

Abstract
Fibrotic disorders account for an increasing burden of disease-associated morbidity and mortality worldwide. Although numerous risk factors have been recognized, the etiologies of many of these clinical syndromes have not been identified, and they are often termed idiopathic or cryptogenic. Here, we provide an evolutionary perspective on fibrosis aimed at elucidating its etiopathogenesis. By asking the ultimate question of “why” this process evolved in multicellular organisms, we hope to uncover proximate explanations for “how” it causes disease in humans. We posit that physiological fibrosis-like reactions evolved as an essential process in host defense against pathogens and in normal wound healing. Based on this premise, we reason that pathological fibrosis is related to one or more of the following: unidentified infectious or noninfectious antigens, autoimmunity, impaired regenerative responses, and the antagonistically pleiotropic action of genes involved in wound healing or development. The importance of genetic susceptibility, epigenetics, aging, and the modern-day environment are highlighted. Consideration of both ultimate and proximate causation goes beyond philosophical cogitations, as it will better inform pathobiological mechanisms of disease and aid in the prevention and treatment of fibrotic diseases.
Imagine not being able to distinguish the real cause from that without which the cause would not be able to act as a cause. It is what the majority appear to do, like people groping in the dark; they call it a cause, thus giving it a name that does not belong to it.
—Plato, Phaedo 99
Introduction
Fibrotic disorders represent an increasing cause of morbidity and mortality worldwide. Since fibrosis is a predominant feature of the pathology of a wide range of diseases across multiple organ systems, fibrotic disorders have been estimated to contribute to an estimated 45% of all-cause mortality in the United States (1). Despite this tremendous clinical impact and advances in our understanding of the pathogenesis of fibrotic processes, effective therapies are limited, and the vast majority of clinical trials have failed. Here, we address the problem of fibrosis from an evolutionary perspective with the hope that this will better inform therapeutic approaches to these complex and intractable disorders. In essence, asking the ultimate teleological question of “why” this process evolved in multicellular organisms may provide us with a more proximate explanation as to “how” it causes or contributes to disease in humans. Understanding both ultimate and proximate causation goes beyond philosophical ideas or theories, as it will better inform pathobiological mechanisms of disease and aid in the prevention and treatment of fibrotic diseases.
“Why”: fibrosis as an evolutionarily conserved adaptive process
A consistent finding among all the fibrotic disorders in humans is the uniform loss of tissue architecture and progressive loss of organ function. It is not immediately obvious why fibrosis might provide any selective advantage, either survival or reproductive, to the organism. However, the tissue morphologic features that can broadly be defined as fibrosis — accumulation of extracellular matrix (ECM) and loss of cellular homeostasis — are seen in normal physiological responses. Importantly, the cellular and molecular mechanisms that participate in physiological responses are also much the same as those identified in pathological contexts. Two evolutionarily conserved adaptive or protective functions for physiological fibrosis are (a) host defense against pathogens and (b) wound healing in response to epithelial injury.
Host defense.
In many ways, fibrosis may be viewed as a component of the innate and adaptive immune response to certain pathogens. Indeed, many of the cells and mediators that constitute classical host responses to infections are seen in fibrotic disorders (2). Thus, a more expanded view of innate and adaptive immunity incorporates the concept that formation of a fibrotic scar may serve a beneficial role to the host by preventing pathogen invasion and/or spread, in addition to the pathogen killing by classical cells of the innate immune response, such as macrophages and neutrophils. Viewed in this way, cells such as fibroblasts and myofibroblasts, which are essential to fibrosis, participate in host defense by containing pathogens in order to more effectively kill them, or as a fail-safe mechanism to prevent their spread when killing fails to completely eliminate them. There are several examples of a fibrotic scar that forms a barrier to prevent pathogen spread: the most common are the thick fibrotic walls surrounding bacterial abscesses and the fibrotic rim around granulomas that contain mycobacterial pathogens. In these examples, an excessive burden of microbes (as with aspiration pneumonia in an alcoholic) or the virulence of a particular microbe (as with Mycobacterium tuberculosis) may not allow killing and eradication of the invading pathogen, and survival of the host organism is dependent on containment of the pathogen by preventing its spread or dissemination.
Wound healing.
The ability to survive wounding in the wild must have strongly influenced the survival of more complex multi-cellular metazoans. After cutaneous wounding, the formation of a fibrotic scar may benefit survival by several mechanisms, including prevention of excessive blood loss, barrier function against the entry of microbes, and formation of a provisional matrix to facilitate regeneration of damaged epithelium. Persistent fibrosis almost always accompanies incomplete or ineffective regeneration across multiple species (3). Interestingly, mammals appear to have lost the remarkable regenerative capacity of certain other species, such as amphibians (4). The reason for this is not clear; however, it is possible that such capacity was not lost, but rather favored in an apparent trade-off during mammalian evolution. Why would fibrosis be favored over regeneration? It could be argued that the bioenergetic investment in complete regeneration of a limb or an organ was too great for complex organisms; thus, limited resources were directed toward creating a fibrotic patchwork to preserve structural integrity that allowed the organism to live, despite loss of functional or reserve capacity. Another evolutionary trade-off may have been to limit the attendant risk of oncogenic transformation of a relatively small number of tissue-resident stem cells and/or progenitors that would be required to undergo a large number of cell divisions, an argument that is particularly relevant to larger, long-lived mammals that seem to have adopted a primarily stem cell–based mechanism for tissue regeneration. This contrasts with the remarkable regenerative capacity observed with limb amputation in amphibians, which rely on dedifferentiation of somatic cells at the wound margins to initiate regeneration without scarring or cancer (5).
“How”: emergence of pathological fibrosis
An evolutionary perspective of fibrosis may aid in understanding not only why, but also how fibrosis develops in pathological contexts. Following from the evolutionary reasoning provided above, a pathological or persistent fibrotic response may be related to (a) the persistence of a hitherto-unidentified pathogen or antigen; (b) the development of autoimmune reactions; (c) an impaired regenerative response that may relate to genetic, epigenetic, or age-related alterations; or (d) the antagonistic, pleiotropic action of selected genes involved in development and wound healing. The first two relate to the host defense function of physiological fibrosis, and the last two to wound healing (Figure 1).
Figure 1. Evolution of fibrosis.
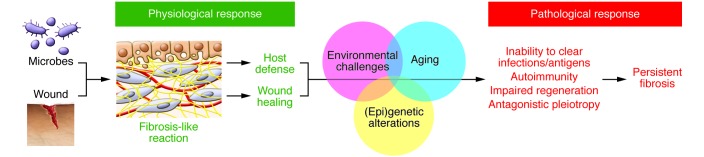
In most multicellular organisms, fibrosis-like reactions serve a useful purpose by limiting pathogen invasion and promoting wound healing. Pathological fibrosis may result from the persistence of extrinsic (unidentified) infections and/or antigens or of intrinsic neoantigens that evoke chronic autoimmune reactions. Impaired tissue regenerative capacity and/or the action of antagonistically pleiotropic genes may trigger perpetual wound healing responses that cause progressive fibrosis. These pathological processes may be instigated by genetic or epigenetic alterations in reparative and immune cells in the setting of modern environmental challenges and in an aging population.
Role of infectious or noninfectious antigens.
Numerous viral and parasitic infections have been identified as causal agents in human fibrotic disorders, in particular liver fibrosis (6); other organ systems such as the heart may also be involved, as exemplified by chronic infection with Trypanosoma cruzi, a well-known cause of cardiac fibrosis and cardiomyopathy (Chagas disease) (7). There appears to be an association of latent or other viral infections among subsets of patients with a progressive and fatal form of lung fibrosis known as idiopathic pulmonary fibrosis (IPF) (8). To date, our understanding of the link between infections and IPF is largely limited to studies showing increased prevalence of latent viruses in the lungs of patients with this disease compared with control populations, or among patients with acute (and typically severe) IPF exacerbations relative to those with stable pulmonary disease (9). Experimental murine models with γ-herpes viruses implicate a potential role for viral infections in the progression and/or exacerbation of lung fibrosis (10, 11). Viruses and other intracellular pathogens can cause apoptosis of alveolar epithelial cells, which is implicated in the pathogenesis of lung fibrosis (12). However, at present, it is unclear to what extent lung fibrosis that may be associated with viral or other infective agents is due to direct effects, rather than a secondary consequence of an injurious immune response provoked by these infections (13). Persistent viral or other infections that evoke immune responses may also predispose for the development of secondary autoimmunity (see below).
The cellular and molecular mechanisms that were hardwired to generate fibrotic responses to infectious pathogens may have been co-opted to respond to noninfectious stressors in our modern environment. Thus, environmental factors such as air pollution may be sufficient to trigger fibrotic responses in the genetically susceptible host. Indeed, noninfectious etiologies have clearly been identified in nonidiopathic causes of liver fibrosis (e.g., alcohol) and lung fibrosis (e.g., asbestosis). Put another way, it is possible that sufficient evolutionary time has not elapsed to allow the host to discriminate between infectious and noninfectious challenges of the present day.
Autoimmunity.
Autoimmunity is defined by the presence of specific receptor-mediated lymphocyte responses to distinct, particular autologous peptides (14). Benign low-level autoimmunity to self-proteins is probably near ubiquitous and may be critical for maintaining immunological homeostasis. In some cases, however, self-reactive immune responses become profoundly dysregulated, and injurious or even life-threatening to the host.
Autoimmune responses often develop as a secondary consequence of adaptive immune host defenses that were initially, and appropriately, directed against extrinsic antigens (e.g., microbes, viruses, and protein toxins or irritants) (14, 15). In certain conditions, particularly when the provocation is chronic and/or fulminant, and conditional on the presence of “permissive” genetic influences, these immune responses can lose their typically exquisite specificity for one or a small, finite number of foreign peptides, instead acquiring more generalized reactivity to other antigens, which may include self-peptides (e.g., epitope spread). In other cases, the extrinsic antigens share primary sequences or conformational homology with self-peptides that then become the collateral targets of misdirected, cross-reactive immune responses (e.g., epitope mimicry). The altered microbiome within damaged lungs (or chronic viral infections) can be highly antigenic, and thus conducive for the development of “secondary” autoimmunity. In addition, normally inert self-proteins, for which immune tolerance was established in early life, can later become immunogenic (e.g., neoantigens) if they are covalently modified by reactive chemicals present in tobacco smoke or by ROS or reactive nitrogen species produced by activated leukocytes.
The overwhelming preponderance of these secondary autoimmune responses appear to be clinically benign, but they are highly pathogenic in an important minority of cases, including carditis, nephritis, neurologic dysfunction associated with group A streptococcal and other microbial infections, paraneoplastic syndromes linked to malignancies, and myriad other tissue-specific autoimmune disorders (15–17). The cascade of injury processes evoked by autoimmune responses can include direct cytotoxicities by autoantibodies or autoreactive T cells; elaboration of numerous proinflammatory, vasoactive, and profibrotic mediators (including IL-4, IL-13, and TGF-β); and recruitment and/or activation of other immune effectors and mesenchymal cells (18–20). The end result of many clinically distinct autoimmune syndromes is tissue fibrosis affecting one or more organs (2).
The importance of recognizing autoimmune processes in the genesis of a fibrotic disorder centers on the potential that directed therapies to interrupt these particular immune processes may be much more effective and/or more readily tolerated than global immunosuppressants. Autoimmune lung diseases can be notoriously refractory to treatment with glucocorticosteroids and other nonspecific immunosuppressants; however, modalities that reduce autoantibody production or physically remove autoantibodies may be more effective (19, 21–24).
Although an autoimmune basis is well appreciated in fibrosis associated with various connective tissue diseases, this has not been established as a pathogenic mechanism in the so-called idiopathic cases, including IPF (25). Interestingly, a recent study showed that high levels of circulating autoantibodies to heat shock protein 70 are associated with poor outcomes in IPF patients (26), and other reports describe clonally expanded and/or highly differentiated lymphocytes in these patients, as well as abnormally high levels of B cell trophic and chemotactic mediators that parallel findings in classic autoantibody diseases (e.g., rheumatoid arthritis, systemic lupus erythematosus, and Sjögren’s syndrome) (27–30). Nonetheless, the precise roles of autoantibodies and neoantigens in the development and/or progression of idiopathic fibrotic disorders, as well as in acute exacerbations of these diseases, deserve further study.
Impaired regeneration.
Epithelial regeneration is a fundamental requirement for normal wound healing, and its impairment may lead to fibrosis. Genetic, epigenetic, and/or age-related alterations may confer susceptibility to fibrosis. Genetic studies in familial cases of IPF indicate a number of pathways related to protein folding and/or trafficking that may adversely affect the regenerative capacity of the epithelium (31). Epigenetic alterations that influence epithelial cell regeneration and fibroblast differentiation or fate may also predispose to fibrotic disease (32, 33).
There is growing recognition that aging is a risk factor for fibrosis. For example, the diagnosis of IPF is rarely made before 50 years of age, and its incidence and prevalence increases sharply with advancing age (34). The biological mechanisms that account for IPF as an age-related disease are not well understood. Several of the recently proposed hallmarks of aging have been linked to the pathogenesis of IPF (35, 36). The switch from regeneration to fibrosis as the primary repair response may represent slow, stochastic changes in cells and/or tissues driven by the accumulation of cellular or molecular damage, as suggested by Kirkwood’s disposable soma theory of aging (37). Alternatively, aging has been theorized to be programmed by genetic and/or metabolic factors, loss of proteostasis, and/or impaired stress responsiveness at more or less predictable ages that may be species specific (38, 39). In either case, it remains to be determined whether genes and/or pathways that have been shown to modulate lifespan in model organisms are tenable therapeutically in specific age-related human fibrotic diseases such as IPF. This is an area of investigation that is likely to reap enormous reward, not only with regard to fibrosis, but for a growing number of diseases in which aging is a known risk factor.
Antagonistic pleiotropy.
The concept of antagonistic pleiotropy was proposed by Williams in 1957 to explain senescence and aging (40); however, this concept may even more aptly apply to a number of complex, age-related diseases (41, 42). If a particular gene mediates beneficial effects in early life but exerts detrimental effects after reproductive age, there will be evolutionary pressure to conserve that gene despite its potential disease-causing effects with aging. We have proposed the concept of antagonistic pleiotropy to explain the role of the ROS-generating enzyme NADPH oxidase 4 (NOX4), which mediates myofibroblast differentiation and normal wound healing in young subjects, but promotes persistent fibrosis with aging (43–45). One explanation for the pleiotropic nature of NOX4 action is the lack of a counter-regulatory response involving NFE2-related factor 2 (NRF2) in aged animals; this NOX4/NRF2 imbalance leads to persistent fibrosis associated with the acquisition of a senescent and apoptosis-resistant myofibroblast phenotype in aged mice, whereas young mice demonstrate the capacity for fibrosis reversibility (44).
The antagonistic pleiotropic action of genes may also apply to numerous developmental genes and/or pathways that are reactivated in age-related fibrotic disease (46). Perhaps this is best illustrated by the canonical Wnt/β-catenin pathway. This developmental pathway, which is essential for organogenesis and normal stem cell function, is upregulated in lungs of patients with IPF and other fibrotic disorders (47–49); importantly, targeting the Wnt/β-catenin pathway protects against fibrosis in preclinical animal models (48–50). The mechanisms that account for the detrimental roles of developmental genes and/or pathways in age-related diseases may involve the permissive effect of specific aging phenotypes, including loss of proteostasis, metabolic reprogramming, immune dysregulation, and alterations in stem cell niches.
Implications for fibrosis research
Can an evolutionary perspective of fibrosis better inform our investigative efforts? Based on the preceding discussion of ultimate causation, perhaps studies of the etiology of idiopathic fibrosis should focus on unidentified infectious agents, or alterations in the tissue microbiome. Alternatively (or additionally), is the modern-day environment to blame? Environmental challenges in an aging individual may be sufficient to trigger autoimmunity, areas that have received little investigative attention. Identification of specific environmental triggers will have major public health value by aiding in the development of preventative, rather than curative, strategies.
Similarly, understanding of human regenerative potential that is likely not lost, but rather suppressed or latent, will unveil new therapeutic opportunities; in particular, understanding mechanisms of dedifferentiation of somatic cells at sites of tissue injury deserve further study. Whether loss of cellular plasticity accounts for age-related propensity to fibrosis is currently unknown. From a clinical standpoint, the health benefits of halting the progression of fibrosis may be almost as important as reverse remodeling to achieve normal organ structure and function, a daunting but possibly attainable proposition. Important ECM-related signaling and epigenetic mechanisms that contribute to the persistence and progression of fibrosis are being uncovered (51, 52). Finally, the identification of antagonistically pleiotropic genes, many of which are linked to embryonic development and/or normal wound healing, may be particularly attractive for therapeutic targeting in chronic diseases of aging, including organ fibrosis.
Acknowledgments
Work in the authors’ laboratories relating to the content and/or concepts proposed herein is supported by NIH grants P01 HL114470 and R01 AG046210 (to V.J. Thannickal), R01 HL102371 (to A. Gaggar), and R01 HL119960 (to S.R. Duncan) and by AHA Grant-in-Aid 14GRNT20180023 (to Y. Zhou).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2014;124(11):4673–4677. doi:10.1172/JCI74368.
References
- 1.Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol. 2004;4(8):583–594. doi: 10.1038/nri1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duffield JS, Lupher M, Thannickal VJ, Wynn TA. Host responses in tissue repair and fibrosis. Annu Rev Pathol. 2013;8:241–276. doi: 10.1146/annurev-pathol-020712-163930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. 2002;298(5601):2188–2190. doi: 10.1126/science.1077857. [DOI] [PubMed] [Google Scholar]
- 4.Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453(7193):314–321. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka EM, Reddien PW. The cellular basis for animal regeneration. Dev Cell. 2011;21(1):172–185. doi: 10.1016/j.devcel.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115(2):209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rossi MA. The pattern of myocardial fibrosis in chronic Chagas’ heart disease. Int J Cardiol. 1991;30(3):335–340. doi: 10.1016/0167-5273(91)90012-E. [DOI] [PubMed] [Google Scholar]
- 8.Tang YW, et al. Herpesvirus DNA is consistently detected in lungs of patients with idiopathic pulmonary fibrosis. J Clin Microbiol. 2003;41(6):2633–2640. doi: 10.1128/JCM.41.6.2633-2640.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Molyneaux PL, Maher TM. The role of infection in the pathogenesis of idiopathic pulmonary fibrosis. Eur Respir Rev. 2013;22(129):376–381. doi: 10.1183/09059180.00000713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mora AL, et al. Lung infection with gamma-herpesvirus induces progressive pulmonary fibrosis in Th2-biased mice. Am J Physiol Lung Cell Mol Physiol. 2005;289(5):L711–L721. doi: 10.1152/ajplung.00007.2005. [DOI] [PubMed] [Google Scholar]
- 11.McMillan TR, et al. Exacerbation of established pulmonary fibrosis in a murine model by gammaherpesvirus. Am J Respir Crit Care Med. 2008;177(7):771–780. doi: 10.1164/rccm.200708-1184OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sisson TH, et al. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am J Respir Crit Care Med. 2010;181(3):254–263. doi: 10.1164/rccm.200810-1615OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meneghin A, Hogaboam CM. Infectious disease, the innate immune response, and fibrosis. J Clin Invest. 2007;117(3):530–538. doi: 10.1172/JCI30595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marrack P, Kappler J, Kotzin BL. Autoimmune disease: why and where it occurs. Nat Med. 2001;7(8):899–905. doi: 10.1038/90935. [DOI] [PubMed] [Google Scholar]
- 15.Wucherpfennig KW. Mechanisms for the induction of autoimmunity by infectious agents. J Clin Invest. 2001;108(8):1097–1104. doi: 10.1172/JCI14235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pordeus V, Szyper-Kravitz M, Levy RA, Vaz NM, Shoenfeld Y. Infections and autoimmunity: a panorama. Clin Rev Allergy Immunol. 2008;34(3):283–299. doi: 10.1007/s12016-007-8048-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wraith DC, Nicholson LB. The adaptive immune system in diseases of the central nervous system. J Clin Invest. 2012;122(4):1172–1179. doi: 10.1172/JCI58648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Monaco C, Andreakos E, Kiriakidis S, Feldmann M, Paleolog E. T-cell-mediated signalling in immune, inflammatory and angiogenic processes: the cascade of events leading to inflammatory diseases. Curr Drug Targets Inflamm Allergy. 2004;3(1):35–42. doi: 10.2174/1568010043483881. [DOI] [PubMed] [Google Scholar]
- 19.Browning JL. B cells move to centre stage: novel opportunities for autoimmune disease treatment. Nat Rev Drug Discov. 2006;5(7):564–576. doi: 10.1038/nrd2085. [DOI] [PubMed] [Google Scholar]
- 20.Mayadas TN, Tsokos GC, Tsuboi N. Mechanisms of immune complex-mediated neutrophil recruitment and tissue injury. Circulation. 2009;120(20):2012–2024. doi: 10.1161/CIRCULATIONAHA.108.771170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sem M, Molberg O, Lund MB, Gran JT. Rituximab treatment of the anti-synthetase syndrome: a retrospective case series. Rheumatology (Oxford). 2009;48(8):968–971. doi: 10.1093/rheumatology/kep157. [DOI] [PubMed] [Google Scholar]
- 22.Keir GJ, et al. Severe interstitial lung disease in connective tissue disease: rituximab as rescue therapy. Eur Respir J. 2012;40(3):641–648. doi: 10.1183/09031936.00163911. [DOI] [PubMed] [Google Scholar]
- 23.Cancro MP, D’Cruz DP, Khamashta MA. The role of B lymphocyte stimulator (BLyS) in systemic lupus erythematosus. J Clin Invest. 2009;119(5):1066–1073. doi: 10.1172/JCI38010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knechtle SJ, Kwun J, Iwakoshi N. Prevention trumps treatment of antibody-mediated transplant rejection. J Clin Invest. 2010;120(4):1036–1039. doi: 10.1172/JCI42532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Noble PW, Barkauskas CE, Jiang D. Pulmonary fibrosis: patterns and perpetrators. J Clin Invest. 2012;122(8):2756–2762. doi: 10.1172/JCI60323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kahloon RA, et al. Patients with idiopathic pulmonary fibrosis with antibodies to heat shock protein 70 have poor prognoses. Am J Respir Crit Care Med. 2013;187(7):768–775. doi: 10.1164/rccm.201203-0506OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herazo-Maya JD, et al. Peripheral blood mononuclear cell gene expression profiles predict poor outcome in idiopathic pulmonary fibrosis. Sci Transl Med. 2013;5(205):205ra136. doi: 10.1126/scitranslmed.3005964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilani SR, et al. CD28 down-regulation on circulating CD4 T-cells is associated with poor prognoses of patients with idiopathic pulmonary fibrosis. PLoS One. 2010;5(1):e8959. doi: 10.1371/journal.pone.0008959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xue J, et al. Plasma B lymphocyte stimulator and B cell differentiation in idiopathic pulmonary fibrosis patients. J Immunol. 2013;191(5):2089–2095. doi: 10.4049/jimmunol.1203476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feghali-Bostwick CA, et al. Cellular and humoral autoreactivity in idiopathic pulmonary fibrosis. J Immunol. 2007;179(4):2592–2599. doi: 10.4049/jimmunol.179.4.2592. [DOI] [PubMed] [Google Scholar]
- 31.Kropski JA, Lawson WE, Young LR, Blackwell TS. Genetic studies provide clues on the pathogenesis of idiopathic pulmonary fibrosis. Dis Model Mech. 2013;6(1):9–17. doi: 10.1242/dmm.010736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hardie WD, Glasser SW, Hagood JS. Emerging concepts in the pathogenesis of lung fibrosis. Am J Pathol. 2009;175(1):3–16. doi: 10.2353/ajpath.2009.081170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanders YY, Hagood JS, Liu H, Zhang W, Ambalavanan N, Thannickal VJ. Histone deacetylase inhibition promotes fibroblast apoptosis and ameliorates pulmonary fibrosis in mice. Eur Respir J. 2014;43(5):1448–1458. doi: 10.1183/09031936.00095113. [DOI] [PubMed] [Google Scholar]
- 34.Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174(7):810–816. doi: 10.1164/rccm.200602-163OC. [DOI] [PubMed] [Google Scholar]
- 35.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thannickal VJ. Mechanistic links between aging and lung fibrosis. Biogerontology. 2013;14(6):609–615. doi: 10.1007/s10522-013-9451-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kirkwood TB. Understanding the odd science of aging. Cell. 2005;120(4):437–447. doi: 10.1016/j.cell.2005.01.027. [DOI] [PubMed] [Google Scholar]
- 38.Prinzinger R. Programmed ageing: the theory of maximal metabolic scope. How does the biological clock tick? EMBO Rep. 2005;6(Spec No):S14–S19. doi: 10.1038/sj.embor.7400425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Labbadia J, Morimoto RI. Proteostasis and longevity: when does aging really begin? F1000Prime Rep. 2014;6:7. doi: 10.12703/P6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williams GC. Pleiotropy, natural selection, and the evolution of senescence. Evolution. 1957;11:398–411. doi: 10.2307/2406060. [DOI] [Google Scholar]
- 41.Carter AJ, Nguyen AQ. Antagonistic pleiotropy as a widespread mechanism for the maintenance of polymorphic disease alleles. BMC Med Genet. 2011;12:160. doi: 10.1186/1471-2350-12-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thannickal VJ. Oxygen in the evolution of complex life and the price we pay. Am J Respir Cell Mol Biol. 2009;40(5):507–510. doi: 10.1165/rcmb.2008-0360PS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hecker L, et al. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med. 2009;15(9):1077–1081. doi: 10.1038/nm.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hecker L, et al. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci Transl Med. 2014;6(231):231ra247. doi: 10.1126/scitranslmed.3008182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thannickal VJ. Aging, antagonistic pleiotropy and fibrotic disease. Int J Biochem Cell Biol. 2010;42(9):1398–1400. doi: 10.1016/j.biocel.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Selman M, Pardo A, Kaminski N. Idiopathic pulmonary fibrosis: aberrant recapitulation of developmental programs? PLoS Med. 2008;5(3):e62. doi: 10.1371/journal.pmed.0050062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chilosi M, et al. Aberrant Wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis. Am J Pathol. 2003;162(5):1495–1502. doi: 10.1016/S0002-9440(10)64282-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Konigshoff M, et al. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J Clin Invest. 2009;119(4):772–787. doi: 10.1172/JCI33950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brack AS, et al. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science. 2007;317(5839):807–810. doi: 10.1126/science.1144090. [DOI] [PubMed] [Google Scholar]
- 50.Henderson WR, et al. Inhibition of Wnt/β-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc Natl Acad Sci U S A. 2010;107(32):14309–14314. doi: 10.1073/pnas.1001520107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou Y, et al. Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J Clin Invest. 2013;123(3):1096–1108. doi: 10.1172/JCI66700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parker MW, et al. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest. 2014;124(4):1622–1635. doi: 10.1172/JCI71386. [DOI] [PMC free article] [PubMed] [Google Scholar]