out the specific effects of prolonged cyclic stretch on the lungs during early postnatal development.
Figure 13 illustrates a proposed model of how MV impacts the lung matrix and microvasculature of Eln+/+ and Eln+/− mice. Under basal conditions, elastic fibers are thinner and reduced in number, collagen-1 is greater, and pulmonary capillaries are fewer in lungs of mutant than of wild-type pups. In response to prolonged MV, lung elastase activity increases and causes degradation of matrix elastin, with an associated increase of fragmented elastic fibers in the lungs of Eln+/+ mice. In contrast, lungs of Eln+/− pups exhibit decreased elastase activity after prolonged MV, with further attenuated elastic fibers bearing increased fibrillin, which is the major constituent of microfibrils in the ECM. Lengthy MV of both Eln+/+ and Eln+/− pups leads to diminished lung abundance of VEGF and PDGF, key growth factors that play a central role in the formation of alveoli and pulmonary microvessels. These changes are linked to increased apoptosis, resulting in defective alveolar septation and loss of lung capillaries, manifest as abnormal lung structure and function.
Fig. 13.

A working model depicting how MV leads to impaired formation of alveoli and lung capillaries and evokes ECM remodeling in Eln+/− and Eln+/+ newborn mice. Under basal conditions, the lungs of Eln+/− pups, compared with Eln+/+ littermates, bear attenuated elastic fibers and increased collagen-1 that is especially notable around small blood vessels, which feed a reduced number of pulmonary capillaries. Prolonged MV elicits increased lung elastase activity and thereby increases elastin degradation, leading to fragmented elastic fibers in Eln+/+ pups. In contrast, MV causes no significant increase of lung elastase activity in Eln+/− mice, but elastin synthesis is further reduced, whereas fibrillin production is increased in response to MV. Lung abundance of two key growth factors, VEGF and PDGF, is decreased in both Eln+/+ and Eln+/− mice after lengthy MV, and these changes are associated with increased apoptosis that has been linked to prolonged cyclic stretch of epithelial and endothelial cells, as well as lung fibroblasts. Taken together, the adverse effects of lengthy MV yield reduced numbers of lung capillaries, fewer alveoli, and remodeling of the ECM, resulting in diminished lung growth and abnormal lung function.
Taken together, the results of this study help to validate the critical role of the ECM, and more specifically its elastic fiber composition, in regulating development of the pulmonary capillary network, which in turn may contribute to the pulmonary hypertension that has been reported in mice with elastin haploinsufficiency (8, 30, 38). Moreover, the study underscores the importance of other ECM components, namely collagen and fibrillins, in maintaining the structural and functional integrity of the lungs under baseline conditions and in response to the stress of prolonged cyclic stretch when elastin is deficient. The challenge now is to better understand the specific cellular signaling cues and molecular mechanisms by which the ECM directs alveolarization and angiogenesis in the lung during development and during repair after injury.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute (NHLBI) Grants HL-086631 (R. Bland) and P01-HL-108797 (M. Rabinovitch/R. Bland); fellowship awards from NHLBI (HL-086216 to K. Parai) and Deutsche Forschungsgemeinschaft [1315 (A. Hildendorff), 1425 (S. Preuss), and 1636 (M. Alejandre)]; and the Vera Moulton Wall Center for Pulmonary Vascular Disease at Stanford University.
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: A.H., K.P., R.E., E.F.N., N.J., F.C., J.P., L.M.M., M.A.A.-A., S.K., J.M., N.F.-T., S.M., B.C.S., and R.D.B. performed experiments; A.H., K.P., R.E., J.P., C.M., S.P., M.A.A.-A., S.K., J.M., N.F.-T., S.M., B.C.S., and R.D.B. analyzed data; A.H., R.E., C.M., J.M., N.F.-T., S.M., M.R., and R.D.B. interpreted results of experiments; A.H., R.E., M.A.A.-A., and R.D.B. prepared figures; A.H. and R.D.B. drafted manuscript; A.H., R.E., E.F.N., N.J., F.C., J.P., L.M.M., C.M., S.P., M.A.A.-A., S.K., J.M., N.F.-T., S.M., B.C.S., M.R., and R.D.B. approved final version of manuscript; M.R. and R.D.B. edited and revised manuscript; R.D.B. conception and design of research.
ACKNOWLEDGMENTS
We thank Dr. Dean Li at the University of Utah in Salt Lake City for generosity in providing elastin haploinsufficient mice that were bred to produce the litters of Eln+/− newborn pups that were used for this study. We also thank Dr. Robert Mecham at Washington University in St. Louis for providing several high-quality antibodies that were applied in this study. Last, we thank Michelle Fox and Dr. Michal Roof for assistance in preparing the manuscript.
REFERENCES
- 1.Albertine KH, Kim BI, Kullama LK, Starcher BC, Cho SC, Carlton DP, Bland RD. Chronic lung injury in preterm lambs. Disordered respiratory tract development. Am J Respir Crit Care Med 159: 945–958, 1999. [DOI] [PubMed] [Google Scholar]
- 2.Bland RD, Albertine KH, Carlton DP, Kullama LK, Davis PL, Cho SC, Kim B, Dahl M, Tabatabaei N. Chronic lung injury in preterm lambs: Abnormalities of the pulmonary circulation and lung fluid balance. Pediatr Res 48: 64–74, 2000. [DOI] [PubMed] [Google Scholar]
- 3.Bland RD, Ertsey R, Mokres LM, Xu L, Jacobson BE, Jiang S, Alvira CM, Rabinovitch M, Shinwell ES, Dixit A. Mechanical ventilation uncouples synthesis and assembly of elastin and increases apoptosis in lungs of newborn mice. Am J Physiol Lung Cell Mol Physiol 294: L3–L14, 2008. [DOI] [PubMed] [Google Scholar]
- 4.Bland RD, Mokres LM, Ertsey R, Jacobson BE, Jiang S, Rabinovitch M, Xu L, Shinwell ES, Zhang F, Beasley MA. Mechanical ventilation with 40% oxygen reduces pulmonary expression of genes that regulate lung development and impairs alveolar septation in newborn mice. Am J Physiol Lung Cell Mol Physiol 293: L1099–L1110, 2007. [DOI] [PubMed] [Google Scholar]
- 5.Bland RD, Xu L, Ertsey R, Rabinovitch M, Albertine KH, Wynn KA, Kumar VH, Ryan RM, Swartz DD, Csiszar K, Fong KSK. Dysregulation of pulmonary elastin synthesis and assembly in preterm lambs with chronic lung disease. Am J Physiol Lung Cell Mol Physiol 292: L1370–L1384, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Emery JL, Mithal A. The number of alveoli in the terminal respiratory unit of man during late intrauterine life and childhood. Arch Dis Child 35: 544–547, 1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ewart AK, Morris CA, Atkinson D, Jin W, Sternes K, Spallone P, Stock AD, Leppert M, Keating MT. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat Genet 5: 11–16, 1993. [DOI] [PubMed] [Google Scholar]
- 8.Faury G, Pezet M, Knutsen RH, Boyle WA, Heximer SP, McLean SE, Minkes RK, Blumer KJ, Kovacs A, Kelly DP, Li DY, Starcher B, Mecham RP. Developmental adaptation of the mouse cardiovascular system to elastin haploinsufficiency. J Clin Invest 112: 1419–1428, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hammerschmidt S, Kuhn H, Grasenack T, Gessner C, Wirtz H. Apoptosis and necrosis induced by cyclic mechanical stretching in alveolar type II cells. Am J Respir Cell Mol Biol 30: 396–402, 2004. [DOI] [PubMed] [Google Scholar]
- 10.Hilgendorff A, Parai K, Ertsey R, Jain N, Navarro EF, Peterson JL, Tamosiuniene R, Nicolls MR, Starcher BC, Rabinovitch M, Bland RD. Inhibiting lung elastase activity enables lung growth in mechanically ventilated newborn mice. Am J Respir Crit Care Med 184: 537–546, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hilgendorff A, Parai K, Ertsey R, Juliana Rey-Parra G, Thebaud B, Tamosiuniene R, Jain N, Navarro EF, Starcher BC, Nicolls MR, Rabinovitch M, Bland RD. Neonatal mice genetically modified to express the elastase inhibitor elafin are protected against the adverse effects of mechanical ventilation on lung growth. Am J Physiol Lung Cell Mol Physiol 303: L215–L227, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hislop AA, Wigglesworth JS, Desai R, Aber V. The effects of preterm delivery and mechanical ventilation on human lung growth. Early Hum Dev 15: 147–164, 1987. [DOI] [PubMed] [Google Scholar]
- 13.Hoffman AM, Shifren A, Mazan MR, Gruntman AM, Lascola KM, Nolen-Walston RD, Kim CF, Tsai L, Pierce RA, Mecham RP, Ingenito EP. Matrix modulation of compensatory lung regrowth and progenitor cell proliferation in mice. Am J Physiol Lung Cell Mol Physiol 298: L158–L168, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ingber DE, Folkman J. How does extracellular matrix control capillary morphogenesis? Cell 58: 803–805, 1989. [DOI] [PubMed] [Google Scholar]
- 15.Jakkula M, Le Cras TD, Gebb S, Hirth KP, Tuder RM, Voelkel NF, Abman SH. Inhibition of angiogenesis decreases alveolarization in the developing rat lung. Am J Physiol Lung Cell Mol Physiol 279: L600–L607, 2000. [DOI] [PubMed] [Google Scholar]
- 16.Kroon AA, Delriccio V, Tseu I, Kavanagh BP, Post M. Mechanical ventilation-induced apoptosis in newborn rat lung is mediated via FasL/Fas pathway. Am J Physiol Lung Cell Mol Physiol 305: L795–L804, 2013. [DOI] [PubMed] [Google Scholar]
- 17.Lee HS, Wang Y, Maciejewski BS, Esho K, Fulton C, Sharma S, Sanchez-Esteban J. Interleukin-10 protects cultured fetal rat type II epithelial cells from injury induced by mechanical stretch. Am J Physiol Lung Cell Mol Physiol 294: L225–L232, 2008. [DOI] [PubMed] [Google Scholar]
- 18.Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT. Elastin is an essential determinant of arterial morphogenesis. Nature 393: 276–280, 1998. [DOI] [PubMed] [Google Scholar]
- 19.Li DY, Faury G, Taylor DG, Davis EC, Boyle WA, Mecham RP, Stenzel P, Boak B, Keating MT. Novel arterial pathology in mice and humans hemizygous for elastin. J Clin Invest 102: 1783–1787, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mammoto A, Connor KM, Mammoto T, Yung CW, Huh D, Aderman CM, Mostoslavsky G, Smith LE, Ingber DE. A mechanosensitive transcriptional mechanism that controls angiogenesis. Nature 457: 1103–1108, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mammoto T, Jiang E, Jiang A, Mammoto A. Extracellular matrix structure and tissue stiffness control postnatal lung development through the lipoprotein receptor-related protein 5/Tie2 signaling system. Am J Respir Cell Mol Biol 49: 1009–1018, 2013. [DOI] [PubMed] [Google Scholar]
- 22.Margraf LR, Tomashefski JF, Bruce MC, Dahms BB. Morphometric analysis of the lung in bronchopulmonary dysplasia. Am Rev Resp Dis 143: 391–400, 1991. [DOI] [PubMed] [Google Scholar]
- 23.McGowan SE. Extracellular matrix and the regulation of lung development and repair. FASEB J 6: 2895–2904, 1992. [PubMed] [Google Scholar]
- 24.Milla C, Yang S, Cornfield DN, Brennan ML, Hazen SL, Panoskaltsis-Mortari A, Blazar BR, Haddad IY. Myeloperoxidase deficiency enhances inflammation after allogeneic marrow transplantation. Am J Physiol Lung Cell Mol Physiol 287: L706–L714, 2004. [DOI] [PubMed] [Google Scholar]
- 25.Mokres LM, Parai K, Hilgendorff A, Ertsey R, Alvira CM, Rabinovitch M, Bland RD. Prolonged mechanical ventilation with air induces apoptosis and causes failure of alveolar septation and angiogenesis in lungs of newborn mice. Am J Physiol Lung Cell Mol Physiol 298: L23–L35, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nave AH, Mizikova I, Niess G, Steenbock H, Reichenberger F, Talavera ML, Veit F, Herold S, Mayer K, Vadasz I, Weissmann N, Seeger W, Brinckmann J, Morty RE. Lysyl oxidases play a causal role in vascular remodeling in clinical and experimental pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol 34: 1446–1458, 2014. [DOI] [PubMed] [Google Scholar]
- 27.Paulsson M,. Basement membrane proteins: structure, assembly, and cellular interactions. Crit Rev Biochem Mol Biol 27: 93–127, 1989. [DOI] [PubMed] [Google Scholar]
- 28.Pierce RA, Albertine KH, Starcher BC, Bohnsack JF, Carlton DP, Bland RD. Chronic lung injury in preterm lambs: Disordered pulmonary elastin deposition. Am J Physiol Lung Cell Mol Physiol 272: L452–L460, 1997. [DOI] [PubMed] [Google Scholar]
- 29.Scherle W. A simple method for volumetry of organs in quantitative stereology. Mikroskopie 26: 57–60, 1970. [PubMed] [Google Scholar]
- 30.Shifren A, Durmowicz AG, Knutsen RH, Faury G, Mecham RP. Elastin insufficiency predisposes to elevated pulmonary circulatory pressures through changes in elastic artery structure. J Appl Physiol 105: 1610–1619, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shifren A, Durmowicz AG, Knutsen RH, Hirano E, Mecham RP. Elastin protein levels are a vital modifier affecting normal lung development and susceptibility to emphysema. Am J Physiol Lung Cell Mol Physiol 292: L778–L787, 2007. [DOI] [PubMed] [Google Scholar]
- 32.Shifren A, Hall CM, Mecham RP, Pierce RA. Effects of elastin haploinsufficiency on compensatory lung growth following pneumonectomy (Abstract). Proc Am Thorac Soc 3: A714, 2006. [Google Scholar]
- 33.Starcher B, Green M, Scott M. Measurement of urinary desmosine as an indicator of acute pulmonary disease. Respiration 62: 252–257, 1995. [DOI] [PubMed] [Google Scholar]
- 34.Takubo Y, Hirai T, Muro S, Kogishi K, Hosokawa M, Mishima M. Age-associated changes in elastin and collagen content and the proportion of types I and III collagen in the lungs of mice. Exp Gerontol 34: 353–364, 1999. [DOI] [PubMed] [Google Scholar]
- 35.Thébaud B, Ladha F, Michelakis ED, Sawicka M, Thurston G, Eaton F, Hashimoto K, Harry G, Haromy A, Korbutt G, Archer SL. Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia-induced lung injury: evidence that angiogenesis participates in alveolarization. Circulation 112: 2477–2486, 2005. [DOI] [PubMed] [Google Scholar]
- 36.Thibeault DW, Mabry SM, Ekekezie II, Truog WE. Lung elastic tissue maturation and perturbations during the evolution of chronic lung disease. Pediatrics 106: 1452–1459, 2000. [DOI] [PubMed] [Google Scholar]
- 37.Wagenseil JE, Mecham RP. New insights into elastic fiber assembly. Birth Defects Res C Embryo Today 81: 229–240, 2007. [DOI] [PubMed] [Google Scholar]
- 38.Wagenseil JE, Nerurkar NL, Knutsen RH, Okamoto RJ, Li DY, Mecham RP. Effects of elastin haploinsufficiency on the mechanical behavior of mouse arteries. Am J Physiol Heart Circ Physiol 289: H1209–H1217, 2005. [DOI] [PubMed] [Google Scholar]
- 39.Wendel DP, Taylor DG, Albertine KH, Keating MT, Li DY. Impaired distal airway development in mice lacking elastin. Am J Respir Cell Mol Biol 23: 320–326, 2000. [DOI] [PubMed] [Google Scholar]
- 40.Witsch TJ, Turowski P, Sakkas E, Niess G, Becker S, Herold S, Mayer K, Vadasz I, Roberts JD Jr, Seeger W, Morty RE. Deregulation of the lysyl hydroxylase matrix cross-linking system in experimental and clinical bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 306: L246–L259, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]