

Abstract
The transmembrane activator and calcium-modulating cyclophilin ligand interactor (TACI) controls differentiation of long-lived plasma cells, and almost 10% of individuals with common variable immunodeficiency (CVID) express either the C104R or A181E variants of TACI. These variants impair TACI function, and TACI-deficient mice exhibit a CVID-like disease. However, 1%–2% of normal individuals harbor the C140R or A181E TACI variants and have no outward signs of CVID, and it is not clear why TACI deficiency in this group does not cause disease. Here, we determined that TACI-deficient mice have low baseline levels of Ig in the blood but retain the ability to mutate Ig-associated genes that encode antigen-specific antibodies. The antigen-specific antibodies in TACI-deficient mice were produced in bursts and had higher avidity than those of WT animals. Moreover, mice lacking TACI were able to clear Citrobacter rodentium, a model pathogen for severe human enteritis, more rapidly than did WT mice. These findings suggest that the high prevalence of TACI deficiency in humans might reflect enhanced host defense against enteritis, which is more severe in those with acquired or inherited immunodeficiencies.
Introduction
The transmembrane activator and calcium modulating cyclophilin ligand interactor (TACI), a member of the TNF receptor family (1), is found predominantly on lymphocytes. TACI signaling causes sustained expression of BLIMP-1 (2), a transcription factor that orchestrates maturation of B cells into long-lived antibody-secreting cells (3, 4) and inhibits apoptosis of plasma cells (5); however, the full spectrum of functions of TACI is not known. Deficiency of TACI in mice and humans causes Ig concentrations in blood to be low (hypogammaglobulinemia), impairs long-lived responses to encapsulated bacteria and influenza (6–8), and can predispose to various combinations of lymphoproliferation, tumor formation, and autoimmunity (9, 10). This constellation of findings is called common variable immunodeficiency (CVID) and is associated with allelic variants of the genes encoding TACI and proteins conveying TACI functions (11, 12).
CVID is the most prevalent primary immunodeficiency disease (13) and differs from other primary immunodeficiency diseases by its late age of onset, usually at ages 20–40 years and sometimes much later, and by its range of manifestations including various combinations of low levels of serum Ig (hypogammaglobulinemia), defective cellular immunity, defective control of lymphocyte proliferation, autoimmunity, and cancer (14, 15). Yet, the most dramatic difference between CVID and other typical primary immunodeficiencies concerns the penetrance of disease associated with allelic variants of TACI and other receptors. In contrast to full penetrance of the null mutations BTK or CD40, which cause X-linked agammaglobulinemia, or hyper-IgM syndrome, respectively, allelic variants of TACI, such as C104R and A181E, which impair TACI function even in the heterozygous state (16), usually exhibit no clinically significant defects in immunity (17). Thus, Salzer et al. (18) found heterozygous C104R or A181E TACI variants in 6.9% of subjects with CVID but also in 2% of 675 normal adults. The later frequency equals the highest prevalence of SNPs found in 1,092 human genomes (19). Other variants that impair signaling by TACI are also found, albeit at lower frequencies, in normal individuals (20). Still more curious, individuals homozygous for C104R exhibit a milder phenotype of CVID and are less likely to develop autoimmunity than are individuals heterozygous for C104R (14, 18, 21), and some respond normally to vaccination despite having hypogammaglobulinemia (22). The vast range of manifestations associated with TACI variants in human subjects, from normal to severe CVID, and the reciprocal impact of gene dosage on the severity of the disease suggest that either background genes exert more impact on phenotype than any TACI allele, or the high prevalence of TACI variants is adaptive in response to environmental factors, or both.
To consider the latter possibility experimentally, we explored the phenotype of TACI deficiency in mice with a fully inbred genetic background. Consistent with the human phenotype of CVID, mice lacking TACI exhibit hypogammaglobulinemia and generate defective long-lived antibody responses to antigen (2, 6, 7). Still, TACI-deficient mice can generate bursts of IgG (2), owing to transient expression of BLIMP-1 caused by DNA double-strand breaks accompanying an Ig isotype class switch (2). Accordingly, we asked whether the antibodies produced by TACI-deficient mice achieve the avidity and function of antibodies produced by normal mice with the same genetic background, whether the antibodies protect against environmental pathogens, and, most importantly, whether differences in TACI-deficient and normal mice might explain the variegated phenotype of TACI variants in man.
Results
TACI deficiency enhances affinity maturation of antigen-specific antibodies.
The ability of antibodies to activate complement and tether phagocytes and hence to clear pathogens depends in part on their avidity for antigen (23). To determine whether TACI deficiency impairs affinity of antibodies produced in response to recently administered antigen, we immunized mice with a model antigen (4-hydroxy-3-nitrophenyacetyl [NP] coupled to an OVA carrier) (2, 24), generated hybridomas 40 days later, and measured the avidity of NP-specific mAbs from the immunized mice. We produced 220 NP-specific hybridomas from inbred TACI-deficient mice and 77 from inbred WT mice of the same genetic background (C57BL/6). The KD of NP-binding IgG was determined by the method of Stevens et al. (25). Twenty-two of 23 NP-specific IgG mAbs from immunized TACI-deficient mice exhibited high affinity, a KD of less than 4 × 10–10 M, while only 4 of 10 mAbs from WT mice exhibited such affinity (Figure 1A). Only 1 NP-specific monoclonal IgG clone was encoded by a non VH1-72 gene; its KD was 1.8 × 10–10 M. These results were confirmed by BioLayer interferometry (BLI) (Figure 1B). Thus, contrary to expectations (8), TACI-deficient mice can produce antigen-specific, high-affinity IgG antibodies in recall responses.
Figure 1. Impact of TACI deficiency on affinity maturation of antibodies produced in response to immunization.

(A) KD of mAbs generated from WT and TACI-deficient mice. KD (y axis) in nM (the reciprocal of avidity is depicted in blue) of monoclonal anti-NP antibodies (mAbs) encoded by the canonical VH1-72 (VH186.2) was determined by ascertaining the concentration at equilibrium at which one-half was bound and one-half unbound (25, 45). mAbs from WT mice had an average KD of 4.6 × 10–10, while mAbs from TACI-deficient mice had an average KD of 2.4 × 10–10 (P = 0.0079, unpaired t test). (B) Comparison of mAb KD determined by real-time measurement of association and dissociation rates and BLI, with KD determined at equilibrium by ELISA. (C) Impact of TACI on the mutation frequency of VH1-72 encoded by anti-NP antibodies. Frequency of sequences with a given number of mutations is plotted. TACI deficiency increased the frequency of independent mutations by more than 2-fold (P < 0.001, contingency analysis). (D) Relationship between frequency of mutations and antibody avidity in response to NP. Frequency of mutations in VH1-72 sequences is depicted in relation to avidity at equilibrium of the corresponding antibodies. Avidity correlated with the number of mutations (P < 0.0001, Wilcoxon matched-pairs, signed-rank test). (E) Localization of aa substitutions in distinct clones encoding VH1-72 heavy chains of anti-NP antibodies obtained from WT and TACI-deficient mice. KD for the corresponding sequences was determined by BLI (values are shown on the right). V(D)J, variable(V), diversity (D) and joining (J) gene segments junction region of the Ig heavy chain exon.
Antibodies of high avidity and affinity in WT mice, and presumably TACI-deficient mice, originate from B cells that have undergone “hypermutation” of Ig variable region exons followed by the selection of clones expressing antibodies of higher affinity. To weigh the relative contributions of these mechanisms, we compared the sequences of the genes encoding heavy-chain exons of NP-specific IgG following PCR amplification from DNA obtained from subcloned hybridomas from TACI-deficient and WT mice that had been immunized with NP-OVA conjugate. From these sequences, we identified and enumerated independent mutations from the VH1-72 germline, the NP-canonical VH1-72 gene fragment. Independent mutations were determined by counting mutations in distinct sequences. Sixty-six percent of VH1-72 sequences from B cells of TACI-deficient mice, but only 32% of VH1-72 sequences from B cells of WT mice, had, on average, more than 10 mutations per sequence (Figure 1, C and D). We counted 312 independent mutations in 7,050 nucleotides sequenced from VH1-72 of TACI-deficient mice (mutation frequency, 4.4%; Supplemental Figure 1D; supplemental material available online with this article; doi:10.1172/JCI74428DS1) and only 164 independent mutations in 6,090 nucleotides from VH1-72 sequences of WT (mutation frequency, 2.7%; Supplemental Figure 1C). Contingency analysis revealed that TACI deficiency significantly increased the frequency of independent mutations (P < 0.0001; Figure 1, C and D). On average, TACI-deficient VH1-72 genes had 8.3 replacement mutations per sequence, while WT VH1-72 had only 5.6 replacement mutations per sequence (contingency analysis, P = 0.0005 by Fisher’s exact test; Figure 1, D and E, and Supplemental Figure 1, A, C, and D). The frequency of replacement mutations in VH1-72 genes obtained from TACI-deficient mice correlated with increased affinity/avidity of the corresponding antibodies, suggesting that the mutated sequences with heightened affinity were efficiently selected in TACI-deficient hosts (Figure 1, D and E).
Our analysis of repeated mutations in distinct B cell clones in TACI-deficient and WT mice suggests that TACI deficiency does not impair antigen selection (Figure 1E). Consistent with that idea, we found accumulation of repeated mutations in the complementarity-determining regions (CDRs) (Supplemental Figure 1, A–F). Of the 312 mutations identified in VH sequences from TACI-deficient mice, 67% were replacement mutations, while in WT mice, 71% of 164 mutations were replacement mutations, both presumably driven by antigen selection. Similarly, W to L substitution at position 33 (Kabat numbering) in VH1-72, a mutation that increases the affinity of anti-NP antibodies by a factor of 10 (26), was as frequent in sequences from TACI-deficient (67%) mice as in those from WT (68%) mice (Supplemental Figure 1).
Light-chain utilization and sequences also suggested that TACI deficiency did not impair antigen selection. Supplemental Figure 1, B, E, and F show that all the IgG NP–specific mAbs isolated used λ light chains. The light chains sequenced from hybridomas derived from TACI-deficient mice used Vλ1 and Vλ2, while the light chains sequenced from hybridomas derived from WT mice used only Vλ1. The Vλ exons showed recurring mutations in the CDR2 regions of both TACI-deficient and WT mice, suggesting that these mutations contribute to antigen selection. In accordance, some of the aa changes are common to both genotypes (Supplemental Figure 1, B, E, and F).
The increased frequency of mutations in the VH genes in clones originating from TACI-deficient mice suggests that each responding B cell clone accumulated more mutations per cell cycle and/or that each clone underwent more cycles in which somatic hypermutation occurred. The increase was not apparently due to increased or protracted expression of Aicda, as TACI-deficient germinal center (GC) B cells expressed the same level of Aid mRNA (1.1-fold more, P = 0.4) as WT GC B cells. Our results thus suggest that an increase in the number of cell cycles during which somatic hypermutation occurs (coupled with effective antigen selection) drives the increase in high-affinity B cell clones and antibodies in TACI-deficient mice.
B cell–intrinsic TACI limits GC B cell expansion.
Somatic hypermutation and selection of B cells expressing receptors with high affinity for antigen take place in GCs, where specialized Th cells, follicular dendritic cells, and antigen provide conditions needed to stimulate and sustain B cell proliferation, somatic hypermutation, and selection. We asked whether and how TACI deficiency impacts on these processes. TACI-deficient and WT mice were immunized with 108 sheep erythrocytes (SRBCs), a standard antigen used to elicit T cell–dependent B cell responses, and 7 days later, the spleens were harvested for study. Figure 2 shows typical images of the GCs of TACI-deficient and WT mice. The GCs of TACI-deficient mice were 3.5-fold larger (P < 0.0001), on average, owing to the presence of more GC B cells, identified by peanut agglutinin (red), and naive B cells, identified by expression of IgD (green) (Figure 2, A and B). Seven days after immunization, the GCs of TACI-deficient mice contained 3.4-fold more activated B cells, marked by expression of CD19, FAS, and GL-7 (identifying a sialic acid glycan on the surface of activated B cells) than did the GCs in WT mice (P < 0.0001); the increase persisted at 14 days, when 2.0-fold more activated B cells were present (P = 0.0012) (Figure 2, C and D).
Figure 2. Impact of TACI deficiency on GC B cells.

(A) Identification of GCs with peanut agglutinin (red) and naive B cells with anti-IgD (green) in frozen tissue sections prepared from spleens 7 days after immunization with SRBCs, counterstained with DAPI (blue). TACI deficiency increased GC and naive B cells. Original magnification, ×200. (B) Area of GCs was measured with ImageJ software. Each point corresponds to the area of 1 GC. On average, GCs in TACI-deficient mice were larger than GCs in C57BL/6 mice (P < 0.0001). (C and D) GCs were identified with anti-CD19, anti–GL7 (50), and anti-FAS (CD95) mAbs and analyzed by FACS. Shown are CD19+ GC cells. GC B cells were increased in TACI-deficient mice (day 7, P < 0.0001; day 14, P = 0.0012). (E) Impact of T cell or B cell TACI deficiency on the number of GC B cells. C57BL/6 mice or Taci-KO (CD45.2) mice immunized with OVA were used as a source of T cells (T-WT and T-KO, respectively) for transfer into WT CD45.1+ mice. CD45.2 B cells isolated from QM or from QM-Taci-KO mice (B-WT and B-KO, respectively) were transferred along with the T cells in the combinations indicated. The number of CD45.2 GC B cells in recipients of CD45.2 TACI-deficient or proficient B or T cells is shown as a proportion of all GC B cells. Multiple comparison tests (1-way ANOVA and Holm-Sidak’s) revealed P = 0.003 for T-KO/B-WT versus T-WT/B-WT; P = 0.001 for T-KO/B-KO versus T-WT/B-WT; and P = 0.0025 for T-WT/B-KO versus T-WT/B-WT.
We next asked whether TACI deficiency in B cells or T cells or both caused expansion of the GC B cell population. To address that question, we tested the GC responses in chimeras generated using allotypic (CD45.2+) B cells from quasi-monoclonal (QM) TACI-sufficient or QM TACI-deficient mice (6) and/or OVA-primed T cells from C57BL/6 (CD45.2+) TACI-deficient or TACI-proficient mice. We adoptively transferred CD45.2 TACI-proficient or -deficient B and T cells into CD45.1 recipients immunized with NP-OVA at the time of transfer, and the fraction of CD45.2 GC B cells was measured 14 days later. As Figure 2E shows, the deficiency of TACI on T cells or B cells (or both) expanded the population of GC B cells. The result further suggests that after exposure to antigen, TACI might govern such interactions between follicular T cells and follicular B cells that delimit the B cell response.
TACI limits GC B cell proliferation.
The greater expansion of GCs and GC B cell populations in TACI-deficient mice after exposure to antigen could reflect increased proliferation, failure of B cells to mature (2), and migration and/or decreased apoptosis. To distinguish between these possibilities, TACI-deficient and WT mice were pulsed with BrdU at various times after exposure to antigen, and the incorporation of BrdU into GC B cells was measured by flow cytometry. Seven days after immunization with SRBCs, a period when GCs should be expanding (27), TACI-deficient mice had 4-fold more GC B cells containing BrdU than did WT mice (P = 0.002) (Figure 3A). In contrast, TACI-deficient and WT mice had the same number of GC B cells containing BrdU 14 days and 28 days after immunization, a time when GCs usually regress (Figure 3A). These results indicate that the presence of TACI restrains B cell proliferation during the period immediately following immunization. However, since TACI-deficient mice had 2.3-fold more GC B cells than did WT mice 14 days after immunization, factors besides proliferation, including failure to migrate or to undergo apoptosis, might contribute to GC cell expansion.
Figure 3. Impact of TACI deficiency on B cell proliferation and apoptosis.

(A) Number of proliferating GC B cells in WT and TACI-deficient mice at 7, 14, or 28 days after immunization with SRBCs. Percentages of BrdU+ GC cells (CD19+GL7+FAS+). TACI deficiency increased proliferation of GC cells 7 days after immunization (P = 0.0002, Holm-Sidak test). (B) Immunofluorescence analysis of GC cell apoptosis. Apoptotic cells were identified by TUNEL, labeled ends were detected with anti-BrdU antibodies (green, upper diagrams and red, lower diagrams), and cells were counterstained with DAPI. Lower diagrams show costaining with anti-GL7 mAbs (green). Results are representative of 6 distinct sections obtained from 2 WT mice and 3 TACI-deficient mice. Original magnification, ×200. (C) Enumeration of apoptotic TUNEL+ GC cells. Taci-KO mice had fewer TUNEL+ cells per GC (P = 0.0012, Mann-Whitney U test). (D) Impact of TACI ligand blockade on B cell apoptosis. WT or Taci-KO splenic B cells were cultured with anti-CD40 and IL-4 in the presence or absence of TACI-Fc to inhibit TACI signaling. After 48 hours, caspase 3+ cells were enumerated by FACS. TACI deficiency or impaired TACI function decreased apoptosis after B cell activation, (P < 0.05, Mann-Whitney U test). (E and F) Relative expression by qPCR of cellular inhibitor of apoptosis (cIap) by GC B cells (E) or Tfh cells (F) 14 days after immunization with SRBCs. Expression of cIap was quantified in cDNA by qPCR reactions using specific primers and SYBR Green incorporation (P < 0.05, Mann-Whitney U test).
TACI promotes GC B cell apoptosis and increases cIAP expression.
Since apoptosis of B cells failing to express a functional Ig or competing poorly for antigen or T cell help normally controls GC expansion, we asked whether TACI deficiency impairs apoptosis of GC B cells. Consistent with that possibility, Figure 3, B and C, shows that 7 days after immunization with SRBCs, TACI-deficient mice had 2.2-fold fewer TUNEL+ cells than did WT mice (P = 0.0055). To confirm that TACI impairs apoptosis, we asked whether inhibition of TACI suppresses caspase 3 activation in stimulated B cells. B cells from WT and TACI-deficient mice were incubated with anti-CD40 antibody and IL-4 (2) for 48 hours in the presence or absence of soluble TACI-Fc, which competes for TACI ligands. As Figure 3D shows, the frequency of apoptotic B cells was reduced in cultures containing TACI-Fc (paired t test, P = 0.04), as was seen in cultures from TACI-deficient mice (P = 0.01). While various pathways might induce apoptosis in stimulated cells, Kanno et al. (28) recently reported that TACI induces apoptosis by increasing the association between TRAF2 and cIAP (cellular inhibitor of apoptosis). Consistent with this report, the GC B cells of TACI-deficient mice immunized with SRBCs contained less cIAP than did GC B cells from the WT mice (P = 0.03 by Mann-Whitney U test) (Figure 3E). The reduction in cIap mRNA was specific to B cells, since TACI-proficient and TACI-deficient T helper follicular (Tfh) cells had similar expression levels of cIap (Figure 3F). Thus, expansion of GC B cells in TACI deficiency reflects, at least in part, impaired apoptosis of stimulated B cells and decreasing cIap expression. If, as Khalil et al. (29) suggested, selection occurs once every cell cycle, then increasing proliferation of GC B cells coupled with decreased apoptosis will improve the chances of selection for mutations of the V region that augment affinity/avidity for antigen.
TACI deficiency increases Tfh recruitment to GCs.
Because T cell help contributes to B cell expansion and selection, we asked whether TACI deficiency changes the population of Tfh cells after immunization. In fact, WT Tfh cells expressed Taci as much as B cells did (Figure 4A). Tfh cells (in TACI-deficient or TACI-proficient mice) also expressed CXCR5, PDL-1, and Bcl6 (Figure 4A). Figure 4B shows that the fraction of Tfh cells was increased before and 5 and 8 days following immunization. The expansion of Tfh cells was due, at least in part, to increased proliferation, since TACI-deficient mice had 2-fold and 2.7-fold more BrdU+ Th cells than did control mice 7 (P = 0.0066) and 14 (P = 0.0046) days after immunization, respectively. The expansion of Th cells was secondary to the lack of TACI on B cells adoptively transferred into a congenic recipient (Figure 4C). These results suggest that B cell–intrinsic TACI deficiency increases the availability of T cell help. In a recently published article, Goenka et al. (30) showed that while the GC lacks BAFF, a TACI ligand, Tfh-derived BAFF contributes to persistence of B cell clones undergoing affinity maturation. Thus, enhanced recruitment of Tfh by TACI-deficient B cells may cause enhanced BAFF production at the T-B cell border, facilitating maintenance of expanded clones and selection of mutated B cells. Since B cells recruit Tfh via increased ICOS-L/ICOS interactions at the T-B follicular border (31), we measured the expression of Icos on cultured splenocytes by quantitative PCR (qPCR). Splenocytes obtained from TACI-deficient mice expressed, on average, 50% more Icos mRNA than did WT GC B cells, but this increase was not statistically significant (P = 0.1). We did not detect differences in the expression of Il4, Il6, or Il10 in TACI-deficient splenocytes or WT cells
Figure 4. Comparison of the phenotype and fraction of Tfh cells in spleens of immunized WT or TACI-deficient mice.

(A) TACI expression by GC cells and Tfh cells. Tfh cells were identified as CD4+CXCR5+ and PD1+ by FACS (upper panel) and tested for expression of Bcl6 or Taci mRNA by qPCR (middle and lower panels, respectively). Tfh cells from TACI-deficient mice expressed as much Bcl6 as those from WT mice (middle panel). Taci expression by T cells was equivalent to that in GC B cells (lower panel). (B) Relative number of Tfh cells in spleens of WT and TACI-deficient mice before (Pre-I) and 5, 8, and 28 days after immunization with SRBCs. Frequencies of Tfh cells in C57BL/6 and Taci-KO mice were compared by 1-way ANOVA and Holm-Sidak multiple comparison tests. Pre-I, P = 0.0015; day 5, P = 0.0004; day 8, P < 0.0001; day 28, P > 0.9999 was nonsignificant. (C) Impact of TACI expression by B and T cells on the size of the Th cell population. Number of Th cells of donor (CD45.2+) or recipient origin (CD45.1+) was determined in chimeras reconstituted as shown in Figure 3 relative to chimeras reconstituted with WT-T and WT-QM B cells, 8 days after immunization with 100 μg NP conjugated with OVA. Increase in the number of CD4+ Th cells was secondary to B cell–intrinsic TACI deficiency. Comparisons by 1-way ANOVA and Holm-Sidak multiple comparison tests. ***P = 0.0001; **P = 0.0013.
TACI deficiency enhances host defense.
Gene variants hindering expression or function of TACI are associated with CVID manifested by a heightened risk of serious infection (14), albeit with profoundly varied penetrance in human subjects (18). Accordingly, we asked whether TACI deficiency and hypogammaglobulinemia impair host defense in mice with a fully inbred background. To address that question, we explored the response to Citrobacter rodentium (C. rodentium), an intestinal pathogen of mice taken to model human enterohemorrhagic and enteropathogenic Escherichia coli (32), which cause severe diarrhea and mortality worldwide. After infection, WT mice exhibit diarrhea, decreased activity, and hunched posture; the levels of bacteria peak at 5 to 15 days and clear entirely at 21 to 28 days (33). Mice deficient in RAG2 and hence lacking the capacity to mount cellular and antibody-mediated responses do not clear C. rodentium and often succumb to sepsis within 16 days after infection (34). The importance of adaptive immunity and immune memory are confirmed by the observation that normal mice previously exposed to the organism have profoundly fewer organisms in the colon 11 days after reinfection (35). Clearance of C. rodentium and prevention of sepsis requires a T cell–dependent B cell response, as an absence of CD4+ T cells and/or B cells causes infected mice to experience persistent infection or death, and administration of immune IgG prevents death (34, 36, 37). Despite the importance of adaptive immunity in clearing C. rodentium and the association of TACI deficiency with CVID, TACI-deficient mice given 109 CFU C. rodentium twice, by gavage, cleared the organism faster than did WT mice (Figure 5A). Two days after reinfection, 3 of 8 TACI-deficient mice, but only 1 of 9 WT mice, cleared the reinfection (Figure 5A). Five days after reinfection, all but 1 TACI-deficient mouse, but only 60% of WT mice, had cleared the organisms (Figure 5A). Consistent with an enhanced memory response, TACI-deficient mice had an increased frequency of C. rodentium–binding B cells (on average, 35%) compared with that seen in WT mice (on average, 27%) 10 days after reinfection (Supplemental Figure 4). These results indicate that TACI-deficient mice mount an enhanced memory response that accelerates clearance of C. rodentium.
Figure 5. Impact of TACI deficiency on resistance to C.

rodentium.C. rodentium (1 × 109) were administered by oral gavage to WT or TACI-deficient mice twice, 45 days apart. (A) Number of C. rodentium in stool 2 and 5 days after the second inoculation. Number of C. rodentium per 1 g of feces was determined by counting colonies in MacConkey agar seeded with serial dilutions of fecal suspensions. Contingency analysis indicated that TACI-deficient mice cleared C. rodentium faster than did WT mice (P = 0.0084). (B) Impact of TACI deficiency on avidity of anti-intimin antibodies in mice infected repeatedly with C. rodentium. Five days after reinfection with C. rodentium, the avidity of anti-intimin antibodies in serum was determined by the concentration of intimin needed to reduce the concentration of unbound anti-intimin IgG by one-half (tested by ELISA). Comparisons between groups were by Mann-Whitney U test (P = 0.0051). (C) Relationship between the average avidity of anti-intimin antibodies and the number of C. rodentium in feces. Number of C. rodentium correlated linearly with the relative avidity of anti-intimin antibodies in serum.
Given the faster clearance of C. rodentium by TACI-deficient mice, we asked whether the response could reflect increased avidity of antibodies elicited by the infecting organism. While the neutralizing targets of anti-Citrobacter antibodies are incompletely known, Ferreira et al. (38) and Ghaem-Maghami et al. (35) showed that antibodies directed against intimin, a surface protein, protect. Accordingly, we tested the average avidity of IgG antibodies directed against intimin in TACI-deficient and WT mice. To estimate the average avidity of specific antibodies in a polyclonal serum, we measured the amount of purified intimin needed to reduce by 50% the binding of anti-intimin antibodies. Figure 5B shows that IgG in sera obtained from TACI-deficient mice 5 days after reinfection with C. rodentium required, on average, 48% less intimin than did WT sera to reduce by 50% the concentration of free anti-intimin in solution (P = 0.01) (Figure 5B), indicating that TACI-deficient mice produce IgG antibodies with increased avidity for intimin. If the avidity of sera IgG determines C. rodentium clearance, we expect to observe a correlative relationship between the KD and the number of microbes in the feces. Figure 5C shows that the concentration of intimin needed to reduce the anti-intimin IgG in solution by half correlated in a linear manner with the number of microbes in the feces 5 days after reinfection.
To determine whether antibodies produced by TACI-deficient mice infected with C. rodentium better enhanced clearance of the organism than antibodies produced by WT mice, we transferred sera from twice-infected animals into RAG1-deficient mice, which were then infected. Figure 6 shows that RAG1-deficient mice given sera from previously infected TACI-deficient mice had 9-fold fewer organisms in the feces than did RAG1-deficient mice given sera from previously infected WT mice and 22-fold fewer organisms than did mice given saline. Thus, despite all the limitations TACI deficiency may impose on immune responses, it enhances the ability of mice to control and clear C. rodentium, in part, owing to enhanced affinity of the antibodies produced.
Figure 6. Number of C.

rodentiumbacteria in feces of RAG1-deficient mice after adoptive transfer of sera from infected TACI-deficient or WT mice. Number of bacteria in feces of RAG1-deficient mice 3 days after infection with 1 × 109 CFU C. rodentium and transfer of 200 μl sera (i.v.) from C57BL/6 mice or TACI-deficient mice, obtained 10 days after reinfection. Control RAG1-deficient mice were administered saline instead of sera and infected with 1 × 109 CFU C. rodentium. Comparisons between groups were by Mann-Whitney U test. P = 0.0157 for Taci-KO versus C57BL/6; P = 0.0176 for TACI versus saline.
Discussion
Primary immunodeficiency diseases such as the DiGeorge syndrome, severe combined immunodeficiency syndrome, sex-linked agammaglobulinemia, and hyper-IgM syndrome are rare, typical genotype frequencies approaching 1 × 106 and almost invariably manifest as severe infection early in life, and, without aggressive medical care, result in early demise (32). In contrast, CVID appears first in adults, exhibits a variegate phenotype (39), and the most frequent genotypes associated with the disease have an aggregate genotype frequency of at least 2% (18). In fact, contrasting with genotypes known to cause most primary immunodeficiencies, those targeting the TACI gene that are most frequently associated with CVID can be found in a large fraction — approximately 2% — of normal individuals (18). Here, we show that the phenotype of TACI deficiency in inbred mice, while resembling CVID in some respects such as baseline hypogammaglobulinemia, includes the ability to produce a burst of IgG of high affinity in response to antigen and infection, allowing affected mice to clear an enteric pathogen that models those that are the most common cause of severe gastroenteritis. The protective impact of TACI deficiency might thus explain the high frequency of mutant alleles of TACI in the normal population.
Our results suggest that deficiency in TACI, whether complete, as in the mice we studied, or partial, as in most human subjects with common variant alleles, does not invariably impose a biological cost, but might be adaptive in communities with a high burden of certain pathogens. Consistent with this concept is the high frequency (2%) of alleles associated with CVID in normal individuals (18) and the fact that the vast majority of humans with CVID have 1 normal TACI allele (18). Martinez-Gallo et al. (40) found that siblings of subjects with CVID carrying the same TACI variants had assayable defects in B cell function but no manifestations of disease (no hypogamaglobulinemia, lymphoproliferation, or infectious complications). Importantly, B cells from these healthy siblings with TACI mutations had appreciably less TACI expression, bound less APRIL, did not increase TACI expression in response to TLR9 activation, and had suboptimal AICDA induction in response to TACI stimulation than did B cells from siblings with fully WT alleles. If mutations in TACI and related genes are necessary, they are clearly not sufficient for CVID to be manifest, and a fuller understanding of the pathogenesis of CVID might depend on identifying other predisposing genes or environmental factors.
The laboratory and clinical phenotypes of TACI deficiency, both in mice and humans, appear contradictory in some respects. The simultaneous presence of hypogammaglobulinemia and increased resistance to enteric infection has been discussed above. A differential propensity for autoimmunity in those with homozygous or heterozygous TACI-mutant alleles would appear to present yet another contradiction. Yong et al. (14) and Salzer et al. (18) found that rare individuals with homozygous TACI mutations have profound B cell deficiencies; yet, these individuals are less likely to develop autoimmune disease than are individuals with heterozygous mutations (21). Our finding that TACI enhances apoptosis of activated B cells in the GC and prior reports that TACI promotes differentiation and survival of plasma cells (2, 5) might together suggest an explanation. A complete absence of TACI function impairs control of GC reactions, perhaps allowing the production of autoantibodies in response to immunization; however, it may also preclude the enduring production of autoantibodies and thus autoimmune disease. Partial TACI deficiency impairs the control of lymphoproliferation as much as its complete absence does, but allows the ongoing production of autoantibodies, as it does not block Ig secretion to the same degree.
Thus, certain TACI-mutant alleles and the deficiency in expression and function of TACI they cause might be adaptive in populations, especially the young, who are vulnerable to enteric or nonenteric pathogens controlled by rapidly produced high-affinity IgG. The cellular mechanism of this protective response, apparently including marked expansion of antigen-responsive GC B cells that allow accelerated affinity maturation, might, however, engender the lymphoproliferative disease, autoimmunity, and cancer that plague those with CVID later in life.
Methods
Mice
WT C57BL/6 mice (CD45.2+), CD45.1, or RAG1-deficient congenic mice were purchased from The Jackson Laboratory. Taci-KO mice have been previously described (2, 6). Taci-KO mice were backcrossed onto a C57BL/6 background for 12 generations. The fraction of loci still heterozygous at the twelfth generation of backcrossing is 1/211 = 4.9 × 10–4. Hence, the genome is over 99.95% identical to C57BL/6, except the regions flanking the deletion of exons 1 and 2 of the Taci gene in mouse chromosome 11 (7). Since contribution of the 129 strains encode a Casp11 variant that causes impaired IL-1β responses to E. coli, C. rodentium, and V. cholerae (41), we confirmed that the mice were fully inbred to this background by skin transplantation. In agreement with their C57BL/6 background, TACI-deficient B cells expressed IgMb (Supplemental Figure 2), and the VH genes isolated from B cells responding to C. rodentium matched C57BL/6 germline alleles (Supplemental Figure 3). Mice were housed in a specific pathogen-free facility at the University of Michigan.
Flow cytometry and antibodies
Fluorochrome-conjugated anti-mouse CD19 (1D3), CD4 (L3T4), CD95 (FAS, JO2), CXCR5 (2G8), B220 (RA3-6B2), IgMb (IF6-78), and T and B cell activation antigen (GL7) antibodies were purchased from BD Biosciences. C. rodentium expressing GFP was a gift of B. Vallance (University of British Columbia, Vancouver, Canada). Phycoerythrin-conjugated (PE-conjugated) anti-PD1 (RMP1-30) antibody was purchased from eBioscience. Data were acquired with a BD FACS Canto II and analyzed using FACSDiva, version 6.1.1 software (both from BD Biosciences).
Apoptosis assay
B cells isolated from spleens using a B cell isolation kit (Miltenyi Biotec) and cultured for 2 days with 5 μg/ml hamster anti-mouse CD40 antibody (HM40-3, BD Biosciences), with or without 2 μg/ml TACI-Fc (Enzo Life Sciences), were stained with anti–active caspase 3 antibody (Abcam). Active caspase 3–positive cells were detected by flow cytometry.
Adoptive transfer
Cell transfer.
QM B cells and T cells were isolated (B cell isolation or pan T cell isolation kit; Miltenyi Biotec) from spleens of nonimmunized mice or from spleens of mice primed with 100 μg OVA (Sigma-Aldrich) 7 days earlier, respectively. Five million B cells and 5 million T cells were injected into the tail vein of congenic C57BL/6 (CD45.1) mice (The Jackson Laboratory). Mice were immunized with 100 μg NP conjugated with OVA (NP-OVA; Biosearch Technologies) in an emulsion prepared with incomplete Freund’s adjuvant (Difco Laboratories).
Sera transfer.
Sera were obtained from 12 Taci-KO or 12 WT mice, infected twice with 1 × 109 C. rodentium, 30 days apart, 10 days after the second inoculation. WT mice sera had, on average, 819.6 μg/ml IgG, and Taci-KO mice sera had, on average, 554.6 μg/ml IgG. Serum from each mouse (200 μl) was transferred into RAG1-deficient mice by i.v. injection into the tail. Ten RAG1-deficient mice received sera from Taci-KO mice or from WT mice. An additional 10 RAG1-deficient mice received 200 μl saline instead. Feces were obtained 3 days after infection and sera transfer and bacteria CFU counted.
Proliferation assay
C57BL/6 or TACI-deficient mice immunized with 2 × 108 SRBCs (Innovative Research) were injected i.p. with 2 mg BrdU 24 hours prior to analysis. BrdU incorporated into cells was detected with FITC-conjugated anti-BrdU antibody according to the manufacturer’s instructions (BD Biosciences).
TUNEL assay
TUNEL assay was performed on 7-micron-thick frozen sections using an In Situ Cell Death Detection Kit (Roche Applied Science) according to the manufacturer’s instructions. Sections were stained with FITC-labeled PNA (Vector Laboratories) or with Alexa Fluor 488–conjugated anti-IgD antibody (BioLegend) and mounted with ProLong Gold Antifade Reagent with DAPI (Invitrogen). Digital images were obtained with a Leica DM6000B microscope and with Leica Application Suite Advanced Fluorescence, version 1.9.0 (both from Leica Microsystems).
Real-time reverse transcriptase-PCR
RNA from sorted GC B cells or Tfh cells was extracted using an RNeasy Plus Mini Kit (QIAGEN). cDNA was obtained with a SuperScript III First-Strand Synthesis System (Invitrogen). qPCR reactions were run on a Mastercycler ep realplex system (Eppendorf), and quantified by FastStart Universal SYBR Green Master (Roche Applied Science) incorporation using the primers listed in the Supplemental Methods.
Quantification of Icos, Cd40l, Il4, Il6, and Il10 was performed by qPCR in cDNA obtained directly from the lysate of 106 splenocytes (lysis buffer consisted of 1% 1 M Tris, pH 8.0 and 2.5% rRNasin in DNase/RNase-free water) after 3 days in culture with 2.5 μg/ml IL-4 (R&D Systems) and 1 μg/ml anti-mCD40 (HM40-3; BioLegend) in RPMI 1640 supplemented with 10% FBS, penicillin, streptomycin, glutamine, and 20 μM 2-mercapto-ethanol. Cells were obtained from Taci-KO or WT mice using TaqMan FAM-MGB probes and primers (Invitrogen). Quantification of Aicda expression was performed by qPCR in cDNA using TaqMan FAM-MGB probes and primers. cDNA was obtained directly from the lysate of 103 cells (lysis buffer was composed of 1% 1 M Tris, pH 8.0 and 2.5% rRNasin in DNase/RNase-free water) obtained from sorted GC B cells from mice (TACI-deficient or WT) injected 10 days earlier with SRBCs. qPCR was performed in triplicate in 3 mice of each strain.
Monoclonal antibodies
Hybridomas were obtained from splenocytes fused with NS-1 partners at the University of Michigan Hybridoma Core. Hybridoma clones producing anti-NP antibodies were identified by ELISA. Isotyping of NP-specific antibodies was also done by ELISA as previously described (42). Hybridomas were generated from 3 TACI-deficient mice and 2 WT C57BL/6 mice. Monoclonality was confirmed by PCR.
Sequencing
DNA obtained from hybridoma clones was amplified using a VH1-72–specific (IMGT; refs. 43, 44) forward primer or universal forward primers for clones containing other VH genes and with a JH4 reverse primer. The nested PCR used the same forward primer and reverse primers located on the J1-J2 intron (see Supplemental Methods). PCR products were isolated from agarose gels using the QIAquick Gel Extraction kit (QIAGEN) and directly sequenced from both ends at the University of Michigan DNA Sequencing Core. Each raw chromatogram was analyzed using FinchTV, version 1.4.0 (Geospiza Inc.).
Affinity measurements
The affinity/avidity to NP of anti–NP IgG was determined by ELISA from the concentration of free anti–NP IgG at equilibrium, with serial dilutions of NP-BSA (100 nM to 0.1 nM). Hybridoma supernatants containing 2 nM IgG were incubated at room temperature overnight with NP-BSA.
The KD was calculated according to the formula below (25, 45):
(Equation 1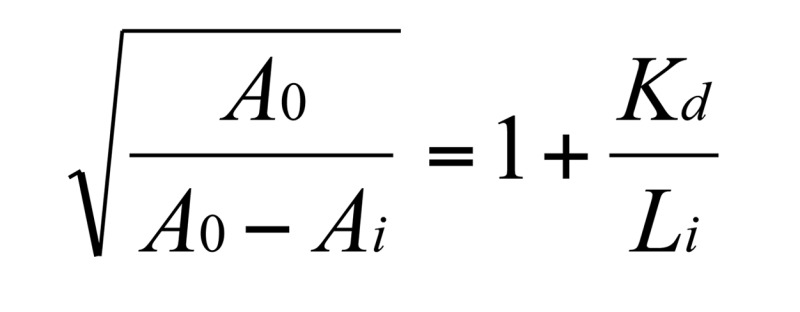 )
)
A0 is OD405 without competitor (NP-BSA); Ai is OD405 with competitor at the concentration of Li; and Li is the concentration of competitor.
The relative affinity/avidity of anti-intimin IgG was deduced from the intimin concentration required to reduce the concentration of nonbound anti-intimin IgG by half.
ELISA
Anti-NP antibodies were measured by ELISA as previously described (42). Anti-intimin antibodies were measured by ELISA on MaxiSorp plates (Nunc) coated with 50 μl of 0.5 μg/ml intimin in PBS. Absorbance was read at 405 nm with a Synergy 2 microplate reader (Biotec Laboratories) and results analyzed using Gen5 Data Analysis Software, version 1.04.5 (BioTek).
Bioluminescence imaging
Bioluminescence imaging (BLI) was run at 25°C with Octet RED (ForteBio) (46–48). KD was deduced from association and dissociation rates of NP-specific Ig (in dilutions of 0.3–10 nM) and measured with super streptavidin sensors (ForteBio) loaded with biotinylated NP4-BSA (Biosearch Technologies). Data were processed with a double-reference subtraction and analyzed with ForteBio Data Analysis Software, version 7.0.
Citrobacter infection
C57BL/6 or TACI-deficient mice were inoculated with 1 × 109 of C. rodentium twice, and the microbe load was determined as previously described (49). Infection was considered cleared when no colonies were detected in the nondiluted feces suspensions.
Statistics
Data are presented as the mean ± SEM in all figures in which error bars are shown. Statistical analysis was performed using GraphPad Prism software, version 6 (GraphPad Software). Distributions of 2 unmatched groups compared using Mann Whitney test. Comparisons of more than 2 groups were by ANOVA. Categorical variables were compared using χ2 or Fisher’s exact test analysis. A P value of less than 0.05 was considered statistically significant.
Study approval
All animal procedures were approved by the University of Michigan Committee on the Use and Care of Animals.
Supplementary Material
Acknowledgments
This work was funded by NIH grants P01 HL079067-01 and R37 HL52297.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2014;124(11):4857–4866. doi:10.1172/JCI74428.
References
- 1.von Bulow GU, Bram RJ. NF-AT activation induced by a CAML-interacting member of the tumor necrosis factor receptor superfamily. Science. 1997;278(5335):138–141. doi: 10.1126/science.278.5335.138. [DOI] [PubMed] [Google Scholar]
- 2.Tsuji S, Cortesão C, Bram R, Platt JL, Cascalho M. TACI deficiency impairs sustained Blimp-1 expression in B cells decreasing long-lived plasma cells in the bone marrow. Blood. 2011;118(22):5832–5839. doi: 10.1182/blood-2011-05-353961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, Liao J, McHeyzer-Williams MG, Calame K. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity. 2003;19(4):607–620. doi: 10.1016/S1074-7613(03)00267-X. [DOI] [PubMed] [Google Scholar]
- 4.Shapiro-Shelef M, Lin KI, Savitsky D, Liao J, Calame K. Blimp-1 is required for maintenance of long-lived plasma cells in the bone marrow. J Exp Med. 2005;202(11):1471–1476. doi: 10.1084/jem.20051611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ou X, Xu S, Lam KP. Deficiency in TNFRSF13B (TACI) expands T-follicular helper and germinal center B cells via increased ICOS-ligand expression but impairs plasma cell survival. Proc Natl Acad Sci U S A. 2012;109(38):15401–15406. doi: 10.1073/pnas.1200386109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mantchev GT, Cortesao C, Rebrovich M, Cascalho M, Bram RJ. TACI is required for efficient plasma cell differentiation in response to T-independent type 2 antigens. J Immunol. 2007;179(4):2282–2288. doi: 10.4049/jimmunol.179.4.2282. [DOI] [PubMed] [Google Scholar]
- 7.von Bulow GU, van Deursen JM, Bram RJ. Regulation of the T-independent humoral response by TACI. Immunity. 2001;14(5):573–582. doi: 10.1016/S1074-7613(01)00130-3. [DOI] [PubMed] [Google Scholar]
- 8.Wolf AI, et al. Protective antiviral antibody responses in a mouse model of influenza virus infection require TACI. J Clin Invest. 2011;121(10):3954–3964. doi: 10.1172/JCI57362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seshasayee D, Valdez P, Yan M, Dixit VM, Tumas D, Grewal IS. Loss of TACI causes fatal lymphoproliferation and autoimmunity, establishing TACI as an inhibitory BLyS receptor. Immunity. 2003;18(2):279–288. doi: 10.1016/S1074-7613(03)00025-6. [DOI] [PubMed] [Google Scholar]
- 10.Warnatz K, Voll RE. Pathogenesis of autoimmunity in common variable immunodeficiency. Front Immunol. 2012;3:210. doi: 10.3389/fimmu.2012.00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salzer U, et al. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet. 2005;37(8):820–828. doi: 10.1038/ng1600. [DOI] [PubMed] [Google Scholar]
- 12.Castigli E, et al. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet. 2005;37(8):829–834. doi: 10.1038/ng1601. [DOI] [PubMed] [Google Scholar]
- 13.CEREDIH: The French PID study group. The French national registry of primary immunodeficiency diseases. Clin Immunol. 2010;135(2):264–272. doi: 10.1016/j.clim.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 14.Yong PF, Thaventhiran JE, Grimbacher B. “A rose is a rose is a rose,” but CVID is Not CVID common variable immune deficiency (CVID), what do we know in 2011? Adv Immunol. 2011;111:47–107. doi: 10.1016/B978-0-12-385991-4.00002-7. [DOI] [PubMed] [Google Scholar]
- 15.Resnick ES, Cunningham-Rundles C. The many faces of the clinical picture of common variable immune deficiency. Curr Opin Allergy Clin Immunol. 2012;12(6):595–601. doi: 10.1097/ACI.0b013e32835914b9. [DOI] [PubMed] [Google Scholar]
- 16.Fried AJ, Rauter I, Dillon SR, Jabara HH, Geha RS. Functional analysis of transmembrane activator and calcium-modulating cyclophilin ligand interactor (TACI) mutations associated with common variable immunodeficiency. J Allergy Clin Immunol. 2011;128(1):226–228. doi: 10.1016/j.jaci.2011.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan-Hammarstrom Q, et al. Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat Genet. 2007;39(4):429–430. doi: 10.1038/ng0407-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salzer U, et al. Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood. 2009;113(9):1967–1976. doi: 10.1182/blood-2008-02-141937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abecasis GR, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mohammadi J, et al. Novel mutations in TACI (TNFRSF13B) causing common variable immunodeficiency. J Clin Immunol. 2009;29(6):777–785. doi: 10.1007/s10875-009-9317-5. [DOI] [PubMed] [Google Scholar]
- 21.Romberg N, et al. CVID-associated TACI mutations affect autoreactive B cell selection and activation. J Clin Invest. 2013;123(10):4283–4293. doi: 10.1172/JCI69854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koopmans W, Woon ST, Brooks AE, Dunbar PR, Browett P, Ameratunga R. Clinical variability of family members with the C104R mutation in transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI). J Clin Immunol. 2013;33(1):68–73. doi: 10.1007/s10875-012-9793-x. [DOI] [PubMed] [Google Scholar]
- 23.Dorner T, Radbruch A. Antibodies and B cell memory in viral immunity. Immunity. 2007;27(3):384–392. doi: 10.1016/j.immuni.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 24.AbuAttieh M, Bender D, Liu E, Wettstein P, Platt JL, Cascalho M. Affinity maturation of antibodies requires integrity of the adult thymus. Eur J Immunol. 2012;42(2):500–510. doi: 10.1002/eji.201141889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stevens FJ. Modification of an ELISA-based procedure for affinity determination: correction necessary for use with bivalent antibody. Mol Immunol. 1987;24(10):1055–1060. doi: 10.1016/0161-5890(87)90073-3. [DOI] [PubMed] [Google Scholar]
- 26.Allen D, Simon T, Sablitzky F, Rajewsky K, Cumano A. Antibody engineering for the analysis of affinity maturation of an anti-hapten response. EMBO J. 1988;7(7):1995–2001. doi: 10.1002/j.1460-2075.1988.tb03038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shlomchik MJ, Weisel F. Germinal center selection and the development of memory B and plasma cells. Immunol Rev. 2012;247(1):52–63. doi: 10.1111/j.1600-065X.2012.01124.x. [DOI] [PubMed] [Google Scholar]
- 28.Kanno Y, Sakurai D, Hase H, Kojima H, Kobata T. TACI induces cIAP1-mediated ubiquitination of NIK by TRAF2 and TANK to limit non-canonical NF-κB signaling. J Recept Signal Transduct Res. 2010;30(2):121–132. doi: 10.3109/10799891003634509. [DOI] [PubMed] [Google Scholar]
- 29.Khalil AM, Cambier JC, Shlomchik MJ. B cell receptor signal transduction in the GC is short-circuited by high phosphatase activity. Science. 2012;336(6085):1178–1181. doi: 10.1126/science.1213368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goenka R, et al. Local BLyS production by T follicular cells mediates retention of high affinity B cells during affinity maturation. J Exp Med. 2014;211(1):45–56. doi: 10.1084/jem.20130505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu H, et al. Follicular T-helper cell recruitment governed by bystander B cells and ICOS-driven motility. Nature. 2013;496(7446):523–527. doi: 10.1038/nature12058. [DOI] [PubMed] [Google Scholar]
- 32.Yee A, De Ravin SS, Elliott E, Ziegler JB. Severe combined immunodeficiency: a national surveillance study. Pediatr Allergy Immunol. 2008;19(4):298–302. doi: 10.1111/j.1399-3038.2007.00646.x. [DOI] [PubMed] [Google Scholar]
- 33.Mundy R, MacDonald TT, Dougan G, Frankel G, Wiles S. Citrobacter rodentium of mice and man. Cell Microbiol. 2005;7(12):1697–1706. doi: 10.1111/j.1462-5822.2005.00625.x. [DOI] [PubMed] [Google Scholar]
- 34.Bry L, Brenner MB. Critical role of T cell-dependent serum antibody, but not the gut-associated lymphoid tissue, for surviving acute mucosal infection with Citrobacter rodentium, an attaching and effacing pathogen. J Immunol. 2004;172(1):433–441. doi: 10.4049/jimmunol.172.1.433. [DOI] [PubMed] [Google Scholar]
- 35.Ghaem-Maghami M, et al. Intimin-specific immune responses prevent bacterial colonization by the attaching-effacing pathogen Citrobacter rodentium. Infect Immun. 2001;69(9):5597–5605. doi: 10.1128/IAI.69.9.5597-5605.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simmons CP, et al. Central role for B lymphocytes and CD4+ T cells in immunity to infection by the attaching and effacing pathogen Citrobacter rodentium. Infect Immun. 2003;71(9):5077–5086. doi: 10.1128/IAI.71.9.5077-5086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maaser C, et al. Clearance of Citrobacter rodentium requires B cells but not secretory immunoglobulin A (IgA) or IgM antibodies. Infect Immun. 2004;72(6):3315–3324. doi: 10.1128/IAI.72.6.3315-3324.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferreira PC, da Silva JB, Piazza RM, Eckmann L, Ho PL, Oliveira ML. Immunization of mice with Lactobacillus casei expressing a beta-intimin fragment reduces intestinal colonization by Citrobacter rodentium. Clin Vaccine Immunol. 2011;18(11):1823–1833. doi: 10.1128/CVI.05262-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chapel H, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112(2):277–286. doi: 10.1182/blood-2007-11-124545. [DOI] [PubMed] [Google Scholar]
- 40.Martinez-Gallo M, Radigan L, Almejun MB, Martinez-Pomar N, Matamoros N, Cunningham-Rundles C. TACI mutations and impaired B-cell function in subjects with CVID and healthy heterozygotes. J Allergy Clin Immunol. 2013;131(2):468–476. doi: 10.1016/j.jaci.2012.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kayagaki N, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341(6151):1246–1249. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- 42.Cascalho M, Ma A, Lee S, Masat L, Wabl M. A quasi-monoclonal mouse. Science. 1996;272(5268):1649–1652. doi: 10.1126/science.272.5268.1649. [DOI] [PubMed] [Google Scholar]
- 43.Giudicelli V, Chaume D, Lefranc MP. IMGT/GENE-DB: a comprehensive database for human and mouse immunoglobulin and T cell receptor genes. Nucleic Acids Res. 2005;33(Database issue):D256–D261. doi: 10.1093/nar/gki010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alamyar E, Giudicelli V, Li S, Duroux P, Lefranc MP. IMGT/HighV-QUEST: the IMGT(R) web portal for immunoglobulin (IG) or antibody and T cell receptor (TR) analysis from NGS high throughput and deep sequencing. Immunome Res. 2012;8(1):26. [Google Scholar]
- 45.Friguet B, Chaffotte AF, Djavadi-Ohaniance L, Goldberg ME. Measurements of the true affinity constant in solution of antigen-antibody complexes by enzyme-linked immunosorbent assay. J Immunol Methods. 1985;77(2):305–319. doi: 10.1016/0022-1759(85)90044-4. [DOI] [PubMed] [Google Scholar]
- 46.Damm-Ganamet KL, Smith RD, Dunbar JB, Jr, Stuckey JA, Carlson HA. CSAR benchmark exercise 2011–2012: evaluation of results from docking and relative ranking of blinded congeneric series. J Chem Inf Model. 2013;53(8):1853–1870. doi: 10.1021/ci400025f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dunbar JB, Jr, et al. CSAR data set release 2012: ligands, affinities, complexes, and docking decoys. J Chem Inf Model. 2013;53(8):1842–1852. doi: 10.1021/ci4000486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li J, Schantz A, Schwegler M, Shankar G. Detection of low-affinity anti-drug antibodies and improved drug tolerance in immunogenicity testing by Octet((R)) biolayer interferometry. J Pharm Biomed Anal. 2011;54(2):286–294. doi: 10.1016/j.jpba.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 49.Kamada N, et al. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science. 2012;336(6086):1325–1329. doi: 10.1126/science.1222195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Naito Y, et al. Germinal center marker GL7 probes activation-dependent repression of N-glycolylneuraminic acid, a sialic acid species involved in the negative modulation of B-cell activation. Mol Cell Biol. 2007;27(8):3008–3022. doi: 10.1128/MCB.02047-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.