

Abstract
Transport of oxygen by red blood cells (rbc) is critical for life and embryogenesis. Here, we determined that provision of the lipid mediator sphingosine 1-phosphate (S1P) to the systemic circulation is an essential function of rbc in embryogenesis. Mice with rbc-specific deletion of sphingosine kinases 1 and 2 (Sphk1 and Sphk2) showed embryonic lethality between E11.5 and E12.5 due to defects in vascular development. Administration of an S1P1 receptor agonist to pregnant dams rescued early embryonic lethality. Even though rbc-specific Sphk1 Sphk2–KO embryos were anemic, the erythropoietic capacity of hematopoietic stem cells (HSCs) was not impaired, suggesting that rbc can develop in the absence of sphingosine kinase activity. Indeed, transplantation of HSCs deficient for Sphk1 and Sphk2 into adult mice produced rbc that lacked S1P and attenuated plasma S1P levels in recipients. However, in adult animals, both rbc and endothelium contributed to plasma S1P. Together, these findings demonstrate that rbc are essential for embryogenesis by supplying the lysophospholipid S1P, which regulates embryonic vascular development via its receptors.
Introduction
Vascular and hematopoietic systems share developmental origins. Early vascular development is initiated by transcription factors such as HIF-1α that respond to low oxygen levels and induce VEGF, a key angiogenic factor (1, 2). On the other hand, hematopoietic stem cells (HSCs) develop from the hemogenic endothelium, which is originally derived from endothelial cells (ECs) of the embryonic vasculature (3). In addition, hypoxia also induces erythropoietin, which acts on HSCs to differentiate into embryonic red blood cells (rbc) (4). Hypoxia, in turn, is alleviated by the delivery of oxygen by rbc, which circulate in newly formed vessels. Indeed, rbc are essential for embryogenesis, since mice that lack rbc die at midgestation (5). In the adult, rbc abnormalities lead to vascular disorders in sickle cell syndrome, hemoglobinopathies, anemias, polycythemias, and infections such as cerebral malaria (6, 7). However, whether rbc regulate embryonic development by secreted factors is not known.
The lysophospholipid mediator S1P, produced from the metabolism of membrane sphingolipids, is secreted into the extracellular environment in vertebrates (8). Due to its poor water solubility, it is bound to chaperones — for example, apolipoprotein M (ApoM) and albumin in the plasma (9). S1P signals via its 5 G protein–coupled receptors to regulate various processes such as vascular development (10) and lymphocyte trafficking (11). For example, the primary vascular network formed by the fusion of new vascular sprouts need S1P signaling via the S1P1 receptor for stabilization and further differentiation (12, 13). However, S1P1 cooperates with other S1P receptors that have distinct signaling properties for regulating vascular development (14). Thus, compound S1P receptor–null mice, i.e., S1pr1 S1pr2–double-KO or S1pr1 S1pr2 S1pr3–triple-KO mice exhibit much more severe vascular developmental anomalies than do single S1pr1–KO mice, which are lethal at E12.5 to E14.5 of development. The source of embryonic S1P that is needed for vascular development is not known. Postnatally, rbc and ECs are thought to supply plasma S1P under homeostasis (15–17). Thus, rbc may serve as the source of S1P for embryonic development. In this study, we developed a mouse model in which sphingosine kinase genes (Sphk1 and Sphk2) (15) were deleted in rbc using erythropoietin receptor-Cre (EpoR-Cre) deleter mice (RBC Sphk dKO mice) (18), which provided an opportunity to assess the importance of rbc-derived S1P during embryonic development. Our results reveal a novel essential function of rbc in embryogenesis, that of S1P provision for vascular development.
Results and Discussion
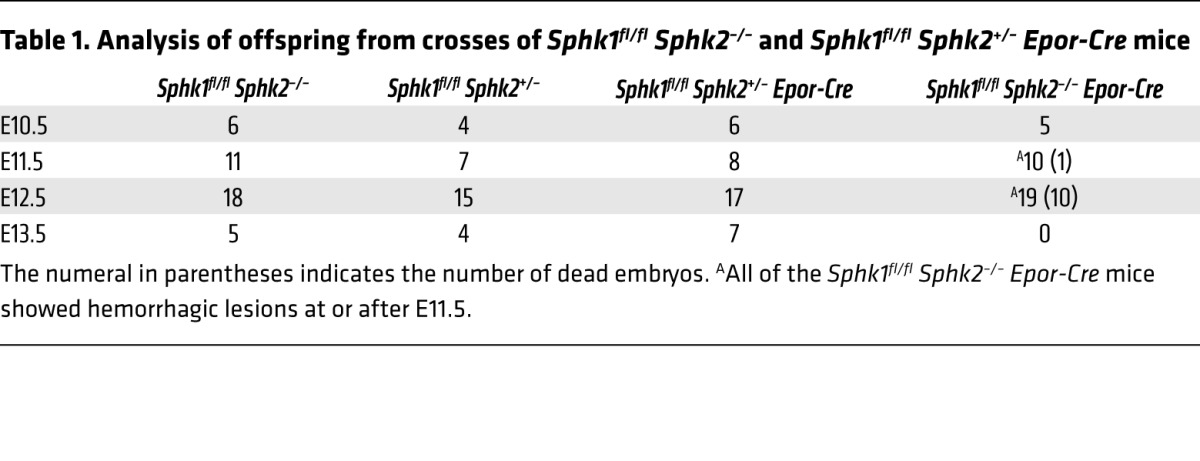
Sphk enzymes are essential for S1P production during embryogenesis (19). Sphk2 mRNA is expressed during primitive, definitive, and adult erythropoiesis, whereas Sphk1 mRNA is induced in adult erythropoiesis (20). This EpoR-Cre deleter mouse strain was shown to delete LoxP site–engineered genes specifically and efficiently in erythrocyte progenitors during early embryonic erythropoiesis (18). Indeed, purified Ter119+ erythrocyte progenitors and embryonic rbc isolated from E12.5 embryos showed almost complete suppression of the Sphk1 transcript, suggesting highly efficient gene deletion in this lineage. In contrast, minimal suppression (~15%) of the Sphk1 transcript was seen in FACS-sorted CD31+ ECs, consistent with the knowledge that EpoR-Cre is expressed at low levels in ECs of the developing vasculature in the mouse (ref. 18 and Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI77685DS1). Heterozygous crosses indicated that RBC Sphk dKO mutations caused embryonic lethality at E11.5 to E12.5. All of the RBC Sphk dKO embryos exhibited hemorrhage, especially in the head region. In addition, cardiac edema, pale yolk sac, and limb morphogenic defects were seen (Figure 1, A and B). No viable RBC Sphk dKO embryos were recovered at E13.5 (Table 1). In contrast, heterozygous embryos were viable and reproduced normally as adult mice, suggesting that complete loss of function of Sphk genes specifically in the rbc lineage causes embryonic lethality.
Figure 1. RBC Sphk dKO causes embryonic lethality, with vascular and cardiac defects.

(A) Photomicrographs of developing embryos. High-power magnification images show the limb buds from the boxed areas of E12.5 embryos. E12.5 RBC Sphk dKO embryos displayed severe hemorrhages (arrows). Original magnification, ×10.5. (B) Hemorrhage of brain vessels (original magnification, ×63) and pale yolk sac (scale bars: 1 mm) observed in E12.5 embryos from RBC Sphk dKO. (C) Vascular morphology in the yolk sac, head region, aorta (section), and hindbrain. Scale bars: 250 μm. Sphk1fl/fl Sphk2+/– mice were used as controls.
Table 1.
Analysis of offspring from crosses of Sphk1fl/fl Sphk2–/– and Sphk1fl/fl Sphk2+/– Epor-Cre mice
To rigorously examine whether embryonic lethality of RBC Sphk dKO mice is due to the loss of expression of Sphk genes in vascular endothelium, we examined whether endothelial-specific deletion of Sphk1 and Sphk2 (EC Sphk dKO) would lead to similar embryonic lethality. We used the Cdh5-Cre-ERT2 deleter strain, which was shown to delete genes specifically in ECs after induction with tamoxifen (21). As shown in Supplemental Figure 2, even though a significant (>75%) reduction of Sphk1 mRNA was achieved in the background of a complete lack of Sphk2 expression in the endothelium in EC Sphk dKO mice, the embryos did not show the hemorrhagic phenotype and were completely viable and appeared normal at E13.5. Indeed, our previous studies showed that global loss of 3 alleles of Sphk (Sphk1–/– Sphk2+/–) is not embryonically lethal and is compatible with normal vascular development (16). These results strongly suggest that Sphk expression in the endothelium is dispensable for embryonic development.
Analysis of the yolk sac blood vessels from E12.5 RBC Sphk dKO embryos (Figure 1C) showed hypersprouting of capillary networks, increased vascular density, and morphogenetic defects of larger vessels. Even though RBC Sphk dKO yolk sac arteries were covered with smooth muscle actin–positive (SMA+) mural cells, some ectopic sprouts were also ensheathed with SMA+ mural cells, resulting in a jagged appearance. In the head region, we observed hypersprouting of small vessels and morphologic abnormalities of the larger structures. The dorsal aorta in the mid-region of the embryo exhibited patchy SMA+ mural cell coverage and hypersprouting and hyperplastic ECs in the non-SMA+ regions. In the embryonic hindbrain, angiogenic vessels showed clear evidence of hypersprouting, excessive branching, and morphologic alterations (Figure 1C). RBC Sphk dKO mice also exhibited cardiac developmental defects. At E12.5, RBC Sphk dKO embryos demonstrated signs of heart failure. The ventricular myocardium of RBC Sphk dKO embryos was severely hypoplastic, often only 1 to 2 cell layers thick compared with a thickness of more than 4 to 5 cell layers in littermate control hearts, and the epicardium was detached from the myocardial surface (Supplemental Figure 3A). Staining for proliferating cells with Ki67 antibody and for apoptotic cells with caspase 3 antibody revealed no significant changes in cardiomyocyte proliferation or apoptosis (Supplemental Figure 3B). Moreover, we found that cardiac differentiation gene expression levels were similar (≤2-fold difference) in control and RBC Sphk dKO heart tissue (Supplemental Figure 3C). These results suggest that the cardiac abnormalities we observed in the RBC Sphk dKO embryos were likely secondary to vascular defects. We speculate that altered hemodynamics in the circulatory system due to vascular developmental defects induced cardiac morphological changes. Thus, RBC Sphk dKO embryos showed clear evidence of severe vascular developmental defects that phenocopied the S1pr1 endothelial–KO mice (22) but were substantially more severe and occurred at an earlier stage of development.
Sphingolipid levels in embryonic rbc and tissues were quantified by liquid chromatography-tandem mass spectrometry (LC-MS/MS). S1P levels in circulating rbc and the embryo proper were almost completely (99% and 95%, respectively) downregulated in the RBC Sphk dKO mice (Figure 2, A and B). Sphingosine and ceramide levels were upregulated, especially in circulating rbc, suggesting that a lack of Sphk enzymes in rbc resulted in a metabolic pileup of precursor sphingolipids. These data strongly suggest that rbc supply almost all of embryonic S1P.
Figure 2. Embryonic rbc–derived S1P acts on the S1P1 receptor to regulate vascular development.

S1P, sphingosine (Sph), and ceramide (Cer) levels in (A) circulating blood cells and (B) embryonic tissues from E12.5 embryos. *P < 0.05; **P < 0.01. n = 3 per group. Data represent the mean ± SD. (C) Representative E13.5 embryos rescued by maternal SEW2871 administration. Untreated RBC Sphk dKO embryos were resorbed at E13.5 and not available for morphological assessment (D) Survival of RBC Sphk dKO embryos at various stages of development with and without administration of maternal SEW2871.
We next examined the hypothesis that circulating S1P activation of S1P receptors in the endothelium is essential for vascular development. Since S1pr1 function is essential for vascular development, while S1pr2 and S1pr3 cooperate with it during embryogenesis (14, 22), we used a pharmacological tool, SEW2871, which specifically activates the S1P1 receptor (23). We administered SEW2871 (10 mg/kg) to pregnant mice from E8.5 to E13.5 of gestation. Maternal SEW2871 administration rescued early E11.5–12.5 lethality in a significant fraction (90%) of embryos (Figure 2, C and D). At E13.5, SEW2871-treated RBC Sphk dKO mice embryos were viable, with a visible heartbeat even though some hemorrhage and edema were seen, suggesting partial rescue of the vascular defects. Inability of SEW2871 to activate S1P2 and S1P3 as well as limited bioavailability of this compound in the embryo following maternal administration likely contributed to the partial rescue. Nevertheless, this result strongly suggests that embryonic rbc–derived circulating S1P acts on the S1P1 receptor in developing vascular ECs as an essential circulating signal.
In addition to widespread hemorrhage, we observed an ~50% reduction in the number of circulating rbc (Supplemental Figure 4A) in RBC Sphk dKO embryos. In addition, RBC Sphk dKO embryos contained only ~50% of Lin–Sca1+c-Kit+ (LSK) hematopoietic progenitor cells (Supplemental Figure 4B). This could be caused by either an intrinsic defect in erythropoiesis or an indirect reduction in rbc numbers due to bleeding. To distinguish between these possibilities, we obtained fetal liver cells from SEW2871-treated E12.5 RBC Sphk dKO embryos and used them in hematopoietic reconstitution assays in irradiated C57Bl/6 recipients. As shown in Supplemental Figure 4C, almost complete (>93%) hematopoietic reconstitution was achieved. Mice that received either WT or RBC Sphk dKO fetal liver cells achieved normal hematocrit levels and similar hematopoietic cell distribution (Figure 3A). In addition, rbc morphology, as determined by both light and transmission electron microscopy, was similar (Figure 3B). While we cannot fully exclude the stage-specific functional roles of Sphk enzymes in primitive and/or fetal definitive erythropoiesis, these results strongly suggest that Sphk enzymes are not needed for adult erythropoiesis and that the midgestation anemia observed in the RBC Sphk dKO embryos was likely due to vascular defects that led to hemorrhage.
Figure 3. Adult erythropoiesis in the absence of Sphk enzymes.

(A) Lethally irradiated WT mice were reconstituted with control (Ctl) or RBC Sphk dKO (KO) fetal liver cells. Peripheral blood cell counts for the transplanted mice. HGB, hemoglobin; HCT, hematocrit; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; RDW-SD, red cell distribution width-standard deviation; RDW-CV, red cell distribution width-coefficient of variation; RET, reticulocytes; PLT, platelets; PDW, platelet distribution width; MPV, mean platelet volume. (B) Eosin staining and transmission electron microscopy images of rbc. Original magnification, ×63 and ×15,000, respectively. Plasma and rbc S1P levels in (C) transplanted mice, (D) WT and Sphk1fl/fl Sphk2+/– Epor-Cre mice, and (E) Sphk1fl/fl Sphk2–/– and EC Sphk dKO mice. n = 3 per group. **P < 0.01. All data represent the mean ± SD.
We also measured rbc and plasma sphingolipid levels in the mice with fetal liver hematopoietic transplants. As shown in Figure 3C, rbc S1P levels were reduced ~85% in the recipients of fetal liver–derived HSCs from RBC Sphk dKO embryos. In contrast, plasma S1P was not reduced to the same extent. Indeed, even in heterozygous RBC Sphk dKO mice (Sphk1fl/fl Sphk2+/– EpoR-Cre), rbc S1P was markedly reduced by ~98%, whereas plasma S1P did not decrease to the same extent (Figure 3D). These data suggest that even though rbc are one of the major sources of circulatory S1P in the postnatal period, alternative sources of S1P exist to maintain plasma S1P levels. This is consistent with previous findings using the inducible Mx-Cre–mediated deletion of Sphk1 and Sphk2 in hematopoietic cells of adult mice, which first identified the rbc as a major source of plasma S1P (15). In addition, Sphk1–/– mice show an ~50% reduction in plasma S1P (16). However, based on indirect in vivo evidence and studies using ECs in vitro, we suggested that vascular endothelium might be an alternative source of plasma S1P (16). Indeed, endothelial-specific KO of the S1P transporter Spns2 reduced plasma S1P levels by ~30% (17). To address this issue, we analyzed plasma S1P levels in adult mice after the Sphk1 and Sphk2 genes were deleted using endothelial-specific Cdh5-Cre-ERT2 deleter mice (21). As shown in Figure 3E, rbc S1P levels were unchanged, whereas plasma S1P levels were reduced by ~30% in EC Sphk dKO mice, suggesting that the endothelium is the alternative source in the adult. Plasma S1P levels are stable under homeostasis, but it is not known how fluctuations in plasma S1P are sensed in vertebrates, which maintain the vascular S1P gradient. Indeed, changes in plasma S1P levels in plasma S1P–less mice and S1P lyase–KO mice are associated with immune cell trafficking defects and inflammatory episodes, respectively (15, 24). Our results indicate that during embryogenesis, rbc are the major source of organismal S1P, which cannot be supplied by other tissues, whereas in the adult, the endothelium serves as an alternate source.
These studies reveal a heretofore unrecognized and essential function of rbc in embryonic development. We found that rbc provision of plasma S1P is needed for vascular plexus stabilization, maturation, and remodeling. We also provide evidence that S1P1 is a target of rbc-derived S1P during vascular development. It is not clear at present how rbc export S1P to the circulation. Although we cannot at this stage exclude the possibility that direct rbc contact with the capillary endothelium is involved in S1P receptor activation, we favor the possibility that secretion of S1P into the embryonic plasma and activation of S1P as a circulating factor are important. This is based on the detailed analysis of embryonic vascular defects in RBC Sphk dKO mice, which show morphological alterations not only in capillaries that are likely to be in direct contact with circulating rbc, but also in larger vessels whose lumens are much larger than the diameter of rbc. Thus, we propose that embryonic rbc secrete S1P as an “erythrocrine” factor that is needed for vascular stabilization, maturation, and remodeling. This represents a yet another signaling connection between erythropoiesis and angiogenesis. We speculate that this and other mechanistic connections may exist between rbc and the endothelium under homeostasis and that such connections may be deregulated in rbc diseases such as malaria, thalassemia, anemia, and polycythemias, which manifest vascular dysfunction and pathology.
Methods
Detailed information can be found in the Supplemental Methods.
Statistics.
All results are expressed as the means ± SD. All data were analyzed using a 2-tailed Student’s t test or 1-way ANOVA with Tukey’s multiple comparison test. A P value less than 0.05 was considered statistically significant.
Study approval.
All animal studies were approved by the IACUC of Weill Cornell Medical College.
Supplementary Material
Acknowledgments
We thank Stuart Orkin for advice regarding the Epor-Cre mice; Todd Evans, Ralph Nachman, Stefano Rivella, and S.K. Dey for critical comments; Lee Cohen-Gould for help with transmission electron microscopy experiments; Jason McCormick for FACS experiments; Sushmita Mukherjee for 2-photon microscopy; Ursula Klingmüller and Ralf H. Adams for the gift of unique mouse strains; and Jacek Bielawski for sphingolipid measurements. This work was supported by NIH grants HL-067330, HL-089934, and CA-077839 (to T. Hla) and intramural grants from the NIDDK, NIH (to R.L. Proia). Sphingolipid measurements were partially supported by Lipidomics Shared Resource, the Hollings Cancer Center, the Medical University of South Carolina (P30 CA138313), and the Lipidomics Core at the SC Lipidomics and Pathobiology COBRE (P20 RR017677).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2014;124(11):4823–4828. doi:10.1172/JCI77685.
References
- 1.Ferrara N, et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380(6573):439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 2.Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359(6398):843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- 3.Chen MJ, et al. Erythroid/myeloid progenitors and hematopoietic stem cells originate from distinct populations of endothelial cells. Cell Stem Cell. 2011;9(6):541–552. doi: 10.1016/j.stem.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orkin SH, Zon LI. Genetics of erythropoiesis: induced mutations in mice and zebrafish. Annu Rev Genet. 1997;31:33–60. doi: 10.1146/annurev.genet.31.1.33. [DOI] [PubMed] [Google Scholar]
- 5.Fujiwara Y, Browne CP, Cunniff K, Goff SC, Orkin SH. Arrested development of embryonic red cell precursors in mouse embryos lacking transcription factor GATA-1. Proc Natl Acad Sci U S A. 1996;93(22):12355–12358. doi: 10.1073/pnas.93.22.12355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caplan LR, Fisher M. The endothelium, platelets, and brain ischemia. Rev Neurol Dis. 2007;4(3):113–121. [PubMed] [Google Scholar]
- 7.Francischetti IM, Seydel KB, Monteiro RQ. Blood coagulation, inflammation, and malaria. Microcirculation. 2008;15(2):81–107. doi: 10.1080/10739680701451516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blaho VA, Hla T. Regulation of mammalian physiology, development, and disease by the sphingosine 1-phosphate and lysophosphatidic acid receptors. Chem Rev. 2011;111(10):6299–6320. doi: 10.1021/cr200273u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christoffersen C, et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc Natl Acad Sci U S A. 2011;108(23):9613–9618. doi: 10.1073/pnas.1103187108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mendelson K, Evans T, Hla T. Sphingosine 1-phosphate signalling. Development. 2014;141(1):5–9. doi: 10.1242/dev.094805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cyster JG, Schwab SR. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol. 2012;30:69–94. doi: 10.1146/annurev-immunol-020711-075011. [DOI] [PubMed] [Google Scholar]
- 12.Gaengel K, et al. The sphingosine-1-phosphate receptor S1PR1 restricts sprouting angiogenesis by regulating the interplay between VE-cadherin and VEGFR2. Dev Cell. 2012;23(3):587–599. doi: 10.1016/j.devcel.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 13.Jung B, et al. Flow-regulated endothelial S1P receptor-1 signaling sustains vascular development. Dev Cell. 2012;23(3):600–610. doi: 10.1016/j.devcel.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kono M, et al. The sphingosine-1-phosphate receptors S1P1, S1P2, and S1P3 function coordinately during embryonic angiogenesis. J Biol Chem. 2004;279(28):29367–29373. doi: 10.1074/jbc.M403937200. [DOI] [PubMed] [Google Scholar]
- 15.Pappu R, et al. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science. 2007;316(5822):295–298. doi: 10.1126/science.1139221. [DOI] [PubMed] [Google Scholar]
- 16.Venkataraman K, et al. Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ Res. 2008;102(6):669–676. doi: 10.1161/CIRCRESAHA.107.165845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fukuhara S, et al. The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J Clin Invest. 2012;122(4):1416–1426. doi: 10.1172/JCI60746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heinrich AC, Pelanda R, Klingmuller U. A mouse model for visualization and conditional mutations in the erythroid lineage. Blood. 2004;104(3):659–666. doi: 10.1182/blood-2003-05-1442. [DOI] [PubMed] [Google Scholar]
- 19.Mizugishi K, Yamashita T, Olivera A, Miller GF, Spiegel S, Proia RL. Essential role for sphingosine kinases in neural and vascular development. Mol Cell Biol. 2005;25(24):11113–11121. doi: 10.1128/MCB.25.24.11113-11121.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kingsley PD, et al. Ontogeny of erythroid gene expression. Blood. 2013;121(6):e5–e13. doi: 10.1182/blood-2012-04-422394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pitulescu ME, Schmidt I, Benedito R, Adams RH. Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat Protoc. 2010;5(9):1518–1534. doi: 10.1038/nprot.2010.113. [DOI] [PubMed] [Google Scholar]
- 22.Liu Y, et al. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Invest. 2000;106(8):951–961. doi: 10.1172/JCI10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jo E, et al. S1P1-selective in vivo-active agonists from high-throughput screening: off-the-shelf chemical probes of receptor interactions, signaling, and fate. Chem Biol. 2005;12(6):703–715. doi: 10.1016/j.chembiol.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 24.Allende ML, et al. Sphingosine-1-phosphate lyase deficiency produces a pro-inflammatory response while impairing neutrophil trafficking. J Biol Chem. 2011;286(9):7348–7358. doi: 10.1074/jbc.M110.171819. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.