

Abstract
BACKGROUND. Vector prime-boost immunization strategies induce strong cellular and humoral immune responses. We examined the priming dose and administration order of heterologous vectors in HIV Vaccine Trials Network 078 (HVTN 078), a randomized, double-blind phase Ib clinical trial to evaluate the safety and immunogenicity of heterologous prime-boost regimens, with a New York vaccinia HIV clade B (NYVAC-B) vaccine and a recombinant adenovirus 5–vectored (rAd5-vectored) vaccine.
METHODS. NYVAC-B included HIV-1 clade B Gag-Pol-Nef and gp120, while rAd5 included HIV-1 clade B Gag-Pol and clades A, B, and C gp140. Eighty Ad5-seronegative subjects were randomized to receive 2 × NYVAC-B followed by 1 × 1010 PFU rAd5 (NYVAC/Ad5hi); 1 × 108 PFU rAd5 followed by 2 × NYVAC-B (Ad5lo/NYVAC); 1 × 109 PFU rAd5 followed by 2 × NYVAC-B (Ad5med/NYVAC); 1 × 1010 PFU rAd5 followed by 2 × NYVAC-B (Ad5hi/NYVAC); or placebo. Immune responses were assessed 2 weeks after the final vaccination. Intracellular cytokine staining measured T cells producing IFN-γ and/or IL-2; cross-clade and epitope-specific binding antibodies were determined; and neutralizing antibodies (nAbs) were assessed with 6 tier 1 viruses.
RESULTS. CD4+ T cell response rates ranged from 42.9% to 93.3%. NYVAC/Ad5hi response rates (P ≤ 0.01) and magnitudes (P ≤ 0.03) were significantly lower than those of other groups. CD8+ T cell response rates ranged from 65.5% to 85.7%. NYVAC/Ad5hi magnitudes were significantly lower than those of other groups (P ≤ 0.04). IgG response rates to the group M consensus gp140 were 89.7% for NYVAC/Ad5hi and 21.4%, 84.6%, and 100% for Ad5lo/NYVAC, Ad5med/NYVAC, and Ad5hi/NYVAC, respectively, and were similar for other vaccine proteins. Overall nAb responses were low, but aggregate responses appeared stronger for Ad5med/NYVAC and Ad5hi/NYVAC than for NYVAC/Ad5hi.
CONCLUSIONS. rAd5 prime followed by NYVAC boost is superior to the reverse regimen for both vaccine-induced cellular and humoral immune responses. Higher Ad5 priming doses significantly increased binding and nAbs. These data provide a basis for optimizing the design of future clinical trials testing vector-based heterologous prime-boost strategies.
TRIAL REGISTRATION. ClinicalTrials.gov NCT00961883.
FUNDING. NIAID, NIH UM1AI068618, AI068635, AI068614, and AI069443.
Introduction
An effective prophylactic HIV vaccine remains a major global health target, especially in developing countries bearing the brunt of the 2.5 million new infections estimated in 2011 (1). Recent HIV vaccine strategies have progressively focused on viral vector–based vaccines in order to induce potent cellular as well as humoral responses. Recombinant adenovirus–vectored (rAd-vectored) HIV vaccines have been extensively studied in preclinical (2) and clinical studies, both alone (3, 4) and in prime-boost regimens preceded by DNA (5), demonstrating excellent immunogenicity. Following the disappointing outcome of the rAd5-vectored Step Study (3) and the more promising results obtained in the RV144 trial (6) with a canarypox-containing regimen, poxvirus vectors have seen a surge in interest over the past few years. Early poxvirus vectors were poorly immunogenic in humans compared with adenovirus-based vaccines (7, 8), but more recent immunogens based on New York vaccinia (NYVAC) or modified vaccinia Ankara (MVA) boosts show promising results in clinical trials (9–13).
The efficient induction of immune responses following vaccination with viral vectors is likely attributable, in part, to their intrinsic adjuvanticity based on the recognition of viral pathogen–associated molecular patterns (PAMPs). While viral vectors take advantage of this mechanism to induce immune responses to their insert, the vaccinated host will invariably mount a response to the carrier as well, making subsequent homologous vector delivery less efficient at boosting the response to the recombinant vaccine antigen. In addition, vectors are frequently based on human pathogens, and vaccine recipients may show preexisting immune responses to the vector that can dampen insert-specific responses (14, 15). Therefore, while repeated vaccination may be necessary to achieve high magnitudes and high response rates of immune responses to the vaccine insert, homologous prime-boost strategies repeatedly administering the same product may result in diminished returns with each subsequent vaccination.
Combining different vectors in heterologous prime-boost HIV-specific regimens represents a promising alternative to homologous boosting, since immune responses to the first vector are not expected to affect the effectiveness of the second, resulting in boosted responses primarily to the recombinant vaccine antigen. The most frequently used heterologous prime-boost modality consists of a DNA prime followed by a vector boost (5, 10, 16), while clinical data on heterologous prime-boost strategies involving 2 vectors are sparse, even though preclinical data show that immune responses in animals primed with an adenovirus vector can be efficiently boosted with a poxvirus vector (17–19). Recent data in nonhuman primates (NHPs) also show that vaccination with an adenovirus prime-poxvirus vector boost resulted in an 83% reduction in the per-exposure probability of infection against repetitive, intrarectal challenges (20). Adenovirus-poxvirus vector combinations have therefore been proposed as a key strategy to move forward in clinical trials. Here, we present human cellular and humoral immunogenicity data of such a combination approach in a randomized, double-blind preventive HIV Vaccine Trials Network trial (HVTN 078). Using previously tested rAd5 (21) and NYVAC (11) vectors, we show not only that the vaccines induce strong cellular and humoral responses, but also that both the order of administration and the priming dose significantly influence the ensuing immune response. Considering the lack of efficacy in the HVTN 505 trial, in which a heterologous DNA prime-rAd5 boost was evaluated (22), optimization of the dose and regimen of future candidate vaccines based on gene delivery with vector combinations will be imperative before advancing to phase IIb clinical trials.
Results
Participant accrual, demographic data, and vaccine safety.
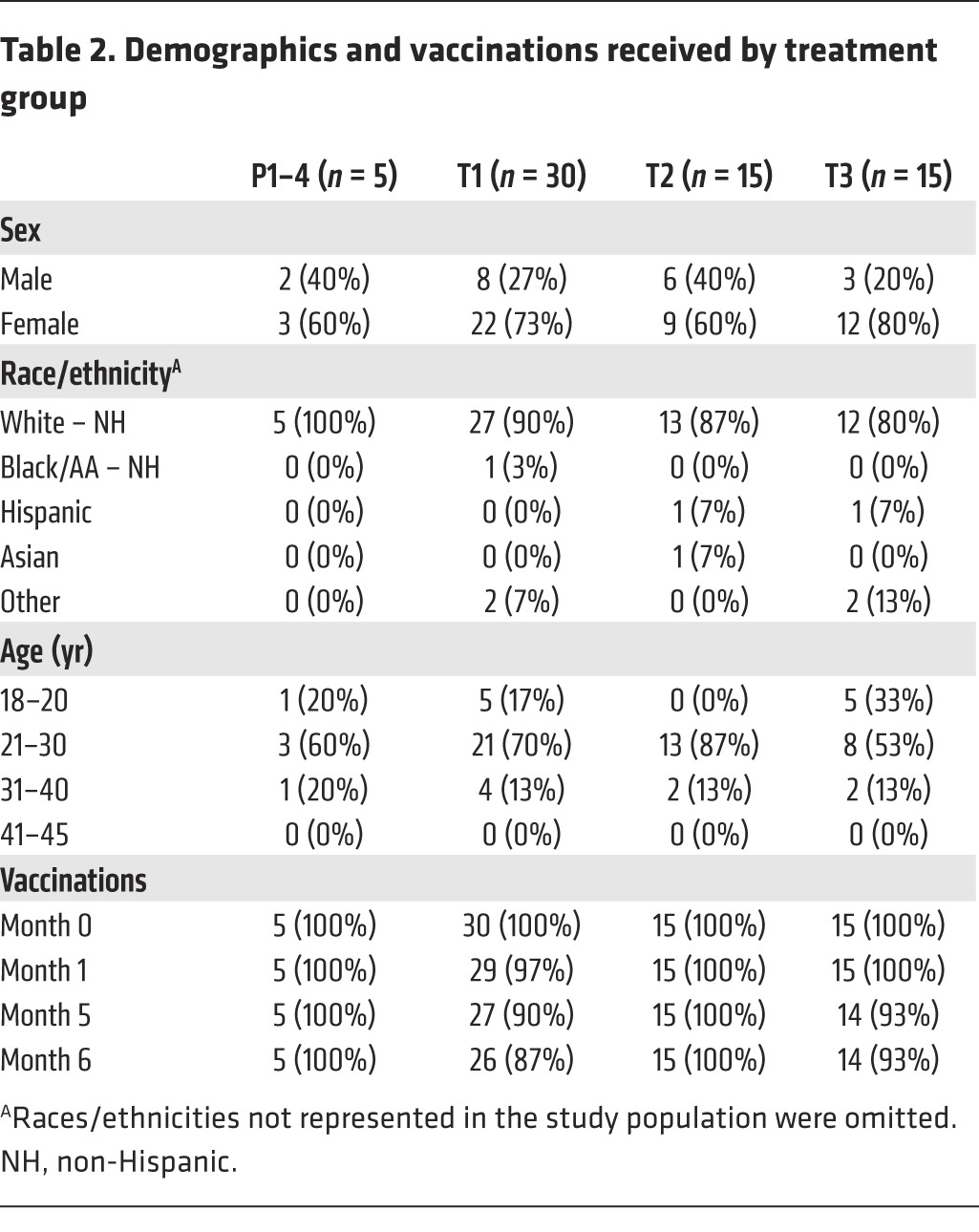
Eighty participants were enrolled, 29% of whom were male and 71% female; 86% were of mixed European descent, 1% each were black or Asian, and 6% were “other”; 5% were of Hispanic ethnicity. Seventy percent of participants were 21–30 years old (yo); 18% were 18–20 yo; and 13% were 31–40 yo. Ninety-eight percent of enrolled participants were retained through the final clinic visit (Figure 1). Allocation of participants to the 4 treatment groups is described in Table 1; demographics and vaccination frequencies per treatment group are detailed in Table 2.
Figure 1. CONSORT statement 2010 flow diagram.

The number of participants enrolled, randomized, followed up, and analyzed is shown for placebo and treatment groups. Two of 12 early terminations occurred prior to the primary immunogenicity visit (visit 10) due to lack of time and commitment to the study. The remaining 7 early terminations due to participants’ refusal resulted predominantly from an extension of the study with version 2 of the protocol; these participants did not consent to the additional visit scheduled in version 2.
Table 1.
HVTN 078 study schema
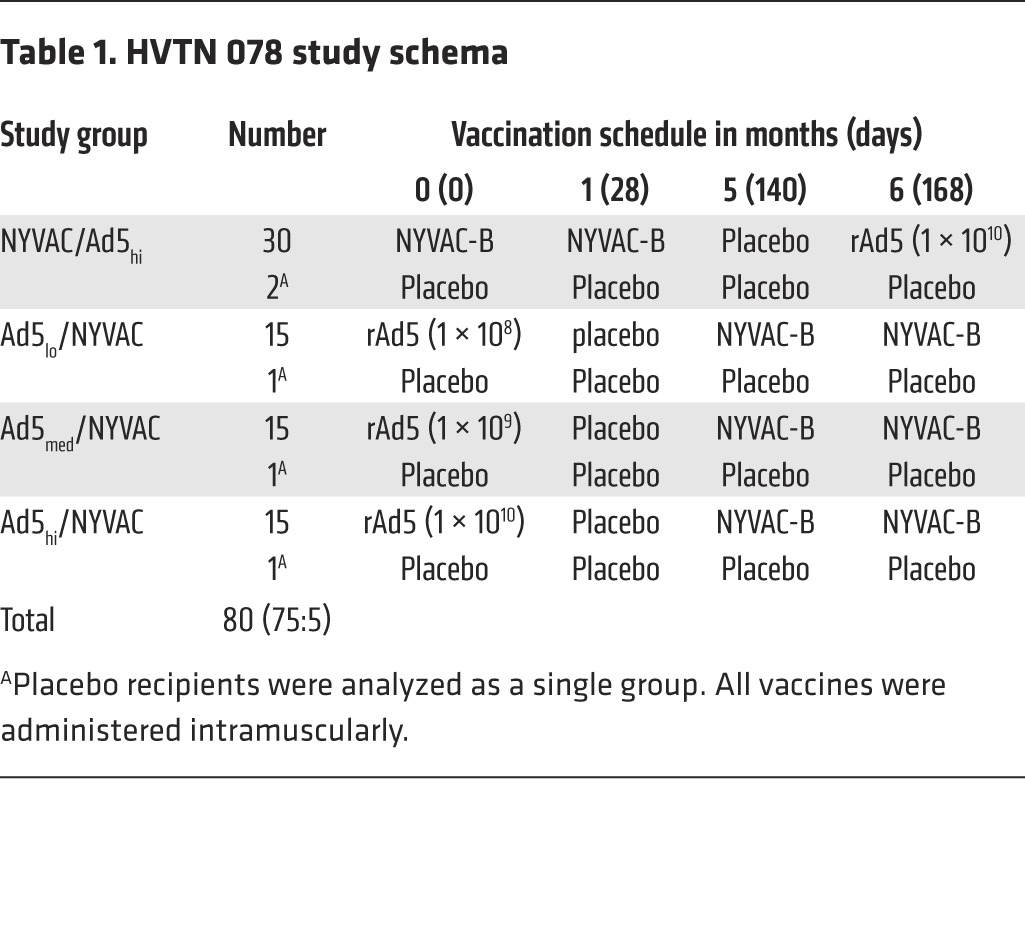
Table 2.
Demographics and vaccinations received by treatment group
The vaccines were safe and well tolerated. Forty percent (n = 32) of participants experienced 1 or more adverse events (AEs) assessed by the investigators as related to the study agents. The incidence of AEs varied by treatment group: 57% (n = 17) of participants in NYVAC/Ad5hi and 47% (n = 7) of participants in Ad5hi/NYVAC, the 2 groups that received the highest rAd5 dose (1010), reported AEs that were assessed as related to the study agents, while only 20% (n = 3) of participants in Ad5lo/NYVAC and 20% (n = 3) of participants in Ad5med/NYVAC reported AEs that were assessed as related to the study agents. No vaccinations were discontinued due to these AEs. As myocarditis and pericarditis have been observed in recipients of vaccinia vaccinations used to protect against smallpox, this protocol excluded individuals with preexisting cardiac risk factors and/or cardiac conditions or ECG findings that could compromise the detection of myocarditis or pericarditis. Participants who reported chest pain, dyspnea, or sensation of palpitations were evaluated by ECG, cardiac troponin, and CK-MB tests. A few participants experienced such symptoms after vaccination and underwent cardiac evaluation; no evidence of myocarditis or pericarditis was found.
Systemic and local reactogenicity were typically mild or moderate, though the NYVAC/Ad5hi and Ad5med/NYVAC groups each had 1 participant with severe systemic reactogenicity after receipt of the fourth and third vaccinations, respectively, and the Ad5lo/NYVAC group had 2 participants with erythema and/or induration of greater than 9 cm in diameter after receipt of the fourth vaccination. Overall, the reactogenicity profile was similar to that observed in prior studies of the NYVAC and rAd5 vaccines used in this study (11, 12, 21).
Vaccination with NYVAC-B and rAd5 elicits HIV envelope–specific binding antibody responses to clades A, B, C, and AE Env.
Heterologous prime-boost vaccine regimens frequently include DNA vaccines, and early studies have shown that DNA is more efficient as a prime than as a boost (23, 24). Regimens combining different vectors, on the other hand, have been rare in humans, and clinical trials did not interrogate the influence of the order in which the vectors were administered (7). We therefore wanted to address (a) whether lower priming doses are as efficient as high doses to elicit immune responses by comparing Ad5lo/NYVAC with Ad5hi/NYVAC and (b) whether the order of administration of NYVAC-B and rAd5 would affect immunogenicity by comparing NYVAC/Ad5hi with Ad5hi/NYVAC.
Both rAd5 and NYVAC-B had previously been tested in humans and have been shown to elicit both humoral and cellular responses (21, 25), but the combination of an adenoviral vector with a poxvirus vector had only been tested in NHPs, in which it showed superior protection from acquisition compared with that of homologous poxvirus vectors (20). The rAd5 vaccine used in the present study contained envelope (Env) protein inserts for clades A, B, and C, increasing the likelihood of inducing cross-clade immune responses. As shown in Figure 2, we detected binding IgG responses to clades A, B, C, and group M consensus gp140, as well as to a clade AE gp120 protein (Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI75894DS1) and Gag p24 (Supplemental Figure 2).
Figure 2. Binding IgG responses elicited in HVTN 078 two weeks after the final vaccination.

IgG titers against consensus clade A Env (ConA) gp140 (A), clade B Env (ConB) gp140 (B), clade C Env (ConC) gp140 (C), and ConS gp140 (D) were calculated for positive responders using AUC (1:50 dilution, 5-fold titration series). Positive responses are shown in red symbols and negative responses in blue. Boxes and whiskers represent the positive responders only (see Methods).
The order in which the 2 vectors were administered only showed minor effects on binding antibody responses at the same rAd5 dose (NYVAC/Ad5hi vs. Ad5hi/NYVAC) (Figure 2). We observed no significant differences between the NYVAC/Ad5hi and Ad5hi/NYVAC groups for Env-specific IgG, although the response rates were consistently lower in the NYVAC/Ad5hi group. On the other hand, we found significantly more Gag-p24-specific IgG responders in the Ad5hi/NYVAC group than in the NYVAC/Ad5hi group (P < 0.0001, Supplemental Figure 2).
Increasing priming doses of rAd5 significantly improve binding IgG responses.
While the order of vector administration had only minor effects on the magnitude of the IgG response, increasing priming doses of rAd5 led to significantly higher response rates of binding Env-specific IgG after the NYVAC-B boost for consensus A Env gp140 (P = 0.0013), consensus C Env gp140 (P < 0.0001), ConS gp140 (P < 0.0001), A244 gp120 (P = 0.0005), and p24 (P < 0.0001), but not for consensus B Env gp140 (P = 0.7), which had high response rates even at the lowest dose (Figure 2 and Supplemental Figures 1 and 2). Such a dose effect was also seen in response magnitudes in positive responders to consensus A gp140 (P < 0.0001), B gp140 (P = 0.002), and C gp140 (P = 0.03), ConS gp140 (P = 0.003), and p24 (P = 0.001).
In light of the findings from the RV144 correlates analysis (26), we also determined the rate and magnitude of V1-V2 IgG responses as well as of IgA responses. Antibody responses to the clade B gp70 V1-V2 case_A2 (used to define the correlate of HIV-1 risk in RV144) were only generated in those groups that included the high dose of rAd5 (24% and 14% for NYVAC/Ad5hi and Ad5hi/NYVAC, respectively; 0% for Ad5lo/NYVAC and Ad5med/NYVAC; Figure 3). We also measured clade C and AE V1-V2 antigens to examine the breadth of V1-V2 responses and found that response rates were higher than those of the clade B construct (up to 50%; Figure 3), but were still rarely detected in the lower-dose rAd5 groups, suggesting that the high dose of Ad5 is necessary to induce responses to V1-V2. We found that the responses to linear V2 sequences were low to absent in 10 vaccinees that were epitope mapped (Supplemental Figure 3). However, antibody responses to cross-clade V3 sequences were the dominant linear IgG response among these vaccinees.
Figure 3. V1-V2–specific IgG and gp140-specific IgA elicited in HVTN 078 two weeks after the final vaccination.

IgG responses to (A) clade B case_A gp70 V1-V2, (B) clade C 1086 V1-V2 tags, and (C) clade AE A244_293F V1-V2 tags were measured as MFI (see Methods). (D) IgA responses to consensus clade B Env gp140 were measured as MFI. Positive responses are shown in red symbols and negative responses in blue. Boxes and whiskers represent the positive responders only (see Methods).
Interestingly, we found that Env-specific IgA production was mainly restricted to NYVAC/Ad5hi (Figure 3D for clade B gp140, P = 0.003 for response rates in NYVAC/Ad5hi vs. Ad5hi/NYVAC; Supplemental Figure 4 for clades A and C as well as ConS gp140).
Neutralizing antibody responses are mainly directed at tier 1 viruses.
Broadly neutralizing antibody (bnAb) responses are a highly desired outcome for an HIV vaccine but are inherently difficult to elicit with current regimens, especially in the absence of protein boosts (27). Here, we assessed whether the combination of 2 highly immunogenic vectors would lead to an increased neutralization potential of the induced antibodies.
Responses against tier 1 viruses were found in the majority of vaccine recipients (Figure 4); response rates were highest for MN.3 (69.3%) followed by SF162.LS (42.1%), BaL.26 (18.4%), Bx08.16 (11.8%), and MW965.26 (14.5%). We observed no response to NP03.13. Neutralizing antibody (nAb) responses to tier 1 viruses were significantly higher in NYVAC/Ad5hi (P = 0.027), Ad5med/NYVAC (P < 0.001), and Ad5hi/NYVAC (P < 0.001) recipients than in placebo recipients. nAb responses to tier 2 viruses were infrequently induced regardless of whether they were measured in the TZM-bl assay or the more sensitive A3R5 assay (data not shown).
Figure 4. Strain-specific nAb responses elicited in HVTN 078.

Six tier 1 Env-pseudotyped viruses (MN.3, SF162.LS, BaL.26, MW965.26, NP03.13, and Bx08.16) were tested in the TZM-bl neutralization assay 2 weeks after the final vaccination. nAb titers are shown for placebo and vaccine recipients. Positive responses are shown in red symbols and negative responses in blue. P, placebo; T1, NYVAC/Ad5hi; T2, Ad5lo/NYVAC; T3, Ad5med/NYVAC; T4, Ad5hi/NYVAC.
nAb responses depend on priming dose and on the order of vector administration.
Increasing doses of the rAd5 prime led to significantly higher rates of Env-specific binding IgG (Figure 2), and nAbs followed the same trend (Figure 4), with the priming dose significantly affecting the response rates for SF162.LS (P < 0.0001), MN.3 (P = 0.0034), and MW965.26 (P = 0.0053). Interestingly, while neither the magnitude nor the response rate for Env-specific IgG differed significantly between the NYVAC/Ad5hi and Ad5hi/NYVAC recipients, the order in which the 2 vectors were administered did affect the neutralization potential of these Abs, with significantly higher neutralization of tier 1 viruses for Ad5hi/NYVAC (P = 0.004, Figure 5). Neutralization was also significantly higher for Ad5med/NYVAC recipients than for NYVAC/Ad5hi (P = 0.048) recipients, but was similar between NYVAC/Ad5hi and Ad5lo/NYVAC (P = 0.3) recipients.
Figure 5. MB of nAb responses elicited in HVTN 078.

Five tier 1 Env-pseudotyped viruses (BaL.26, Bx08.16, MW965.26, SF162.LS, and MN.3) were tested in the TZM-bl neutralization assay 2 weeks after the final vaccination. Data are represented as MB curves, where the x axis represents nAb titers, and the y axis represents the fraction of viruses neutralized. Dashed lines represent subject-specific responses. Solid lines represent group averages.
nAb responses decline by 6 months after the last immunization.
To assess the longevity of the antibody response, we assessed nAb responses for the clade B strain MN.3 at 6 months after the last immunization (5.5 months after the peak time point). MN.3 was chosen for this analysis, since responses to this virus were detectable in all treatment groups at peak. As shown in Figure 6, responses were still detectable in all groups, although the response rate declined significantly in the NYVAC/Ad5hi group (P < 0.001). The reduction in response rates in the Ad5-primed groups was less pronounced, and 86.7% of subjects maintained positive nAb titers at the late time point in the Ad5hi/NYVAC group. While the magnitude of responses declined in all groups, this reduction was only significant for Ad5hi/NYVAC (P = 0.001) recipients. These data suggest that vaccine-induced humoral immune responses were more durable at 1 year after enrollment for the groups receiving the highest dose of rAd5.
Figure 6. Longevity of nAb responses in HVTN 078.

The tier 1 Env-pseudotyped virus MN.3 was tested in the TZM-bl neutralization assay 2 weeks (visit 10) and 6 months (visit 13) after the final vaccination. nAb titers are shown for placebo and vaccine recipients. Positive responses are shown in red symbols and negative responses in blue.
Vaccine-induced seroreactivity in commercially available diagnostic kits is dependent on priming dose and on the order of vector administration.
While the induction of strong cross-clade antibody responses is a major goal for HIV vaccines, those responses may include anti-HIV antibodies that can be detected on commercially available HIV serologic tests in the absence of HIV infection. Therefore, the HVTN diagnostic program uses testing algorithms that differentiate vaccine-induced seroreactivity (VISR) from true HIV infection. We tested all vaccine recipients at the end of the study to assess VISR on a minimum of 3 commercially available kits. This testing determines the likelihood that a participant will be misclassified as infected if they are tested outside of the Network. VISR was assessed using the HVTN algorithm for the evaluation of seroreactivity (EOS), which began with 3 different enzyme immunoassay (EIA) kits. Samples with at least 1 reactive EIA were followed up with Western blotting and RNA PCR to distinguish VISR from natural infection. As shown in Figure 7, the different EIA kits picked up responses in partially overlapping sets of subjects. We found that the response rates for VISR were comparable between the NYVAC/Ad5hi and Ad5hi/NYVAC (P = 0.2) groups, but lower in the Ad5lo/NYVAC and Ad5med/NYVAC (P = 0.09 and P = 0.03 compared with NYVAC/Ad5hi, respectively; P = 0.002 for the Ad5 dose response using the Cochran-Armitage trend test) groups, following the trend of responses observed using the HIV-1–binding antibody multiplex assay (BAMA) (Figure 2). Eighty-five percent of subjects who had a reactive EIA also had a reactive Western blot, but all were negative for viral RNA, suggesting that reactivity was due to vaccination rather than HIV-1 infection.
Figure 7. VISR elicited in HVTN 078.

Sera were tested at the end of the study (1 year after enrollment) using 3 commercially available EIA kits: Abbott Architect HIV Ag/Ab Combo, Bio-Rad Genetic Systems HIV 1/2 Plus O EIA, and Bio-Rad Multispot HIV-1/HIV-2 Rapid Test. The percentage of reactive samples for any test (left) or each of the individual tests is shown. T1, NYVAC/Ad5hi (white bars); T2, Ad5lo/NYVAC (light gray bars); T3, Ad5med/NYVAC (dark gray bars); T4, Ad5hi/NYVAC (black bars).
NYVAC is a stronger boost than a prime for T cell responses.
In addition to humoral responses, we assessed cellular immune responses by intracellular cytokine staining (ICS) in 75 participants for CD4+ T cells (1 filtered due to a high background) and 76 participants for CD8+ T cells. As shown in Figure 8, CD4+ and CD8+ T cell IFN-γ and/or IL-2 responses of high magnitude were observed in all 4 treatment groups.
Figure 8. T cell responses elicited in HVTN 078.

CD4+ (A) and CD8+ (B) T cell responses were measured 2 weeks after the final vaccination by ICS and reported as the percentage of CD4+ or CD8+ T cells producing IFN-γ and/or IL-2 for placebo recipients (combined for groups 1–4) and vaccinees in each treatment group. Positive responses are shown in red symbols and negative responses in blue. Boxes and whiskers represent positive responders only (see Methods). N, NYVAC-B; P, placebo; Ad, rAd5; Lo, 108 rAd5 PFU; Med, 109 rAd5 PFU; Hi, 1010 rAd5 PFU.
Response rates for CD4+ T cells were significantly higher in regimens using NYVAC-B as a boost rather than as a prime (NYVAC/Ad5hi vs. Ad5lo/NYVAC: P = 0.001; NYVAC/Ad5hi vs. Ad5med/NYVAC: P = 0.005; and NYVAC/Ad5hi vs. Ad5hi/NYVAC: P = 0.01; adjusted P values using Fisher’s exact test; Figure 8A). CD4+ T cell magnitudes were also significantly higher after the NYVAC-B boost than after the rAd5 boost (NYVAC/Ad5hi vs. Ad5lo/NYVAC: P < 0.001, NYVAC/Ad5hi vs. Ad5med/NYVAC: P = 0.03; and NYVAC/Ad5hi vs. Ad5hi/NYVAC: P < 0.001; adjusted P values using the Wilcoxon test).
We did not find the response rates for CD8+ T cells to be significantly different (Figure 8B); however, CD8+ T cell magnitudes were significantly higher after the NYVAC-B boost than after the Ad5 boost (NYVAC/Ad5hi vs. Ad5lo/NYVAC: P = 0.02; NYVAC/Ad5hi vs. Ad5med/NYVAC: P = 0.04; and NYVAC/Ad5hi vs. Ad5hi/NYVAC: P = 0.012; adjusted P values using the Wilcoxon test). For both CD4+ and CD8+ T cells, we observed similar levels of medium-to-high response rates across the Env, Gag, and Pol peptide pools, while no or very low response rates were observed for Nef (Supplemental Figure 5).
Cellular immunogenicity of rAd5/NYVAC-B is not significantly affected by an rAd5 priming dose.
DNA vaccines mostly are poorly immunogenic in humans but have a significant effect on immune responses after a heterologous boost, suggesting that a weak prime may be ideal for induction of strong T cell responses. We therefore tested 3 concentrations of rAd5 (108, 109, and 1010 PFU) when used to prime NYVAC-B.
Figure 8 shows that dose escalation of the prime had minimal impact on the response rates and magnitudes for CD4+ and CD8+ T cells; interestingly, while not significantly different, CD8+ T cell response rates slightly increased with increasing doses of rAd5 vector, while CD4+ T cell responses were higher at lower doses.
Functionality of responses is similar across all tested regimens.
We assessed the coexpression of multiple functional markers (polyfunctionality) for all positive responses. Polyfunctional profiles for IFN-γ, IL-2, TNF-α, and granzyme B expression for Gag-specific CD4+ (Figure 9A) and CD8+ T cells (Figure 9B) did not differ significantly between NYVAC-B and Ad5 primes.
Figure 9. Polyfunctionality of Gag-specific T cell responses elicited in HVTN 078.

CD4+ (A) and CD8+ (B) T cell responses to Gag were measured 2 weeks after the final vaccination by ICS and reported as the percentage of CD4+ or CD8+ T cells with 1, 2, 3, or 4 functions (top panels) or the percentage of CD4+ or CD8+ T cells within a functional subset that expressed a given combination of markers as specified by “+” or “–” below the bottom panels.
Discussion
Numerous clinical trials have shown the improved immunogenicity of heterologous prime-boost vaccine strategies, especially for induction of cellular immune responses (reviewed in ref. 28). Following the disappointing results of Ad5-vectored vaccines alone (Step Study; ref. 3) or in combination with DNA prime (HVTN 505; ref. 22) in phase IIb trials, it remains important to evaluate other combinations when they demonstrate broader, stronger, and unique immune responses in preclinical studies. Many factors need to be considered when determining the ideal combination of 2 vaccines used in a heterologous prime-boost regimen; foremost among these are: (a) which vaccine antigen delivery approaches work well together; (b) the order in which they are administered; (c) the most immunogenic dose, which may differ from the optimal dose for each individual vaccine candidate or depend upon the order in which they are given; (d) the interval between the prime and the boost; and (e) the route chosen for delivery. Our data from the HVTN 078 study confirm the preclinical data showing that the combination of an adenovirus and a poxvirus vector is highly immunogenic and demonstrate that NYVAC is a more potent boost than a prime for both cellular and humoral responses. In addition, we show that priming with different doses of rAd5 does not affect T cell responses when followed by NYVAC, but that a high-dose prime is necessary for strong IgG responses, especially to Env V1-V2.
Dose escalation studies are frequently part of early-phase vaccine trials; generally, higher doses are more immunogenic, and the dose escalation aims at finding the highest tolerable dose based on safety considerations. HVTN 078 is to our knowledge the first clinical trial evaluating the optimal priming dose for immunogenicity in regimens involving highly immunogenic heterologous recombinant vaccine vectors with a fixed boosting dose. Since relatively weak immunogens such as DNA can have substantial impact on the magnitude of T cell responses following subsequent boosting with a heterologous vector, low-dose priming may provide an equal or better primary stimulus to the immune system than do higher doses of the priming vector. Indeed, both low-dose (108 PFU) and high-dose (1010 PFU) Ad5 primed T cell responses when followed by NYVAC. Interestingly, the noninferiority of the low-dose prime did not hold for humoral immune responses, for which there was a clear positive dose-effect response to the prime. This was especially pronounced for IgG responses to Env V1-V2, which were mostly absent in the groups primed with low and medium rAd5 doses. The V1-V2 response rates (0%–50%) in this study were lower than those induced by RV144 using the same assay (72%–96% in RV144; ref. 29). Additional studies are needed to examine differences in the epitope-specific responses to the different regions of the HIV-1 envelope included in the V1-V2 antigens.
Heterologous prime-boost strategies using a combination of poxvirus vectors and adenoviral vectors have been proposed as a promising HIV vaccine strategy based on recent data in the NHP model showing 80%–83% per-exposure vaccine efficacy for SIV acquisition (20). The number of exposures necessary for SIV acquisition in this study most significantly correlated with binding antibody titers. Interestingly, in both our study as well as in the NHP study, the benefit of a poxvirus vector boost rather than a prime boost was least pronounced for Env-specific binding antibodies, in line with the minor benefit of the poxvirus vector boost for the observed per-exposure efficacy in the NHP study.
Serum IgA responses were significantly more frequent in the NYVAC/Ad5hi group; yet, based on the results of the RV144 trial, in which specific plasma Env IgA was found to be a direct correlate of risk (26, 30), the benefit of particular Env IgA responses in the blood was unclear. We found that Gag-specific antibodies were nearly absent in the NYVAC/Ad5hi group, possibly due to an N-terminal Gly → Ala substitution in the NYVAC-B Gag that prevents the formation and release of virus-like particles from transfected cells and therefore may have diminished the impact of the prime.
In our study, rAd5/NYVAC-B induced CD4+ and CD8+ T cell responses of significantly greater magnitude and CD4+ T cell responses in significantly more vaccinees than did NYVAC-B/rAd5. Although cellular responses may not primarily have an effect on HIV acquisition, data from NHP studies as well as from the Step Study show that increased vaccine-induced Gag-specific T cell responses are associated with a reduced viral load set-point (20, 31). Along with responses to the other proteins contained in the HVTN 078 vaccines, we found that Gag-specific CD4+ and CD8+ T cell responses were more frequent and of higher magnitude in the NYVAC-B–boosted groups.
Assessment of VISR on commercially available diagnostic kits indicated that up to 67% of participants receiving the highest dose of adenovirus were at risk of being misclassified as HIV infected if they were to be tested outside of the Network system. Seroreactivity rates measured by these diagnostic kits were overall lower than those measured using our sensitive BAMA; but VISR mostly followed the response rates for binding Abs — both rates being highest in the Ad5hi/NYVAC group. To protect participants from the potential social harms that can result from a misdiagnosis, the HVTN counsels and strongly encourages vaccine trial participants who exhibit VISR at the end of the vaccine study to continue to receive HIV diagnostic testing within the Network system.
Overall, vaccine-induced HIV-specific immune responses in HVTN 078 were high, with 100% response rates for binding and nAbs and over 85% for both T cell subsets. Of note, CD4+ T cell responses dominated in this trial, which is unusual for vaccine regimens including rAd5. This enrichment of CD4+ T cell responses is likely due to the inclusion of NYVAC-B; like other poxvirus vectors, it preferentially induces this subset (7, 10). CD8+ T cell responses were somewhat lower in HVTN 078 compared with those measured in HVTN 054, in which a single dose of the same rAd5 was given; this may be explained in part by the time point chosen for immunogenicity assessment. While 2 weeks after immunization seems to be optimal for poxvirus vectors and was used as the primary immunogenicity time point in HVTN 078, responses to rAd5 usually peak after 3 or 4 weeks, the primary immunogenicity time point assessed in HVTN 054 (32). Additional limitations of this study include its modest sample size, which leaves open the possibility of infrequent vaccine-associated AEs that might be observed in a larger study; the precision of the observed trend in the immunogenicity data; and the enrollment of participants at only 1 site with limited demographics, where self-reporting of safety outcomes may differ from that in other populations.
Taken together, the HVTN 078 data provide support for further characterization of heterologous vector combinations in clinical trials. A high dose of a priming adenovirus vector followed by a poxvirus vector would be a preferred regimen based on our data, as increased nAb titers may provide additional benefit in preventing HIV infection in people compared with the NHP model, and stronger cellular immune responses can provide a second line of defense to contain virus replication should breakthrough infection occur (31). Moving forward, adenovirus vectors of low seroprevalence, such as Ad26 or Ad35, or nonhuman adenovirus vectors based on chimpanzee- or gorilla-derived adenoviruses should replace the rAd5 vector used in this trial; alternatively, other heterologous immunogens, such as DNA, could replace the adenovirus prime. NYVAC represents a promising partner for these heterologous prime-boost strategies, and further studies evaluating combinations with alternate poxvirus vectors will determine which strategy is most immunogenic. The dichotomy in the benefit of higher priming doses for antibody, but not T cell, responses will need to be confirmed in additional studies, and a new trial testing the influence of the order of vaccine administration using a DNA-protein strategy is currently being planned within the HVTN.
Methods
Study participants.
Eighty male and female study participants were enrolled by clinical staff at the HVTN site in Lausanne, Switzerland, from 2009 to 2011. Subjects were required to meet the following criteria for enrollment: age 18–45 years; good general health; completion of a questionnaire assessing an understanding of the study and the nature of participation; being willing and able to provide informed consent; willingness to receive HIV test results; being amenable to HIV risk reduction counseling; and assessed by clinic staff as being at low risk for HIV infection. Pregnant women were excluded, and volunteers who could become pregnant agreed to consistently use effective contraception and not seek pregnancy through alternative methods. Laboratory inclusion criteria, tested within 8 weeks prior to study enrollment, included negative HIV-1 and HIV-2 serum antibody tests; aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), and creatinine levels below the institutional upper limits of normal; negative blood tests for chronic hepatitis B and C; normal urine; hemoglobin greater than or equal to 11.0 g/dl for volunteers who were born female and greater than or equal to 13.0 g/dl for volunteers who were born male; wbc count between 3,300 and 12,000 cells/mm3; total lymphocyte count greater than or equal to 800 cells/mm3; remaining differential either within the institutional normal range or with site physician approval; platelet counts between 125,000 and 550,000/mm3; and an Ad5 nAb titer less than 1:18 (33). Figure 1 shows a CONSORT statement flow chart of study enrollment, allocation, and safety analysis (34).
Study procedures.
HVTN 078 was a single-center, randomized, double-blind phase Ib clinical trial to evaluate the safety and immunogenicity of heterologous prime-boost vaccine regimens (NYVAC-B/rAd5 vs. rAd5/NYVAC-B) in healthy, HIV-1–uninfected, Ad5 seronegative adult participants. All participants (75 received vaccine and 5 placebo) received 4 injections of NYVAC-B (NYVAC vector containing HIV-1BX08 gp120 and HIV-1IIIB gag-pol-nef at a dose of 1 × 107 PFU), rAd5 (HIV-1 rAd serotype 5 [rAd5] vector vaccine VRC-HIVADV014-00-VP [HIV-1HXB2/NL4-3 Gag-Pol fusion; HIV-192RW020, HIV-1HXB2/Bal, and HIV-197ZA012 Env] at 3 different doses [1 × 1010, 1 × 109, and 1 × 108 PFU]), NYVAC-B placebo (0.9% NaCl), or rAd5 placebo (final formulation buffer [FFB]), according to the randomized treatment assignment, as a 1-ml intramuscular injection in the nondominant deltoid. The randomization sequence was obtained by computer-generated random numbers and provided to the site by a central data monitoring center; randomization was not stratified and was done in 5 blocks of size 16. The pharmacist at the Lausanne site with responsibility for dispensing the appropriate vaccine was responsible for maintaining the security of the randomization code and did not participate in the clinical assessment of participants. To maintain blinding, the pharmacist placed an overlay on the syringe containing the study products. The vaccination schedule and distribution into 4 treatment groups are described in Table 1.
Immune responses were measured at visits 2 (baseline), 10 (2 weeks after the fourth [final] immunization scheduled at month 6), and 13 (6 months after the fourth immunization). Of the 80 participants, at visit 10 there were 2 terminations, 1 missed visit, and 1 out-of-window sample collection; therefore, data from this primary immunogenicity time point are available for up to 76 participants. At visit 13, there were 2 terminations, and 1 sample was not collected.
Sample size calculation.
Sample sizes for this study were primarily powered for safety evaluation. The sample size of 30 vaccine recipients in group 1 and 15 vaccine recipients per group in groups 2–4 provided a 90% chance of observing at least 1 serious AE if the true rate of such an event were at least 8% and 15%, respectively; there was a 90% chance that we would not observe at least 1 serious AE if the true rate were no more than 0.35% and 0.69%, respectively. The precision to estimate immunogenicity was somewhat limited and therefore not a primary objective of the study. For any observed response rate, the width of a 2-sided 95% confidence interval was at most 40% for group 1 and 55% for groups 2–4. These later calculations assumed a 10% missing data rate in immunogenicity endpoints for various reasons, including study subjects terminating their participation in the study early, problems in shipping specimens, or low cell variability of processed samples. Two placebo recipients in group 1 and 1 per group in groups 2–4 were randomized in each group for the purpose of blinding in the assessment of safety and immunogenicity endpoints.
Sample processing.
Serum for humoral assays was obtained from serum-separating tubes (SSTs) and frozen at –80°C until use. Peripheral blood mononuclear cells (PBMCs) for cellular assays were isolated and cryopreserved from heparin-anticoagulated whole blood within 6 hours of venipuncture, as described previously (35). PBMCs were thawed and cultured overnight at 37°C, 5% CO2 in R10 (RPMI 1640 [Gibco], 10% FBS [Gemini Bioproducts], 2 mM L-glutamine [Gibco], 100 μg/ml streptomycin sulfate [Gibco], and 100 U/ml penicillin G [Gibco]). The cells were then counted prior to stimulation using the Guava ViaCount Kit (Millipore), according to the manufacturer’s instructions.
Binding antibody responses.
Serum HIV-1–specific IgG responses (1:50 dilution) against 6 HIV proteins (clades A, B, and C gp140; group M consensus gp140; clade AE gp120 [A244gp120gDneg/293F/mon]; and clade B Gag p24) and 3 V1-V2 antigens (AE.A244 V1-V2 Tags/293F, C.1086C_V1_V2 Tags, and gp70_B.CaseA_V1_V2) were measured 2 weeks after the fourth vaccination by a validated HIV-1–binding antibody multiplex assay (BAMA) as previously described (36). Antibody measurements were acquired on a Bio-Plex instrument (Bio-Rad), and the readout was expressed as the mean fluorescence intensity (MFI). The positive control in each assay was HIV-positive sera, and the negative control was HIV-negative human sera and blank beads. Samples with a blank bead MFI greater than 5,000 after 1 re-testing were excluded. Samples were determined to be positive if both the MFI and blank-subtracted MFI were more than 3-fold over the baseline MFI and blank-subtracted MFI, respectively, and if the blank-subtracted MFI values were above an antigen-specific cutoff that equaled the antigen-specific average MFI plus 3 standard deviations of 60 seronegative plasma samples. Antibody titers were calculated for positive responders using AUC (1:50 dilution, 5-fold titration series). Binding antibody data were available for 76 participants.
Serum IgG (1:250 dilution) from a subset of those vaccinees with the highest nAb responses were further epitope mapped at V2 and V10 using linear peptide microarrays, as previously described (37, 38).
nAb assay.
nAbs against HIV-1 were measured as a function of reductions in Tat-regulated luciferase (Luc) reporter gene expression in either TZM-bl cells or A3R5 cells. Serum samples were tested 2 weeks after the fourth vaccination (primary immunogenicity time point) for their ability to neutralize Env variants that exhibited either a highly sensitive tier 1 neutralization phenotype or a less sensitive tier 2 neutralization phenotype that was more typical of most circulating strains (39). Six tier 1 variants (clade B: MN.3, SF162.LS, Bal.26, and Bx08; clade C: MW965.26; clade E: NP03.13) were assayed as Env-pseudotyped viruses in TZM-bl cells to obtain 50% inhibitory dose (ID50) neutralization titers as previously described (40, 41). In addition, 6 tier 2 Env-pseudotyped viruses (clade B: 6535.3, PVO.4, QH0692.42, RHPA4259.7; clade C: CAP45.2.00.G3 and TV1.21) were assayed in TZM-bl cells using a single 1:10 dilution of serum sample. Seven additional tier 2 variants (clade B: RHPA, SC22.3C2, CH77; clade C: CAP45.2.00.G3, Ce1086_B2, Ce1176_A3, Du151.2) were assayed as Env.IMC.LucR viruses (42) using a single 1:10 dilution of serum in the more sensitive A3R5 cell line (43). Both assays have been formally optimized and validated (44, 45) and were performed in compliance with good clinical laboratory practices (GCLPs). Responses against tier 1 viruses were considered positive if the titer was greater than or equal to 10, where a titer was defined as the serum dilution that reduced the relative luminescence units (RLU) by 50% relative to the RLU in the virus control wells (cells plus virus only) after subtraction of the background RLU (cells only). Responses against tier 2 viruses were considered positive if the percentage of neutralization at the single tested serum dilution was greater than or equal to 50%, where the percentage of neutralization was determined by calculating the difference in average RLU between test wells containing postimmune samples and test wells containing preimmune samples from the same participant. At the primary immunogenicity time point, data for nAbs against tier 1 viruses were available from 76 participants for BaL.26, Bx08.16, MW965.26, and SF162.LS; 75 participants for MN.3; and 42 participants for NP03.13. Data for nAbs against tier 2 viruses at the primary immunogenicity time point were available from 71 participants for A3R5 and 33 participants for TZM-bl. In addition, serum samples from 75 participants at 6 months after the fourth vaccination (longevity time point) were tested for nAbs against MN.3 in the TZM-bl assay.
VISR.
HIV infection was assessed at multiple time points during the study (months 3, 6, 9, and 12) using the HVTN in-study diagnostic algorithm, which uses a single EIA test, the Bio-Rad Genetic Systems HIV 1/2 Plus O EIA. VISR was assessed at the end of the study (month 12, i.e., 1 year following enrollment) using a diagnostic algorithm that included 3 different EIA tests: Abbott Architect HIV Ag/Ab Combo, Bio-Rad Genetic Systems HIV 1/2 Plus O EIA, and Bio-Rad Multispot HIV-1/HIV-2 Rapid Test. Both diagnostic algorithms require Western blotting (Bio-Rad Genetic Systems HIV-1 Western Blot) and RNA PCR (Abbott m2000 HIV-1 Real-Time PCR) to be run on samples that have a reactive EIA to distinguish vaccine-induced responses from actual infection. VISR data were available for 74 participants for the 3 EIAs and 26 participants for Western blotting.
ICS assay.
ICS was performed on cryopreserved PBMCs by flow cytometry to examine HIV-1–specific vaccine-induced CD4+ and CD8+ T cell responses at 2 weeks after the fourth (last) vaccination. Cytokine production was assessed after stimulation with potential T cell epitope (PTE) global peptide pools (46) representing 15-mer peptides from Gag, Nef, Pol, and Env (Bio-Synthesis) at 1 μg/ml as previously described (32, 47). The 6-hour stimulation included brefeldin A (10 μg/ml; Sigma-Aldrich) and anti-CD28/anti-CD49d (each at 1 μg/ml; BD Biosciences). Staphylococcal enterotoxin B (SEB) (Sigma-Aldrich) was used as a positive control, and peptide diluent (DMSO at a final concentration of 1%) was used as a negative control. Cells were stained with LIVE/DEAD Fixable Aqua Dead Cell Stain (Invitrogen), then fixed, permeabilized, and stained intracellularly with fluorescently labeled antibodies against CD3, CD4, CD8, IFN-γ, IL-2, TNF-α, granzyme B, CD57, and perforin (32). Data were acquired on an LSRII (BD Biosciences) and analyzed using FlowJo software (Tree Star Inc.). Positivity of the ICS responses of individual cytokines or cytokine combinations was determined by a 1-sided Fisher’s exact test applied to each peptide pool–specific response versus the negative control response, with a discrete Bonferroni adjustment for the multiple comparisons due to testing against multiple peptide pools. Peptide pools with adjusted P values less than α = 0.00001 were considered positive. If at least 1 peptide pool for a specific HIV-1 protein was positive, then the overall response to the protein was considered positive (47). Data were filtered if background responses (DMSO control) were greater than 0.1% cytokine secretion, or if fewer than 5,000 events occurred within the CD4+ or CD8+ T cell subpopulations. Data were available for 76 participants for CD8+ T cell responses; data for 1 participant were filtered due to a high background for CD4+ T cell responses.
Statistics.
For samples to be included in the immunogenicity evaluation of the tested vaccine regimens, their corresponding blood draw date had to be within the allowable visit window specified by the protocol. Confidence intervals for response rates were calculated with the score test method (48). Fisher’s exact tests and Wilcoxon rank-sum tests were used to compare response rates and response magnitudes among vaccine groups. The Cochran-Armitage trend test (49, 50) for response rates and the Jonckheere-Terpstra test (51) for response magnitudes were used to assess the effect of increasing Ad5 dosages among Ad5lo/NYVAC and Ad5hi/NYVAC groups on vaccine-induced immune responses. Two-sided P values adjusted by the Holm method (52) were reported for the primary immunogenicity endpoint (ICS). Two-sided P values without any multiplicity adjustment were reported for all other exploratory immunogenicity endpoints, and a P value of less than 0.05 was considered significant. Box plots were used to summarize the distribution of various immune responses, where the mid-line of the box denotes the median, and the ends of the box denote the 25th and 75th percentiles, with whiskers extended to the extreme data points that are no more than 1.5 times the interquartile range or, if no values meet this criterion, to the data extremes. Where there are both positive and negative responses, these summaries in the box plots refer to the positive responses.
Magnitude and breadth (MB) plots (53) were used to display the potency and breadth of nAb responses over a panel of viruses. The x axis of an M-B curve represents the threshold of neutralization that was considered positive, and the y axis represents the fraction of isolates neutralized. The AUC-MB was calculated as the average of the log10 nAb titer over the panel of isolates. The Wilcoxon rank-sum test was used to test for a difference in AUC-MB distribution between 2 independent groups. Similar MB curves were used to compare responses in the Vax003, Vax004, and RV144 HIV-1 vaccine efficacy trials (54).
Study approval.
The study protocol was approved by the institutional ethics committee of the Centre Hospitalier Universitaire Vaudois (CHUV) (Lausanne, Switzerland) and by Swissmedic, the Swiss Agency for Therapeutic Products (Bern, Switzerland). All study participants provided written informed consent prior to participation.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the NIAID of the NIH under award numbers UM1AI068618, AI068635, AI068614, and AI069443. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
This trial was conducted by the HVTN. We gratefully acknowledge the participation and support of many colleagues and staff on the protocol team and at the site and are particularly grateful for the participation of the 80 study participants. We thank Stephen Voght for technical editing of the manuscript. We thank Stephen De Rosa and Donald Carter for oversight of flow cytometric experiments; Terri Stewart, Kevin Hawkins, Jane Vasilyeva, Paul Newling, Vicki Ashley, Judith Lucas, and Yong Lin for technical assistance; Marcella Sarzotti-Kelsoe and Michael Stirewalt for quality assurance oversight; and Daryl Morris for the polyfunctionality analysis. We thank the James B. Pendleton Charitable Trust for their generous equipment donation.
Footnotes
Note regarding evaluation of this manuscript: Manuscripts authored by scientists associated with Duke University, The University of North Carolina at Chapel Hill, Duke-NUS, and the Sanford-Burnham Medical Research Institute are handled not by members of the editorial board but rather by the science editors, who consult with selected external editors and reviewers.
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2014;124(11):4843–4856. doi:10.1172/JCI75894.
References
- 1. (UNAIDS) JUNPoHA. WHO Library Cataloguing-in-Publication Data. 2nd ed. Geneva, Switzerland; 2012. [Google Scholar]
- 2.Tan WG, et al. Comparative analysis of simian immunodeficiency virus Gag-specific effector and memory CD8(+) T cells induced by different adenovirus vectors. J Virol. 2013;87(3):1359–1372. doi: 10.1128/JVI.02055-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buchbinder SP, et al. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet. 2008;372(9653):1881–1893. doi: 10.1016/S0140-6736(08)61591-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barouch DH, et al. Characterization of humoral and cellular immune responses elicited by a recombinant adenovirus serotype 26 HIV-1 Env vaccine in healthy adults (IPCAVD 001). J Infect Dis. 2013;207(2):248–256. doi: 10.1093/infdis/jis671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Churchyard GJ, et al. A phase IIA randomized clinical trial of a multiclade HIV-1 DNA prime followed by a multiclade rAd5 HIV-1 vaccine boost in healthy adults (HVTN204). PLoS One. 2011;6(8):e21225. doi: 10.1371/journal.pone.0021225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rerks-Ngarm S, et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med. 2009;361(23):2209–2220. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- 7.Keefer MC, et al. A phase I trial of preventive HIV vaccination with heterologous poxviral-vectors containing matching HIV-1 inserts in healthy HIV-uninfected subjects. Vaccine. 2011;29(10):1948–1958. doi: 10.1016/j.vaccine.2010.12.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Egan MA, et al. Induction of human immunodeficiency virus type 1 (HIV-1)-specific cytolytic T lymphocyte responses in seronegative adults by a nonreplicating, host-range-restricted canarypox vector (ALVAC) carrying the HIV-1MN env gene. J Infect Dis. 1995;171(6):1623–1627. doi: 10.1093/infdis/171.6.1623. [DOI] [PubMed] [Google Scholar]
- 9.Harari A, et al. An HIV-1 clade C DNA prime, NYVAC boost vaccine regimen induces reliable, polyfunctional, and long-lasting T cell responses. J Exp Med. 2008;205(1):63–77. doi: 10.1084/jem.20071331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goepfert PA, et al. Phase 1 safety and immunogenicity testing of DNA and recombinant modified vaccinia Ankara vaccines expressing HIV-1 virus-like particles. J Infect Dis. 2011;203(5):610–619. doi: 10.1093/infdis/jiq105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bart PA, et al. EV01: a phase I trial in healthy HIV negative volunteers to evaluate a clade C HIV vaccine, NYVAC-C undertaken by the EuroVacc Consortium. Vaccine. 2008;26(25):3153–3161. doi: 10.1016/j.vaccine.2008.03.083. [DOI] [PubMed] [Google Scholar]
- 12.McCormack S, et al. EV02: a Phase I trial to compare the safety and immunogenicity of HIV DNA-C prime-NYVAC-C boost to NYVAC-C alone. Vaccine. 2008;26(25):3162–3174. doi: 10.1016/j.vaccine.2008.02.072. [DOI] [PubMed] [Google Scholar]
- 13.Hayes P, et al. Safety and immunogenicity of DNA prime and modified vaccinia ankara virus-HIV subtype C vaccine boost in healthy adults. Clin Vaccine Immunol. 2013;20(3):397–408. doi: 10.1128/CVI.00637-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Priddy FH, et al. Safety and immunogenicity of a replication-incompetent adenovirus type 5 HIV-1 clade B gag/pol/nef vaccine in healthy adults. Clin Infect Dis. 2008;46(11):1769–1781. doi: 10.1086/587993. [DOI] [PubMed] [Google Scholar]
- 15.Frahm N, et al. Human adenovirus-specific T cells modulate HIV-specific T cell responses to an Ad5-vectored HIV-1 vaccine. J Clin Invest. 2012;122(1):359–367. doi: 10.1172/JCI60202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diaz-Montero CM, et al. Phase 1 studies of the safety and immunogenicity of electroporated HER2/CEA DNA vaccine followed by adenoviral boost immunization in patients with solid tumors. J Transl Med. 2013;11:62. doi: 10.1186/1479-5876-11-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Casimiro DR, et al. Heterologous human immunodeficiency virus type 1 priming-boosting immunization strategies involving replication-defective adenovirus and poxvirus vaccine vectors. J Virol. 2004;78(20):11434–11438. doi: 10.1128/JVI.78.20.11434-11438.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Betts G, et al. Optimising immunogenicity with viral vectors: mixing MVA and HAdV-5 expressing the mycobacterial antigen Ag85A in a single injection. PLoS One. 2012;7(12):e50447. doi: 10.1371/journal.pone.0050447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ratto-Kim S, et al. Heterologous prime-boost regimens using rAd35 and rMVA vectors elicit stronger cellular immune responses to HIV proteins than homologous regimens. PLoS One. 2012;7(9):e45840. doi: 10.1371/journal.pone.0045840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barouch DH, et al. Vaccine protection against acquisition of neutralization-resistant SIV challenges in rhesus monkeys. Nature. 2012;482(7383):89–93. doi: 10.1038/nature10766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peiperl L, et al. Safety and immunogenicity of a replication-defective adenovirus type 5 HIV vaccine in Ad5-seronegative persons: a randomized clinical trial (HVTN 054). PLoS One. 2010;5(10):e13579. doi: 10.1371/journal.pone.0013579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hammer SM, et al. Efficacy trial of a DNA/rAd5 HIV-1 preventive vaccine. N Engl J Med. 2013;369(22):2083–2092. doi: 10.1056/NEJMoa1310566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sin JI, Bagarazzi M, Pachuk C, Weiner DB. DNA priming-protein boosting enhances both antigen-specific antibody and Th1-type cellular immune responses in a murine herpes simplex virus-2 gD vaccine model. DNA Cell Biol. 1999;18(10):771–779. doi: 10.1089/104454999314917. [DOI] [PubMed] [Google Scholar]
- 24.Park SH, Yang SH, Lee CG, Youn JW, Chang J, Sung YC. Efficient induction of T helper 1 CD4+ T-cell responses to hepatitis C virus core and E2 by a DNA prime-adenovirus boost. Vaccine. 2003;21(31):4555–4564. doi: 10.1016/S0264-410X(03)00499-7. [DOI] [PubMed] [Google Scholar]
- 25.Harari A, et al. NYVAC immunization induces polyfunctional HIV-specific T-cell responses in chronically-infected, ART-treated HIV patients. Eur J Immunol. 2012;42(11):3038–3048. doi: 10.1002/eji.201242696. [DOI] [PubMed] [Google Scholar]
- 26.Haynes BF, et al. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N Engl J Med. 2012;366(14):1275–1286. doi: 10.1056/NEJMoa1113425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mascola JR, Montefiori DC. The role of antibodies in HIV vaccines. Annu Rev Immunol. 2010;28:413–444. doi: 10.1146/annurev-immunol-030409-101256. [DOI] [PubMed] [Google Scholar]
- 28.Lu S. Heterologous prime-boost vaccination. Curr Opin Immunol. 2009;21(3):346–351. doi: 10.1016/j.coi.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zolla-Pazner S, et al. Vaccine-induced IgG antibodies to V1V2 regions of multiple HIV-1 subtypes correlate with decreased risk of HIV-1 infection. PLoS One. 2014;9(2):e87572. doi: 10.1371/journal.pone.0087572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tomaras GD, et al. Vaccine-induced plasma IgA specific for the C1 region of the HIV-1 envelope blocks binding and effector function of IgG. Proc Natl Acad Sci U S A. 2013;110(22):9019–9024. doi: 10.1073/pnas.1301456110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Janes H, et al. Vaccine-induced gag-specific T cells are associated with reduced viremia after HIV-1 infection. J Infect Dis. 2013;208(8):1231–1239. doi: 10.1093/infdis/jit322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Rosa SC, et al. HIV-DNA priming alters T cell responses to HIV-adenovirus vaccine even when responses to DNA are undetectable. J Immunol. 2011;187(6):3391–3401. doi: 10.4049/jimmunol.1101421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aste-Amezaga M, et al. Quantitative adenovirus neutralization assays based on the secreted alkaline phosphatase reporter gene: application in epidemiologic studies and in the design of adenovector vaccines. Hum Gene Ther. 2004;15(3):293–304. doi: 10.1089/104303404322886147. [DOI] [PubMed] [Google Scholar]
- 34.Schulz KF, Altman DG, Moher D, Group C. CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. PLoS Med. 2010;7(3):e1000251. doi: 10.1371/journal.pmed.1000251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bull M, et al. Defining blood processing parameters for optimal detection of cryopreserved antigen-specific responses for HIV vaccine trials. J Immunol Methods. 2007;322(1-2):57–69. doi: 10.1016/j.jim.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tomaras GD, et al. Initial B-cell responses to transmitted human immunodeficiency virus type 1: virion-binding immunoglobulin M (IgM) and IgG antibodies followed by plasma anti-gp41 antibodies with ineffective control of initial viremia. J Virol. 2008;82(24):12449–12463. doi: 10.1128/JVI.01708-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tomaras GD, et al. Polyclonal B cell responses to conserved neutralization epitopes in a subset of HIV-1-infected individuals. J Virol. 2011;85(21):11502–11519. doi: 10.1128/JVI.05363-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gottardo R, et al. Plasma IgG to linear epitopes in the V2 and V3 regions of HIV-1 gp120 correlate with a reduced risk of infection in the RV144 vaccine efficacy trial. PLoS One. 2013;8(9):e75665. doi: 10.1371/journal.pone.0075665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seaman MS, et al. Tiered categorization of a diverse panel of HIV-1 Env pseudoviruses for assessment of neutralizing antibodies. J Virol. 2010;84(3):1439–1452. doi: 10.1128/JVI.02108-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li M, et al. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J Virol. 2005;79(16):10108–10125. doi: 10.1128/JVI.79.16.10108-10125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Montefiori DC. Evaluating neutralizing antibodies against HIV, SIV, and SHIV in luciferase reporter gene assays. Curr Protoc Immunol. 2005;Chapter 12:Unit 12.11. doi: 10.1002/0471142735.im1211s64. [DOI] [PubMed] [Google Scholar]
- 42.Edmonds TG, et al. Replication competent molecular clones of HIV-1 expressing Renilla luciferase facilitate the analysis of antibody inhibition in PBMC. Virology. 2010;408(1):1–13. doi: 10.1016/j.virol.2010.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McLinden RJ, et al. Detection of HIV-1 neutralizing antibodies in a human CD4(+)/CXCR4(+)/CCR5(+) T-lymphoblastoid cell assay system. PLoS One. 2013;8(11):e77756. doi: 10.1371/journal.pone.0077756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sarzotti-Kelsoe M, et al. Optimization validation of the TZM-bl assay for standardized assessments of neutralizing antibodies against HIV-1. J Immunol Methods. 2014;409C:131–146. doi: 10.1016/j.jim.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sarzotti-Kelsoe M, et al. Optimization validation of a neutralizing antibody assay for HIV-1 in A3R5 cells. J Immunol Methods. 2014;409:147–160. doi: 10.1016/j.jim.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li F, et al. Peptide selection for human immunodeficiency virus type 1 CTL-based vaccine evaluation. Vaccine. 2006;24(47–48):6893–6904. doi: 10.1016/j.vaccine.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 47.Horton H, et al. Optimization and validation of an 8-color intracellular cytokine staining (ICS) assay to quantify antigen-specific T cells induced by vaccination. J Immunol Methods. 2007;323(1):39–54. doi: 10.1016/j.jim.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Agresti A, Coull BA. Approximate is better than ‘exact’ for interval estimation of binomial proportions. Am Stat. 1998;52(17):119–126. [Google Scholar]
- 49.Chochran WG. Some methods for strengthening the common chi-squared tests. Biometrics. 1954;10(4):417–451. doi: 10.2307/3001616. [DOI] [Google Scholar]
- 50.Armitage P. Tests for linear trends in proportions and frequencies. Biometrics. 1955;11(3):375–386. doi: 10.2307/3001775. [DOI] [Google Scholar]
- 51.Jonckheere AR. A distribution-free k-sample test again ordered alternatives. Biometrika. 1954;41:133–145. [Google Scholar]
- 52.Holm S. A simple sequentially rejective multiple test procedure. Scand J Statist. 1979;6(2):65–70. [Google Scholar]
- 53.Huang Y, Gilbert PB, Montefiori DC, Self SG. Simultaneous evaluation of the magnitude and breadth of a left and right censored multivariate response, with application to HIV vaccine development. Stat Biopharm Res. 2009;1(1):81–91. doi: 10.1198/sbr.2009.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Montefiori DC, et al. Magnitude and breadth of the neutralizing antibody response in the RV144 and Vax003 HIV-1 vaccine efficacy trials. J Infect Dis. 2012;206(3):431–441. doi: 10.1093/infdis/jis367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.