

Abstract
Familial hyperkalemic hypertension (FHHt) is a monogenic disease resulting from mutations in genes encoding WNK kinases, the ubiquitin scaffold protein cullin 3 (CUL3), or the substrate adaptor kelch-like 3 (KLHL3). Disease-associated CUL3 mutations abrogate WNK kinase degradation in cells, but it is not clear how mutant forms of CUL3 promote WNK stability. Here, we demonstrated that an FHHt-causing CUL3 mutant (CUL3 Δ403–459) not only retains the ability to bind and ubiquitylate WNK kinases and KLHL3 in cells, but is also more heavily neddylated and activated than WT CUL3. In cells, activated CUL3 Δ403–459 depleted KLHL3, preventing WNK degradation, despite increased CUL3-mediated WNK ubiquitylation; therefore, CUL3 loss in kidney should phenocopy FHHt in murine models. As predicted, nephron-specific deletion of Cul3 in mice did increase WNK kinase levels and the abundance of phosphorylated Na-Cl cotransporter (NCC). Over time, however, Cul3 deletion caused renal dysfunction, including hypochloremic alkalosis, diabetes insipidus, and salt-sensitive hypotension, with depletion of sodium potassium chloride cotransporter 2 and aquaporin 2. Moreover, these animals exhibited renal inflammation, fibrosis, and increased cyclin E. These results indicate that FHHt-associated CUL3 Δ403–459 targets KLHL3 for degradation, thereby preventing WNK degradation, whereas general loss of CUL3 activity — while also impairing WNK degradation — has widespread toxic effects in the kidney.
Introduction
Familial hyperkalemic hypertension (FHHt; also called pseudohypoaldosteronism type II) is a rare monogenic disease characterized by hyperkalemia and hypertension (1). The disease can result from mutations in 4 genes: the atypical kinases WNK1 and WNK4, and the cullin RING ligase (CRL) family members kelch-like 3 (KLHL3) and cullin 3 (CUL3). The products of these genes all appear to modulate the thiazide-sensitive Na-Cl cotransporter (NCC; encoded by SLC12A3). While activation or suppression of ion channels or transporters other than NCC may also contribute to the phenotype, there is strong evidence that excessive NCC activity (2), as reflected by hyperphosphorylation and increased abundance (3), plays an essential and nonredundant role.
Disease-causing WNK1 and WNK4 mutations increase WNK kinase activity, which is directed largely toward 2 other homologous kinases: serine threonine kinase 39 (SPAK; encoded by STK39) and oxidative stress responsive 1 (OSR1; encoded by OXSR1). These kinases, in turn, phosphorylate and activate NCC directly, causing both hyperkalemia and hypertension. The described mutations in WNK1 are intronic (and hence noncoding) mutations; these increase expression of the kinase-active form of WNK1 along the distal convoluted tubule (DCT), a nephron segment where its expression levels are typically low (4), thereby activating SPAK and NCC (4). WNK4 mutations are missense mutations in 1 of 2 coiled-coiled motifs. At least some WNK4 mutations appear to cause the disease by disrupting WNK4’s ability to interact with KLHL3. KLHL3 is a BR-C, ttk, and bab (BTB) motif–containing protein that interacts with CUL3 and a RING ligase to form an E3 ubiquitin ligase complex. KLHL3 acts as an adaptor for substrates; binding of WNK4 or WNK1 to the complex leads to their ubiquitylation, tagging them for degradation, likely via the proteasome (5–8). Similarly, the disease-causing KLHL3 mutations are missense mutations, generating KLHL3 that cannot interact with either CUL3 or WNK kinases (depending on the site of the mutation). This provides a plausible hypothesis for the disease pathogenesis: increased WNK kinase abundance activates SPAK and NCC (1).
The CUL3 mutations that cause FHHt result in loss of exon 9 during splicing, leading to a CUL3 protein with a 57–amino acid deletion (Δ403–459) (9). These are autosomal-dominant mutations, which may occur de novo (9). Uchida and colleagues reported that disease-causing CUL3 mutations generate a protein that degrades WNK4 less efficiently (5). This would suggest that WNK4, KLHL3, and CUL3 mutations all impair the ability of the CRL pathway to ubiquitylate and degrade the WNK kinase via similar loss-of-function mechanisms. Here, we present data indicating that the disease-causing mutant CUL3 instead exhibits enhanced ubiquitin ligase activity, leading to KLHL3 degradation. These effects caused target-specific loss of ubiquitylating activity by degrading the scaffolding protein and thereby increasing WNK kinase abundance. As systemic constitutive Cul3 knockout is embryonic lethal (10), we elected to test the hypothesis that loss of Cul3 would mimic human disease by deleting it only along the nephron and only in adult mice, thereby avoiding systemic and developmental effects. The results showed that loss of CUL3 activity along the nephron did lead to an increase in WNK kinase and NCC abundance, as expected, but it also led to substantial tubule cell toxicity and renal fibrosis. We propose a model to account for these observations.
Results
CUL3 Δ403–459 binds and ubiquitylates KLHL3 and WNK4.
It has been proposed that disease-causing CUL3 mutations abrogate CUL3’s ability to bind to the substrate adaptor KLHL3 and facilitate WNK4 ubiquitylation (5, 11). However, the location of the Δ403–459 mutation is not in any known functional domain (9). Thus, to test whether CUL3 Δ403–459 retains the ability to associate with KLHL3, we immunoprecipitated KLHL3 from HEK293 cells with WT CUL3 and CUL3 Δ403–459. CUL3 Δ403–459 not only retained the ability to interact with KLHL3, it appeared to exhibit a greater affinity for the substrate adaptor (Figure 1A). As controls, we confirmed that CUL3 Δ51–67, a mutant known to lack binding to BTB domain proteins (12), did not associate with KLHL3, and that activating mutant CUL3 K712R, which retains its binding abilities, associated normally. We confirmed that the apparent enhanced binding activity was not restricted to KLHL3, as CUL3 Δ403–459 also exhibited enhanced association with 2 other BTB proteins (Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI76126DS1).
Figure 1. CUL3 and CUL3 Δ403–459 bind and facilitate ubiquitylation of KLHL3 and WNK4.

(A) Immunoprecipitation results showing cotransfection of MYC-tagged KLHL3 with WT CUL3, CUL3 Δ403–459, CUL3 Δ51–67 (which cannot bind BTB proteins), and CUL3 K712R (an activated CUL3 known to bind normally). (B) Ubiquitylation of WNK4 by WT CUL3 and CUL3 Δ403–459. The CUL3 and KLHL3 substrate WNK4 were transfected with either WT CUL3 or CUL3 Δ403–459 and HA-ubiquitin. WNK4 ubiquitylation was greater in the presence of CUL3 Δ403–459. (C) WT CUL3 and CUL3 Δ403–459 were transfected with the substrate adaptor KLHL3 and HA-tagged ubiquitin. CUL3 facilitated ubiquitylation of KLHL3, but CUL3 Δ403–459 was more efficient than WT CUL3. (D) HeLa cells were transfected with WT CUL3, CUL3 Δ403–459, and/or KLHL3 as indicated and stained for fluorescent microscopy. WT CUL3 was predominantly nuclear (single arrow), whereas the mutant was mainly localized to the cytoplasm (double arrow). KLHL3, a cytoplasmic protein, showed the ability to recruit WT CUL3 to the cytoplasm (triple arrow); it did not change CUL3 Δ403–459 localization. Scale bar: 50 μm. A similar pattern was observed in mDCT cells (Supplemental Figure 7). (E) The CUL3 substrate cyclin E (MYC tagged) and HA-tagged ubiquitin were transfected. An increase in ubiquitylated cyclin E was observed in the presence of CUL3 Δ403–459. In all cases, results are representative of at least 3.
Wakabayashi and colleagues reported that CUL3 Δ403–459 exhibited a reduced ability to degrade WNK4 in cells (5). Although they suggested that this reflected a loss of ubiquitylating activity, this was not tested directly. Thus, we tested the ability of CUL3 and CUL3 Δ403–459 to ubiquitylate WNK4 by coexpressing WNK4, ubiquitin, and either CUL3 or CUL3 Δ403–459. The results confirmed that CUL3 is capable of facilitating WNK4 ubiquitylation in cells (7). Surprisingly, however, CUL3 Δ403–459 was more effective than WT CUL3 in facilitating WNK4 ubiquitylation, not less so (Figure 1B); note that the conditions used for this experiment assured that WNK4 abundance was high. As exogenous BTB-containing proteins (such as KLHL3) were not added to the reaction mixtures, we speculated that the ubiquitylation of WNK4 resulted from the presence of an endogenous adaptor. To confirm this, we used CUL3 Δ51–67 (which does not bind BTB proteins) as an additional control. CUL3 Δ51–67 did not ubiquitylate WNK4; as we could not detect KLHL3 in HEK293 cells at baseline (see below), the nature of this adaptor remains unknown.
Because Ohta and colleagues previously reported that CUL3 is capable of ubiquitylating its own substrate adaptor, KLHL3 (7), we tested whether CUL3 Δ403–459 retains its ability to ubiquitylate KLHL3. We first confirmed that CUL3 facilitated ubiquitylation of KLHL3, but also found that CUL3 Δ403–459 ubiquitylated KLHL3 more effectively than did WT CUL3 (Figure 1C); this suggested that mutant CUL3 is a generalized gain-of-function mutation. To provide further evidence, we studied the localization of CUL3 in HeLa cells. In these cells, CUL3 must be recruited from the nucleus to the cytoplasm, where it is neddylated and activated (13). Here, we confirmed that CUL3 was predominantly nuclear in unstimulated HeLa cells and recruited to the cytoplasm when KLHL3 was cotransfected. In contrast, even at baseline, CUL3 Δ403–459 was predominantly cytoplasmic, without cotransfection of KLHL3 (Figure 1D), which suggests that it is activated. To confirm that this increased ubiquitylating activity is a general phenomenon, we compared the ability of WT CUL3 and CUL3 Δ403–459 to facilitate ubiquitylation of a canonical CUL3 target, cyclin E (10). As with WNK4, CUL3 Δ403–459 facilitated more ubiquitylation of cyclin E than did CUL3 (Figure 1E).
CUL3 Δ403–459 reduces KLHL3 and increases WNK kinase abundance in cells.
These results suggest that disease-causing mutant CUL3 exhibits enhanced activity toward both WNK4 and KLHL3, but they do not indicate how this affects WNK kinase abundance and NCC activity, as ubiquitylation has diverse effects. As the experiments in Figure 1 were designed to keep protein abundance high to test for binding sensitivity, they were not informative regarding the effects of mutant CUL3 on WNK kinase and KLHL3 abundance. We therefore tested whether KLHL3 suppresses WNK kinase–stimulated NCC activity in Xenopus oocytes. In these experiments, it was necessary to stimulate NCC activity with a kidney-enriched activating WNK1 isoform (14) or with WNK3 to observe effects of KLHL3 or CUL3. The results showed that WT KLHL3, but not disease-causing KLHL3 R528H, substantially reduced NCC activity (Figure 2A), consistent with previously suggested models (5–8, 11). Similarly, KLHL3 coexpressed with CUL3, but not with CUL3 Δ403–459, reduced NCC activity (Figure 2B). This result seems to suggest that CUL3 Δ403–459 lacks function, and yet CUL3 Δ403–459 retained ubiquitin ligase activity, as shown above. Thus, we further examined the mechanisms involved. In HEK293 cells, KLHL3 expression reduced WNK4 and WNK3 abundance; the effect on WNK1 was not significant (Figure 2, C–E, and Supplemental Figure 2). In this setting, WT CUL3 overexpression had no substantial effect on WNK kinase abundance, although it did reduce KLHL3 abundance slightly (Figure 2C), consistent with its previously reported ability to ubiquitylate KLHL3. In contrast, overexpression of disease-causing CUL3 Δ403–459 increased WNK4 abundance substantially and reduced KLHL3 abundance strikingly (Figure 2C and Supplemental Figure 2). Similar effects were also observed in the presence of WNK1 and WNK3 (Figure 2, D and E). As KLHL3 (or a homolog) is required for CUL3 to degrade substrates efficiently (5), this suggests that CUL3 Δ403–459 increases WNK kinase abundance because it degrades the obligate WNK substrate adaptor.
Figure 2. Effects of KLHL3 and CUL3 on NCC activity and WNK kinase abundance.

(A) Effects of WT KLHL3 (K3) and the KLHL3 R528H mutant (K3 mut) on NCC activity in oocytes. In the presence of WNK1, WT KLHL3, but not the KLHL3 mutant, inhibited activity. Significance by ANOVA with Dunnett test. n = 3 frogs. (B) In the presence of KLHL3, CUL3 inhibited NCC activity, stimulated by either WNK1 or WNK3 (WNK), whereas CUL3 Δ403–459 (CUL3 Δ) did not. Pooled results from 2 experiments with WNK1 and 1 with WNK3 (>20 oocytes per experiment; n = 3 frogs per group; significance by ANOVA with Dunnett test; for individual data, see Supplemental Figure 8). (C–E) Immunoblots showing effects of KLHL3 alone, or with WT or mutant CUL3, on WNK4 (C), WNK3 (D), and WNK1 (E) abundance in HEK293 cells.
CUL3 Δ403–459 is activated and promotes KLHL3 degradation.
To function in ubiquitylation reactions, CUL3 must be activated by, and bound to, NEDD8 (10), a process called neddylation. A higher-resolution immunoblot clearly demonstrated that endogenous CUL3 runs as a doublet. MLN4924, a nonspecific neddylation inhibitor, collapsed the higher band into the lower one (Figure 3A), which confirmed that the higher band was the neddylated form. To test whether CUL3 Δ403–459 is more active because it is more heavily neddylated than CUL3, we immunoprecipitated FLAG-tagged CUL3 from HEK293 cells and blotted for NEDD8. Under basal conditions, CUL3 Δ403–459 was much more heavily neddylated than CUL3 (Figure 3B). To confirm that CUL3 degradation of KLHL3 requires ubiquitylation, we tested the effects of MLN4924, which inhibits CUL3 activation by neddylation. The abundance of KLHL3 was increased by MLN4924 treatment (Figure 3C and Supplemental Figure 3). Ubiquitylation typically targets proteins for degradation via the proteasome. We therefore tested whether inhibiting the proteasome would inhibit CUL3-mediated WNK kinase degradation. The results showed that proteasomal inhibition increased both WNK4 abundance and the relative abundance of the neddylated CUL3 form (Figure 3D). It did not increase KLHL3 abundance, however (see Discussion).
Figure 3. Neddylation of WT and mutant CUL3.

(A) The neddylation inhibitor MLN4924 collapsed the upper band of CUL3. (B) Increased neddylation of CUL3 Δ403–459, compared with WT, precipitated from HEK293 cells and blotted for NEDD8. The CUL3 constructs were tagged with FLAG, and immunoprecipitation was performed with an anti-FLAG antibody. Anti-FLAG and actin immunoblots show input; note that mutant CUL3 was expressed at lower levels than WT. (C) Inhibiting neddylation with MLN4924 reduced KLHL3 degradation in the presence of both WT and mutant CUL3. (D) Inhibiting proteasomal degradation with MG132 increased WNK4 abundance in the presence of KLHL3 and CUL3. Similar results were obtained with CUL3 Δ403–459. Immunoprecipitation was not performed for A, C, and D.
KLHL3 and CUL3 are differentially expressed along the nephron.
We next examined sites of KLHL3 and CUL3 expression in mouse kidney. Quantitative PCR confirmed that Klhl3 mRNA was highly expressed along the DCT (Figure 4A and ref. 15), with substantial expression along the thick ascending limb (TAL) and collecting duct (CD). The pattern of Cul3 expression was quite different, with modest and relatively uniform expression along all segments (Figure 4A). Controls confirmed correct tubule identification (Supplemental Figure 4).
Figure 4. Sites of expression of KLHL3 and CUL3 along the normal nephron.

(A) Klhl3 and Cul3 expression along the nephron, analyzed by quantitative PCR. Proximal tubule (PT), TAL, DCT, CD, and glomerulus (Glom) are compared with total kidney. Values are mean ± SEM from 3 independent experiments. (B–D) IF of KLHL3, WNK4, and CUL3 in cortical segments (DCT identified by NCC; B and C are consecutive sections); CUL3 was enriched in proximal tubules. (E and F) KLHL3 (E) and WNK4 (F) in TAL (identified by NKCC2); these are also consecutive sections. (G) KLHL3 in the medulla, where its expression along the thin limb (TL) and TAL (identified by NKCC2) was robust. (H) KLHL3 at the junction of a DCT, connecting tubule (CNT), and cortical collecting duct; CNT and cortical collecting duct (CCD) were identified by AQP2. Intercalated cells (IC) were enriched. (I) Medullary collecting duct, showing that KLHL3 was highly expressed in intercalated cells, as indicated by H+-ATPase expression. (J) Expression sites for KLHL3 and CUL3. OM, outer medulla; OS, outer stripe; IS, inner stripe; IM, inner medulla; C, cortex; MD, macula densa; MCD, medullary connecting duct; CTAL, cortical TAL; MTAL, medullary TAL. Intercalated cells are denoted by boxes. Scale bars: 20 μm.
At the protein level, KLHL3 was abundant in DCT cells, as was WNK4 (Figure 4, B and C). In contrast, CUL3 expression was diffuse throughout the nephron (Figure 4D), consistent with the PCR results, with lower levels in the DCT (antibody specificity was confirmed in mice with kidney-specific Cul3 knockout; Supplemental Figure 5). KLHL3 was also highly expressed along the TAL, as identified by NKCC2 (encoded by SLC12A1); in the TAL, WNK4 and NKCC2 colocalized at the apical membrane, whereas WNK4 and NCC did not colocalize in the DCT (Figure 4, B, C, E, and F). KLHL3 was also detected in thin ascending limbs, and at lower levels in principal cells of the cortical collecting ducts, as identified by double labeling for aquaporin 2 (AQP2) (Figure 4, G and H). Intercalated cells, identified by H+-ATPase, appeared to be enriched in KLHL3, and although the abundance was low in medullary CD principal cells, it remained high in intercalated cells (Figure 4I). Sites of KLHL3 and CUL3 expression along the nephron are shown schematically in Figure 4J.
Mice with kidney-specific Cul3 deletion exhibit abundant WNK kinase and NCC phosphorylation.
Disease-causing KLHL3 mutants lack the ability to interact either with CUL3 or with WNK kinases, leading to WNK kinase accumulation and NCC activation (5–8, 16). CUL3 knockdown in cells led to an increase in WNK1 abundance (7), and transfection of dominant-negative CUL3 1–400 increased WNK4 in oocytes (8). This suggested that Cul3 knockout should phenocopy FHHt. To test this, we generated mice in which Cul3 could be deleted along the nephron, under the control of doxycycline. We bred mice with loxP sites surrounding exons 4–7 of Cul3 (Cul3fl/fl) (10) to mice carrying Pax8-rTA and TRE-LC1 (17). The resulting offspring were screened and selected for Cul3fl/fl homozygosity and Pax8-rTA/TRE-LC1 heterozygosity. These mice were born at the expected rate and appeared normal at birth and throughout development. Doxycycline treatment in these mice induced kidney-specific Cul3 deletion (referred to herein as KS-Cul3–/– mice).
Initial experiments indicated that Cul3 deletion led to kidney damage within several weeks (see below), so we examined the KS-Cul3–/– phenotype after short-term deletion. Mice maintained on a normal salt diet (0.49% NaCl) were treated with doxycycline for 7 days, and blood and kidneys were collected for analyses. Western blot analysis revealed substantial reductions in CUL3 after only 7 days of doxycycline, along with significant increases in the abundance of WNK4, WNK1, and WNK3 and the phosphorylated form of NCC (pNCC) (Figure 5, A and B). Although the abundance of total NCC was not different, the abundance of larger–molecular weight complexes of NCC, typically assumed to be dimers, was higher in KS-Cul3–/– mice (Figure 5A). In contrast, the abundance of pNKCC2 and AQP2 was significantly reduced (Figure 5, A and B). KLHL3 was not affected. KS-Cul3–/– mice displayed mild hypernatremia and a tendency toward hypokalemia, but neither the hematocrit nor the aldosterone concentration was significantly different (Figure 5, C–F). As the possible hypokalemia, and any undetected extracellular fluid (ECF) volume depletion, might have activated NCC as a compensatory effect, we then repeated the experiments using mice maintained on a high-salt diet (8% NaCl) for 1 week prior to starting doxycycline treatment and throughout the entire 7-day treatment period. Under these conditions, WNK4 and pNCC levels were again significantly higher in KS-Cul3–/– mice (Figure 5, G and H). In contrast to our findings with normal-salt diet, pNKCC2 was not significantly altered (although there was a trend toward an increase; see Discussion), and AQP2 showed a dramatic increase in expression. Interestingly, in 1 mouse in which Cul3 deletion was incomplete (Figure 5G), the abundance of pNCC and WNK4 appeared lower than that in mice with more complete Cul3 deletion. KS-Cul3–/– mice also displayed hypernatremia, but the plasma potassium concentration was similar to that of controls (Figure 5, I and J); both hematocrit and aldosterone were also similar between treatment groups (Figure 5, K and L), suggestive of absent ECF volume depletion.
Figure 5. WNK kinase abundance, plasma electrolytes, hematocrit and aldosterone in mice with short-term Cul3 deletion.

Control and doxycycline-treated (KS-Cul3–/–) mice were fed (A–F) control diet or (G–L) high-salt diet. (A and G) Immunoblotting of CUL3, pNCC, total NCC (showing monomeric and likely dimeric forms), WNK4, WNK1, WNK3, KLHL3, pNKCC2, and AQP2, as indicated. Arrow in G denotes a mouse in which Cul3 deletion was incomplete (see text for details). (B and H) Quantification of A and G, respectively. *P < 0.05 vs. control, ANOVA with Dunnett test. (C–F and I–L) Plasma sodium concentration (C and I), plasma potassium concentration (D and J), hematocrit (E and K), and plasma aldosterone concentration (F and L). Plots show individual values and mean ± SEM; differences were determined by unpaired t test.
Chronic KS-Cul3–/– mice exhibit polyuria, salt wasting, and salt-sensitive hypotension.
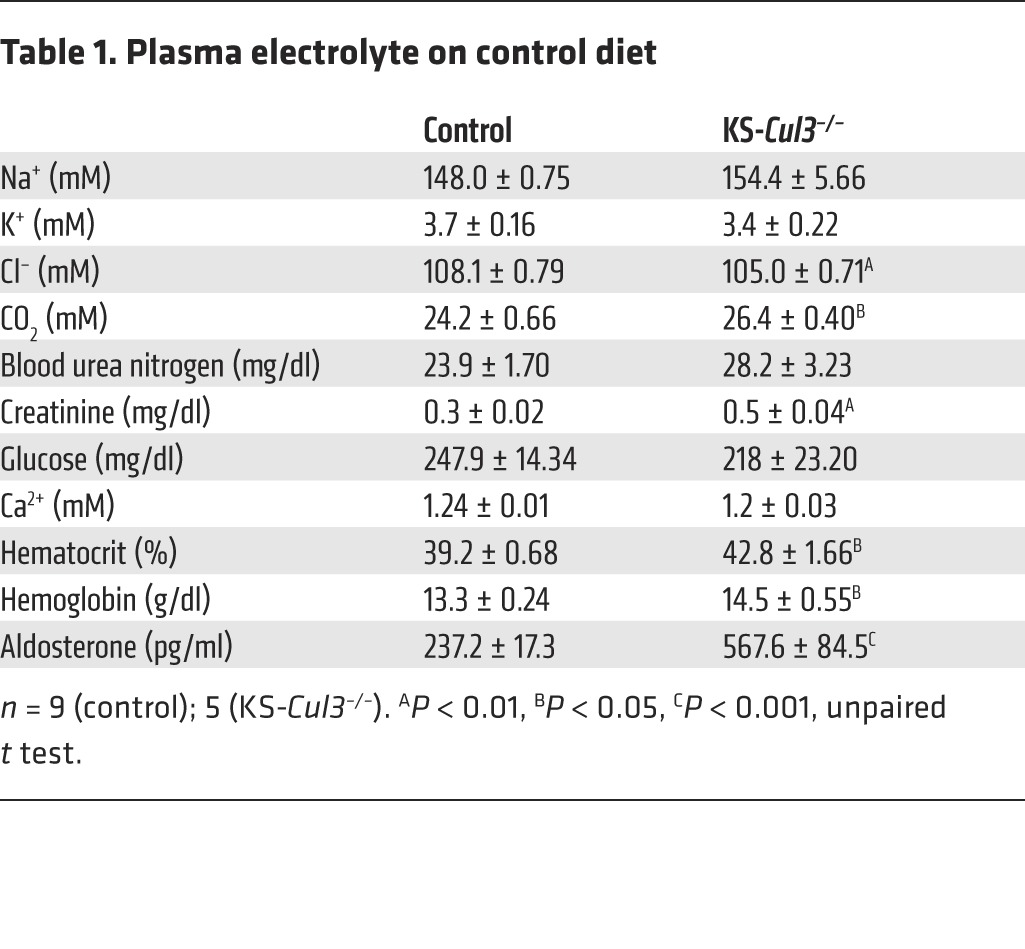
To test whether longer-term Cul3 deletion mimics the FHHt phenotype, we studied another group of mice 6 weeks after doxycycline treatment. At this time point, Western blotting of kidney tissue again confirmed substantial loss of CUL3 in KS-Cul3–/– mice (Figure 6A), but the mice appeared normal, although smaller. The abundance of WNK1, WNK3, and WNK4 was greater, with WNK4 being affected most dramatically (Figure 6A). As in the mice with short-term deletion, pNCC was more abundant in KS-Cul3–/– mice than in controls, and the total NCC abundance appeared slightly increased, but this was not significant when corrected for actin (Figure 6B). There was also evidence that SPAK and perhaps OSR1 were activated (Figure 6B), as indicated by an isoform switch (18). In these mice, although KLHL3 abundance was not increased (data not shown), KLHL3 assumed a punctate appearance in DCT and TAL segments, and WNK4 appeared very abundant and more widely distributed than in controls (Figure 6C). Despite the increased pNCC, the plasma [K+] was not higher in the KS-Cul3–/– mice (it tended to be lower), and the mice exhibited hypochloremic alkalosis (Table 1). In these mice, there were also signs of ECF volume contraction, with higher aldosterone and hematocrit (Figure 6, D and E). Finally, the urine volume was dramatically increase and urine osmolality was substantially lower in KS-Cul3–/– mice (Figure 6, F and G). Western blotting revealed that AQP2 was almost completely absent in KS-Cul3–/– mice (Figure 6H).
Figure 6. WNK kinase abundance, hematocrit and aldosterone in mice with chronic Cul3 deletion.

Mice were studied 6 weeks after control or doxycycline (KS-Cul3–/–) treatment. (A) Differences in WNK1 (P = 0.028), WNK3 (P = 0.009), WNK4 (P = 0.002), and CUL3 (P = 0.0002) were all significant (ANOVA with Dunnett test) after normalization to GAPDH, as actin varied between groups. (B) Differences in pNCC (P ≤ 0.001), NCC (P < 0.05), full-length SPAK (P < 0.001), kidney-specific SPAK (P < 0.05) and OSR1 (P < 0.01) were significant (ANOVA with Dunnett test). (C) IF showing more enhanced KLHL3 appearance and substantially increased WNK4 abundance (representative of 3). Scale bars: 50 μm. (D–G) Effects of KS-Cul3–/– on plasma aldosterone (D), hematocrit (E), 24-hour urine excretion (F), and urine osmolality (G). Plots show individual values with mean ± SEM; differences were determined by unpaired t test. (H) AQP2 was almost completely absent from mice with long-term Cul3 disruption. Note that these samples were from a different set of animals from that in A and B.
Table 1.
Plasma electrolyte on control diet
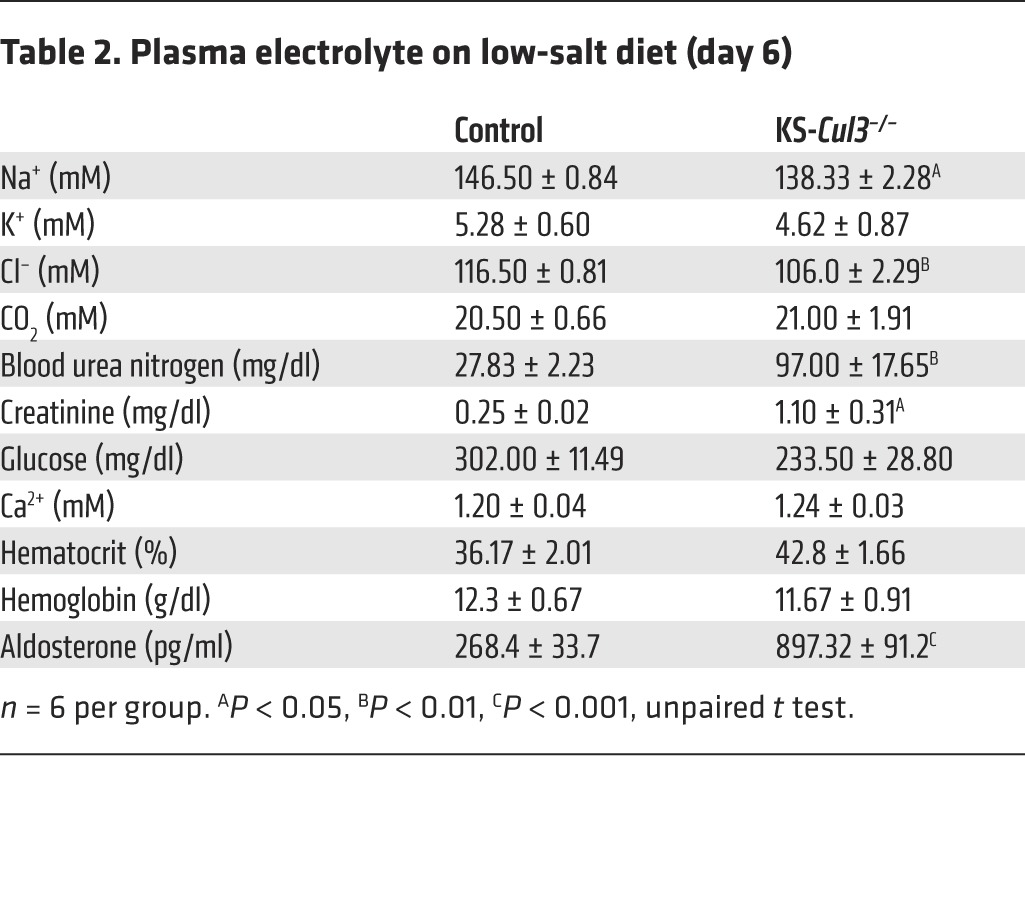
KS-Cul3–/– mice weighed less than control animals and displayed profound polyuria (Figure 7, A and B). To test for salt wasting, we performed metabolic cage studies at baseline and also subjected the mice to dietary salt deprivation. The difference in BW between control and KS-Cul3–/– mice at baseline was magnified by dietary salt restriction, as the KS-Cul3–/– mice lost weight, while control mice gained it (Figure 7A). When placed on a low-salt diet, the urinary sodium and chloride excretion rates declined more slowly in KS-Cul3–/– mice than in controls (Figure 7, C and D), confirming salt wasting. KS-Cul3–/– mice also exhibited potassium wasting and profound hypercalciuria (Figure 7, E and F), even though the plasma calcium concentration remained normal (Table 2). These data suggested that the mice were wasting both salt and water, but pNCC levels were elevated, and pNKCC2 expression was unchanged at baseline (Figure 6B); ENaC abundance also did not appear to be affected (data not shown). As this did not explain the salt wasting with hypercalciuria at baseline, we also examined pNKCC2 expression by immunofluorescence (IF). Cortical pNKCC2 was maintained, but pNKCC2 was essentially absent in the renal medulla (Figure 7G), which likely explains these effects, although kidney damage (see below) limited quantitative interpretation of the IF data. The mean arterial pressure (MAP), measured by radiotelemetry, was not different in the 2 groups under basal conditions, KS-Cul3–/– mice exhibited a gradual decline when challenged with dietary salt restriction (Figure 7I).
Figure 7. BW, urinary electrolytes, urine osmolality, and MAP in control and KS-Cul3–/– mice during low salt intake.

(A) BW of control and KS-Cul3–/– mice on a normal salt diet (NL) and for 6 days after switching to a low-salt diet (L1–L6) (n = 5–6 per group). P < 0.001, repeated-measures ANOVA. (B–F) Urinary electrolyte and water excretion in control and KS-Cul3–/– mice at baseline and after switching to low-salt diet (n = 6–7, all females aged 5 months). P < 0.001 for all differences, 2-way repeated-measures ANOVA. (G) Near absence of pNKCC2 in medulla from KS-Cul3–/– mice. Scale bars: 100 μm. (H) MAP, obtained by telemetry in male control and KS-Cul3–/– mice at 4 months of age, after long-term Cul3 disruption. The switch from normal to low dietary salt was considered day 0 (n = 3 per group for telemetry).
Table 2.
Plasma electrolyte on low-salt diet (day 6)
Chronic KS-Cul3–/– causes kidney damage and inflammation.
During harvest of kidneys for Western blotting, we observed that KS-Cul3–/– kidneys were smaller than WT kidneys and misshapen. KS-Cul3–/– kidneys were denser than WT kidneys (Figure 8A). Consistent with the development of renal damage, plasma creatinine was higher in these mice (Figure 8B). Histological analysis revealed patchy tubulointerstitial inflammation and fibrosis, with signs of inflammation including neutrophils and white cell casts (Figure 8C). These changes were observed as early as 11 days after the initiation of doxycycline treatment (data not shown) and appeared to be concentrated in the medulla, with extensions into the cortex (possibly representing medullary rays). We suspected that Cul3 deletion activated cyclin-dependent kinases in kidney tubules, leading to kidney damage, as CUL3 normally suppresses cyclin E (10, 19), a cell cycle regulator important for restraining cell growth. Cul3 deletion from liver causes cell proliferation by activating cyclin E (20), so we examined cyclin E abundance in kidneys. As anticipated, the abundance of cyclin E was substantially greater in KS-Cul3–/– mice than in controls (Figure 8D).
Figure 8. Kidney weight, plasma creatinine, cyclin E expression, and kidney morphology in control and KS-Cul3–/– mice.

(A) Kidneys were significantly heavier and (B) plasma creatinine significantly higher in KS-Cul3–/– mice than in controls. (C) KS-Cul3–/– mice developed renal inflammation and fibrosis. Ovals denote glomeruli. Atrophic tubules (large arrows) and mononuclear inflammatory cells (small arrows) are illustrated. Inset shows a white cell cast (large arrow) and neutrophils (small arrows). Original magnification, ×400; ×200 (inset). (D) Cyclin E was increased in KS-Cul3–/– mice (P < 0.001), and there was a switch in apparent molecular size (blot quantified by Ponceau staining; see Supplemental Figure 9). (E) Simplified model of CUL3 effects. In WT cells, KLHL3 binds WNK kinases and CUL3, permitting ubiquitylation (ubiq) of WNK kinases, degrading WNK kinases, and restraining NCC. It also ubiquitylates cyclin E (via a different BTB protein). In Cul3-deficient cells, KLHL3 binds to WNK4, but cannot foster degradation as there is no CUL3, so WNK and pNCC increase. Disruption of Cul3 in the kidney also leads to kidney damage, possibly via increased expression of cyclin E. In CUL3-mediated FHHt, the mutant protein ubiquitylates KLHL3 strongly, leading to its degradation. Even though CUL3 retains the ability to ubiquitylate WNK kinases, this does not occur, as KLHL3 is missing and cannot complex CUL3 with WNK kinases. Several components and details of the CRL pathway are omitted for clarity.
Discussion
Mutations in WNK1, WNK4, KLHL3, and CUL3 cause FHHt, a disease of hypertension and hyperkalemia. Vascular actions of CUL3 may contribute to the hypertension, as Pelham and colleagues proposed that CUL3 regulates vascular tone via an effect on RhoA/Rho kinase signaling (21). Yet FHHt is dominated by hyperkalemia, which suggests that the kidney plays a key role, and NCC activation along the distal nephron is now viewed as an essential feature (1). Although many mechanistic details about the disease remain unclear, WNK1, WNK4, KLHL3, and CUL3 all appeared to affect the abundance and activity of NCC and to modulate the response to dietary salt and potassium challenge. As noted above, most data suggest that mutations in either WNK1 or WNK4 increase the abundance and activity of NCC by activating the intermediary kinase SPAK (4, 22, 23). In contrast, KLHL3 is an adaptor protein that binds to WNK kinases and, in association with CUL3, facilitates their ubiquitylation and degradation (5–8). Some disease-causing WNK4 mutants lack the ability to associate with KLHL3 normally, whereas some disease-causing KLHL3 mutants lack the ability to form complexes with WNK kinases or CUL3; it has been suggested that such mutations abrogate WNK1 and WNK4 ubiquitylation, increasing WNK kinase abundance and activating NCC (5–8). Wakabayashi and colleagues showed that CUL3 Δ403–459 also increases WNK kinase abundance relative to CUL3 when expressed in cells and suggested that this mutation also generates a protein that lacks the ability to ubiquitylate proteins effectively (i.e., loss of function; ref. 5).
Here, we confirmed that CUL3 reduced NCC activity in oocytes (8) and showed that CUL3 Δ403–459 did not. We also confirmed that CUL3 Δ403–459 degraded WNK kinases less effectively than did WT CUL3 (5), but this was not the result of a simple loss of function. Instead, CUL3 Δ403–459 exhibited increased ability to bind and to degrade substrates. To explain how such a gain-of-function mutation could impair CUL3 actions on WNK kinases, we reflected on another property of CUL3. Ohta and colleagues showed that CUL3 can ubiquitylate and foster the degradation of its own substrate adaptor, KLHL3 (7). We showed in cells that introducing mutant CUL3 Δ403–459 did lead to a striking increase in WNK kinase abundance, but this was associated with an equally striking reduction in KLHL3. CUL3 Δ403–459 was also more heavily neddylated at baseline, providing further evidence of its activation. Based on these results, we suggest that CUL3 mutations cause FHHt by degrading KLHL3 in the distal nephron; as WNK kinase ubiquitylation is dependent on KLHL3 (7), this impairs WNK kinase degradation (Figure 8E).
We were able to mimic the effect of CUL3 Δ403–459 (both increasing WNK kinase abundance and decreasing KLHL3 abundance) in the same experimental system; we also showed that the effect of CUL3 Δ403–459 on KLHL3 was dependent on neddylation, because neddylation inhibitors blocked it, and that the effects on WNK kinases were dependent on the proteasome. As the gain of function observed with CUL3 Δ403–459 appears to be a general phenomenon, and as CUL3 is widely expressed, we propose that CUL3-initiated FHHt is a distal tubule disease, largely because KLHL3 is so highly expressed in this segment (at least 20-fold higher than other segments; Figure 4 and ref. 15). Boyden and colleagues suggested that FHHt-causing CUL3 mutations must not affect all, or even many, CUL3 actions, as that “would undoubtedly produce very broad phenotypes” (9). In fact, such a broad phenotype does result from systemic CUL3 deletion, which is lethal (10), or from renal deletion, which we found here to cause kidney damage and fibrosis. Instead, Boyden and colleagues suggested that the CUL3 mutation would likely disrupt its ability to ubiquitylate a specific substrate within the KLHL3 pathway (9). The current results suggest that the affected substrate is likely to be KLHL3 itself, but we detected enhanced, rather than reduced, ubiquitylation. We do not know whether CUL3 Δ403–459 activity is increased with respect to all CUL3 substrates, but our results showed that this effect was not restricted to KLHL3. It was thus surprising that KLHL3 was not more abundant in the KS-Cul3–/– mice; conversely, cyclin E, another CUL3 substrate, was more abundant. This may reflect the relatively low CUL3 activity in DCT cells at baseline, which may also permit KLHL3 abundance to be very high. Low activity could result from low CUL3 abundance in the DCT (Figure 4), from preservation of CUL3 in its unneddylated state, or from other mechanisms that protect KLHL3 from ubiquitylation.
Examples of similar regulated feedback loops are known. Kelch-like ECH-associated protein 1 (KEAP1) is a BTB-containing substrate adaptor that targets the nuclear factor-erythroid 2 p45-related factor 2 (NRF2; encoded by NFE2L2) for ubiquitylation and degradation through the actions of CUL3. Yet oxidative stress can make KEAP1 itself, rather than NRF2, the primary target for CUL3-dependent ubiquitylation, leading to an increase in NRF2 (24). It is interesting to note that KEAP1 degradation is not affected by inhibition of the proteasome, highly reminiscent of our observed lack of effect of MG132 on KLHL3 (Figure 4D). The importance of regulatory ubiquitylation of adaptor proteins by cullins was first observed in yeast, where F-box adaptor proteins are targeted by the same CUL1 that mediates ubiquitylation of their substrates (25, 26). It is therefore possible that the effects of CUL3 to target either WNK kinases or KLHL3 actively may be modulated physiologically.
Our results indicate that FHHt-causing CUL3 Δ403–459 lacks the ability to mediate CUL3-KLHL3-RING E3 ubiquitin ligase activity toward WNK kinases because the obligate adaptor protein KLHL3 is degraded; this therefore leads to WNK kinase accumulation (Figure 8E). Therefore, we predicted that loss of CUL3 function in the kidney would phenocopy FHHt, since CUL3-KLHL3-RING E3 ubiquitin ligase activity would also be disrupted in this situation. Indeed, after short- or long-term disruption of Cul3, WNK kinase and pNCC levels were elevated, as expected. Yet KS-Cul3–/– mice exhibited profound salt, potassium, and calcium wasting (as explained by significantly reduced renal medullary pNKCC2) and polyuria (as shown by the near-complete loss of renal AQP2). As this salt and water wasting might have contributed to the increased WNK4 and pNCC as compensatory responses, we analyzed expression after short-term Cul3 disruption on both normal-salt and high-salt diets (the latter to avoid ECF volume contraction). The mice consuming a normal-salt diet did have mild (albeit not significant) hypokalemia, which might stimulate NCC, but plasma potassium concentration was entirely normal when the mice consumed a high-salt diet. Furthermore, the plasma aldosterone concentration and hematocrit were nearly the same between treatment groups. Our results suggest that the effects of Cul3 deletion on WNK kinase abundance are primary, and not the result of compensatory processes, and are entirely consistent with the recent report by Sohara and colleagues showing effects of KLHL3 R528H knockin (16). The striking increase in AQP2 abundance in the mice fed high-salt diet was likely the expected effect of tonicity on vasopressin and suggests that, at this early stage, the mice can activate AQP2 normally. The reduction in pNKCC2 abundance in the mice on normal-salt diet, but not high-salt diet, may have resulted from the combination of mild ECF volume contraction and CUL3 lack, as dietary salt restriction reduced pNCC abundance in chronic KS-Cul3–/– mice (data not shown). The mechanisms for these anomalous effects remain to be determined. According to our model (Figure 8E), the DCT is exquisitely sensitive to FHHt-causing mutations in CUL3 because of the extremely high expression of KLHL3 along this segment. The proportional reduction in KLHL3 levels may have more profound consequences on ion transport along the DCT than along other segments. In KS-Cul3–/– mice, on the other hand, Cul3 is deleted along the entire nephron, so interactions between CUL3 and all BTB proteins are lost, which led to significant defects along multiple segments, not just the DCT. Effects on other segments, which led notably to reductions in AQP2 and medullary pNKCC2, overrode the increased pNCC expression, resulting in salt wasting and polyuria rather than an FHHt phenotype.
An unexpected finding was the development of kidney damage in KS-Cul3–/– mice, characterized by inflammation and fibrosis. Tissue damage was also described when Cul3 was deleted from the liver (20), so these effects may be relevant in other contexts. Loss-of-function CUL3 mutations in humans are associated with papillary renal cell carcinoma (27), which suggests that general loss of CUL3 activity is highly detrimental to cells. As kidney inflammation and fibrosis are essential in a host of progressive kidney diseases, KS-Cul3–/– mice should provide an interesting experimental model for future studies; the current results suggest that CUL3 activating molecules might slow the progression of chronic kidney disease.
Together, our present results suggest that disease-causing CUL3 Δ403–459 exhibits enhanced ubiquitylating activity and is heavily neddylated at baseline. Such a gain-of-function mutation is common in autosomal-dominant diseases, such as CUL3-initiated FHHt. This gain of function likely targets the DCT because of the extraordinarily high levels of KLHL3 expression there, as KLHL3 abundance appeared to be exquisitely sensitive to degradation by CUL3 Δ403–459. The current results also demonstrate an essential role for CUL3 in kidney tubules, where it appears to maintain cellular quiescence and health. Manipulation of CUL3 effects in the kidney may provide new avenues to prevent or treat human disease.
Methods
Antibodies and reagents.
The polyclonal NCC (28); anti-NKCC2 (29); pNKCC2 (30); WNK1, WNK3, and WNK4 (31); C-SPAK (32); pT53-NCC (18); OSR1 (33); and CUL3 (10) antibodies have been previously described. To further validate our WNK4 antibody, we tested it on Wnk4–/– mice (Supplemental Figure 6 and ref. 34). 2 KLHL3 antibodies were used. On cell blots, we used an antibody from Abcam (catalog no. 66655), but this gave poor signals on IF and in kidney. Thus, we generated a polyclonal antibody in rabbits, directed against the 19 N-terminal amino acids of mouse KLHL3; it recognized a band of the correct size on blots from transfected cells, but not untransfected cells (see Figure 2). For CUL3, we also used 2 antibodies: the first was described previously (10); the second was from Cell Signaling. Both gave similar signals on IF and WB. MLN 4924 (catalog no. A-1139; Activebiochem) for CUL3 (Invitrogen) was purchased. For binding and ubiquitylation assays, we used anti-FLAG M2 monoclonal antibody (Sigma-Aldrich), anti-actin polyclonal antibody (Sigma-Aldrich), anti-HA monoclonal antibody (Covance), and anti-MYC polyclonal antibody (catalog no. sc-789; Santa Cruz Biotechnology). Anti–cyclin E antibodies were a gift from J. Roberts (Fred Hutchinson Cancer Research Center, Seattle, Washington, USA). Human KLHL3 cDNA was purchased from GeneCopeia Inc. (catalog no. EX-A1395-M11) and subcloned into the pMO-MYC vector using PCR.
Immunoprecipitation, Western blotting, and ubiquitylation assay.
Transfected HEK293 cells were harvested in 1 mM EDTA in PBS and sonicated for 10 seconds in radioimmunoprecipitation assay (RIPA) buffer (1% NP-40; 1% sodium deoxycholate; 0.1% SDS; 150 mM NaCl; 0.01 M sodium phosphate buffer, pH 7.2; and 2 mM EDTA). For immunoprecipitation assays, the clarified lysates were incubated with primary antibody and protein A–sepharose beads for 2 hours at room temperature. Beads were spun down and washed twice with RIPA buffer. Protein samples were separated by electrophoresis on 10% polyacrylamide gels, transferred to PVDF membranes (Bio-Rad), and incubated overnight in primary antibodies. Proteins were visualized using HRP-conjugated secondary antibodies and enhanced chemiluminescence, followed by digital acquisition using an Alpha Innotech Fluorochem SP system.
For ubiquitylation assays, HEK cells were cotransfected with HA-tagged ubiquitin expression vector. After 36 hours, cells were lysed on ice in RIPA buffer plus 10 mM N-ethylmaleimide and proteinase inhibitors. After harvesting, cysteine was added to a final concentration of 0.1%. Immunoprecipitation was carried out as above; proteins were separated on 6% SDS-polyacrylamide gel transferred onto nitrocellulose and incubated with the appropriate primary antibody.
Cell IF.
HeLa and mDCT cells were seeded on coverslips and transfected with FLAG-tagged CUL3, FLAG-tagged CUL3 Δ403–459, or MYC-tagged KLHL3. Cells were fixed in 4% paraformaldehyde and washed for 1 minute in a buffer containing 1% Triton X-100, 2 mM EGTA, and 5 mM PIPES before being incubated at 4°C overnight with anti-MYC polyclonal antibody (catalog no. sc-789; Santa Cruz Biotechnology) and anti-FLAG M2 monoclonal antibody (Sigma-Aldrich). Cells were then incubated in Alexa Fluor 488– and Alexa Fluor 568–conjugated secondary antibodies, followed by staining with DAPI. Cells were visualized and photographed using a Zeiss AXIO Imager M2 microscope and AxioVision software.
Cell culture and transfections.
The Flp-In T-REx HEK cell line was described previously (35). HEK293, HeLa, and mDCT cells were maintained in DMEM supplemented with 10% FBS. Cells were transiently transfected using calcium phosphate precipitation or Lipofectamine 2000 (Ambion, Invitrogen) as previously described (10).
Xenopus oocyte experiments.
Techniques for these experiments were as previously described (36–38).
Animals.
The inducible renal tubule–specific Cul3 knockout mice (KS-Cul3–/–) were generated by use of a Tet-On/Cre-loxP system (17). Pax8-rtTA transgenic mice, which express the reverse tetracycline-dependent transactivator (rtTA) in all proximal and distal tubules and in the entire collecting duct system of both embryonic and adult kidneys, were bred with TRE-LC1 transgenic mice, which express Cre recombinase under the control of an rtTA response element. Double-transgenic Pax8-rtTA/TRE-LC1 mice (Pax8/LC1), which allow for doxycycline-induced, renal tubule–specific, Cre-mediated recombination, were bred with mice homozygous for the floxed allele Cul3fl/fl (19) to obtain Cul3fl/+ Pax8/LC1 mice, which were then interbred to obtain Cul3fl/fl Pax8/LC1 mice. To induce Cre-mediated excision at the Cul3 allele, doxycycline hyclate was administered at a dose of 2 mg/ml in 5% sucrose in deionized water for 7 or 14 days. Control mice received 5% sucrose as drinking water. KS-Cul3–/– mice were then compared with littermate KS-Cul3fl/fl mice treated with vehicle. For some studies, knockout mice were compared with littermates that did not express 1 of the transgenes, but were instead treated with doxycycline. We detected no physiological differences between Cul3fl/fl mice treated with vehicle and control mice, consistent with the lack of leakiness of this system (17).
PCR genotyping.
Crude genomic DNA extracts were prepared from tail snips by heating at 95°C for 45 minutes in NaOH (pH 12.0), followed by neutralization with Tris-HCl (pH 5.0). 4 μl extract was used directly in PCR reactions as described previously (19). Final determination of genotype was performed by Western blotting of kidney protein lysates after sacrifice.
Dietary manipulation.
For mice in which Cul3 was disrupted in the short term, custom research diets with varying salt levels (Harlan Teklad) were used. Mice aged 2–3 months were placed on control diet (0.49% NaCl, 0.8% K+) or high-salt diet (8% NaCl, 0.8% K+) and subjected to plasma electrolyte analysis and kidney collection for Western blotting. For blood pressure measurements, mice were first placed on control diet, and blood pressure was measured continuously for 4 days. They were then switched to salt-deficient diet (0.01% NaCl, 0.8% K+), at which point blood pressure was measured continuously for up to 8 days.
Metabolic cage studies.
5-month-old mice were acclimated to metabolic cages (Hatteras Instruments) for 3 days prior to urine collection. A gel diet containing 0.49% NaCl (normal salt) was provided, and this was switched to salt-deficient diet (0.02% NaCl) as indicated. Urine was collected under water-saturated light mineral oil. Urinary sodium and potassium were determined by flame photometry, urinary chloride was determined colormetrically (Pointe Scientific), and urinary calcium was measured by o-Cresolphthalein Complexone method (Pointe Scientific), according to the manufacturer’s protocol (except samples were diluted 1:2. Urinary creatinine was measured by modified Jaffe method (Pointe Scientific) on samples diluted 1:10.
Analysis of blood electrolytes and plasma aldosterone.
Whole blood was collected via terminal cardiac puncture (under anesthesia) and placed into heparinized Eppendorf tubes. 80 μl whole blood was then immediately added to a Chem8+ cartridge and analyzed using i-STAT analyzer (Abbot Point of Care Inc.). Plasma aldosterone levels were measured by ELISA (IBL-America).
Blood pressure measurement.
Blood pressure was measured by radiotelemetry using PA-C10 transmitters (Data Sciences Inc.). Briefly, under anesthesia, a midline neck incision was made, and the catheter from the radiotelemetry transmitter was inserted in the carotid artery. The body of the telemetry device was placed in a subcutaneous pocket on the right flank. Recordings were started 7 days after surgery, with recording segments of 20 seconds every 10 minutes. Hourly averages were calculated for MAP.
Kidney Western blot analyses.
Kidneys were removed, snap frozen in liquid nitrogen, and homogenized in chilled homogenization buffer containing protease and phosphatase inhibitors (50 mM Tris-HCl, pH 7.4; 1 mM EDTA; 1 mM EGTA; 0.3 M sucrose; 1 mM PMSF; 8.5 mM leupeptin; 1 mM orthovanadate; 50 mM sodium fluoride; 1 mg/ml aprotinin; and 1 mM dithiothreitol) on ice using a Potter Homogenizer. The homogenate was centrifuged at 4,000 g for 15 minutes at 4°C, and the supernatant was separated on 3%–8% NuPage Tris acetate gel at 20 mA. After transfer to PVDF, membranes were cut to allow parallel blotting with anti–β-actin or GAPDH to allow normalization. The blot was blocked with Blotto-T (5% nonfat dried milk in PBS-T) and incubated with the primary antibody for 1 hour, then washed and incubated with secondary antibody (HRP-coupled, diluted 1:4,000; Zymed) and detected using the Western Lightning kit (Perkin Elmer). Densitometry using ImageJ software (NIH), normalized to β-actin or GAPDH, was performed to compare relative protein concentrations.
Kidney IF.
Kidneys were fixed by in vivo perfusion (39). 5-μm mouse paraffin kidney sections were dewaxed, boiled in citrate buffer (pH 6) for antigen retrieval, and blocked with 5% skim milk–PBS or bovine serum albumin–PBS prior to incubation with primary antibodies. Cryostat sections were treated with 0.5% Triton X-100 in PBS for 30 minutes. Primary antibodies were applied for 1 hour at room temperature followed by overnight incubation at 4°C. Costaining was performed with NCC and NKCC2 (39), separated by washing steps. Fluorescent dye–conjugated (Cy-2 or Cy-3) secondary antibodies were applied for detection, and sections were analyzed with a confocal microscope (LSM 5 Exciter; Zeiss).
Nephron segment PCR.
Mouse renal tubule segments were isolated by microdissection as described previously (40). RNA was extracted from segments using RNA extraction kit (Invitek) according to the manufacturer’s protocol, and cDNA was synthesized by reverse transcription (Tetro Reverse Transcriptase; Promega). Quantitative PCR was performed using HOT FIREPol EvaGreen qPCR Mix Plus (Solis BioDyne) and primers specific for Klhl3 (forward, 5′-GCCATGGAGTACCACCTCCT-3′; reverse, 5′-ACCAACCACAATCATGACCTTG-3′) and Cul3 (forward, 5′-GCACCATGTCGAATCTGAGC-3′; reverse, 5′-TTCATCCATGGTCATCGGAAA-3′). Gene expression analysis was performed applying the ΔΔCt method and normalized against Actb.
Statistics.
Unpaired 2-tailed t tests were used to compare 2 groups. Differences among a single control group and multiple experimental groups were analyzed by 1-way ANOVA followed by Dunnett multiple-comparison test. Time course differences were analyzed by repeated-measures ANOVA. Unless otherwise indicated, data are shown as mean ± SEM. A P value less than 0.05 was considered significant.
Study approval.
Animal studies were approved by the Oregon Health and Science University Institutional Animal Care and Usage Committee (protocol IS918).
Supplementary Material
Acknowledgments
This work was supported by NIH grants R01 DK51496 (to D.H. Ellison), K01 DK076617 and R01 DK098141 (to J.A. McCormick), and R01 GM082940 (to J.D. Singer) as well as by Merit Review grant 1I01BX002228-01A1 from the Department of Veterans Affairs (to D.H. Ellison). C. Zhang was supported by the Shanghai Municipal Education Commission and a Shanghai Jiao Tong University K.C. Wong Medical Fellowship Fund. E.R. Argaiz was supported by a scholarship from Conacyt and is a graduate student in the biomedical science PhD program of Universidad Nacional Autónoma de México. D.H. Ellison received a Marie Curie Fellowship from the European Union during part of these studies. A.S. Terker is supported by a Predoctoral Fellowship from the American Heart Association (13PRE14090030). The authors appreciate the interpretation of the renal morphology and advice provided by Megan Troxell, and the helpful discussions with Juliette Hadchouel.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2014;124(11):4723–4736. doi:10.1172/JCI76126.
References
- 1.Pathare G, Hoenderop JG, Bindels RJ, San-Cristobal P. A molecular update on Pseudohypoaldosteronism type II. Am J Physiol Renal Physiol. 2013;305(11):F1513–F1520. doi: 10.1152/ajprenal.00440.2013. [DOI] [PubMed] [Google Scholar]
- 2.Mayan H, Vered I, Mouallem M, Tzadok-Witkon M, Pauzner R, Farfel Z. Pseudohypoaldosteronism type II: marked sensitivity to thiazides, hypercalciuria, normomagnesemia, and low bone mineral density. J Clin Endocrinol Metab. 2002;87(7):3248–3254. doi: 10.1210/jcem.87.7.8449. [DOI] [PubMed] [Google Scholar]
- 3.Isobe K, et al. Development of enzyme-linked immunosorbent assays for urinary thiazide-sensitive Na-Cl cotransporter measurement. Am J Physiol Renal Physiol. 2013;305(9):F1374–F1381. doi: 10.1152/ajprenal.00208.2013. [DOI] [PubMed] [Google Scholar]
- 4.Vidal-Petiot E, et al. WNK1-related familial hyperkalemic hypertension results from an increased expression of L-WNK1 specifically in the distal nephron. Proc Natl Acad Sci U S A. 2013;110(35):14366–14371. doi: 10.1073/pnas.1304230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wakabayashi M, et al. Impaired KLHL3-mediated ubiquitination of WNK4 causes human hypertension. Cell Rep. 2013;3(3):858–868. doi: 10.1016/j.celrep.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 6.Shibata S, Zhang J, Puthumana J, Stone KL, Lifton RP. Kelch-like 3 and Cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of WNK4. Proc Natl Acad Sci U S A. 2013;110(19):7838–7843. doi: 10.1073/pnas.1304592110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohta A, et al. The CUL3-KLHL3 E3 ligase complex mutated in Gordon’s hypertension syndrome interacts with ubiquitylates WNK isoforms; disease-causing mutations in KLHL3 WNK4 disrupt interaction. Biochem J. 2013;451(1):111–122. doi: 10.1042/BJ20121903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu G, Peng JB. Disease-causing mutations in KLHL3 impair its effect on WNK4 degradation. FEBS Lett. 2013;587(12):1717–1722. doi: 10.1016/j.febslet.2013.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boyden LM, et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. 2012;482(7383):98–102. doi: 10.1038/nature10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singer JD, Gurian-West M, Clurman B, Roberts JM. Cullin-3 targets cyclin E for ubiquitination and controls S phase in mammalian cells. Genes Dev. 1999;13(18):2375–2387. doi: 10.1101/gad.13.18.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uchida S, Sohara E, Rai T, Sasaki S. Regulation of with-no-lysine kinase signaling by Kelch-like proteins. Biol Cell. 2014;106(2):45–56. doi: 10.1111/boc.201300069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Canning P, et al. Structural basis for Cul3 protein assembly with the BTB-Kelch family of E3 ubiquitin ligases. J Biol Chem. 2013;288(11):7803–7814. doi: 10.1074/jbc.M112.437996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meyer-Schaller N, et al. The human Dcn1-like protein DCNL3 promotes Cul3 neddylation at membranes. Proc Natl Acad Sci U S A. 2009;106(30):12365–12370. doi: 10.1073/pnas.0812528106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chávez-Canales M, et al. The WNK-SPAK-NCC cascade revisited: WNK1 stimulates the activity of the NaCl cotransporter via SPAK [published online ahead of print August 11, 2014]. Hypertension. doi: 10.1161/HYPERTENSIONAHA.114.04036. doi:10.1161/HYPERTENSIONAHA.114.04036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Louis-Dit-Picard H, et al. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat Genet. 2012;44(4):456–460. doi: 10.1038/ng.2218. [DOI] [PubMed] [Google Scholar]
- 16.Susa K, et al. Impaired degradation of WNK1 WNK4 kinases causes PHAII in mutant KLHL3 knock-in mice [published online ahead of print May 12, 2014]. Hum Mol Genet. doi: 10.1093/hmg/ddu217. 10.1093/hmg/ddu217. [DOI] [PubMed] [Google Scholar]
- 17.Traykova-Brauch M, et al. An efficient and versatile system for acute and chronic modulation of renal tubular function in transgenic mice. Nat Med. 2008;14(9):979–984. doi: 10.1038/nm.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCormick JA, et al. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab. 2011;14(3):352–364. doi: 10.1016/j.cmet.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McEvoy JD, Kossatz U, Malek N, Singer JD. Constitutive turnover of cyclin E by Cul3 maintains quiescence. Mol Cell Biol. 2007;27(10):3651–3666. doi: 10.1128/MCB.00720-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kossatz U, et al. The cyclin E regulator cullin 3 prevents mouse hepatic progenitor cells from becoming tumor-initiating cells. J Clin Invest. 2010;120(11):3820–3833. doi: 10.1172/JCI41959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pelham CJ, et al. Cullin-3 regulates vascular smooth muscle function and arterial blood pressure via PPARγ and RhoA/Rho-kinase. Cell Metab. 2012;16(4):462–472. doi: 10.1016/j.cmet.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lalioti MD, et al. Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat Genet. 2006;38(10):1124–1132. doi: 10.1038/ng1877. [DOI] [PubMed] [Google Scholar]
- 23.Yang SS, et al. Molecular pathogenesis of pseudohypoaldosteronism type II: generation and analysis of a Wnk4(D561A/+) knockin mouse model. Cell Metab. 2007;5(5):331–344. doi: 10.1016/j.cmet.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 24.Zhang DD, Lo S-C, Sun Z, Habib GM, Lieberman MW, Hannink M. Ubiquitination of Keap1, a BTB-Kel substrate adaptor protein for Cul3, targets Keap1 for degradation by a proteasome-independent pathway. J Biol Chem. 2005;280(34):30091–30099. doi: 10.1074/jbc.M501279200. [DOI] [PubMed] [Google Scholar]
- 25.Galan JM, Peter M. Ubiquitin-dependent degradation of multiple F-box proteins by an autocatalytic mechanism. Proc Natl Acad Sci U S A. 1999;96(16):9124–9129. doi: 10.1073/pnas.96.16.9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou P, Howley PM. Ubiquitination and degradation of the substrate recognition subunits of SCF ubiquitin-protein ligases. Mol Cell. 1998;2(5):571–580. doi: 10.1016/S1097-2765(00)80156-2. [DOI] [PubMed] [Google Scholar]
- 27.Ooi A, et al. CUL3 and NRF2 mutations confer an NRF2 activation phenotype in a sporadic form of papillary renal cell cardinoma. Cancer Res. 2013;73(7):2044–2051. doi: 10.1158/0008-5472.CAN-12-3227. [DOI] [PubMed] [Google Scholar]
- 28.Bostanjoglo M, et al. 11Beta-hydroxysteroid dehydrogenase, mineralocorticoid receptor, and thiazide-sensitive Na-Cl cotransporter expression by distal tubules. J Am Soc Nephrol. 1998;9(8):1347–1358. doi: 10.1681/ASN.V981347. [DOI] [PubMed] [Google Scholar]
- 29.Schmitt R, Klussmann E, Kahl T, Ellison DH, Bachmann S. Renal expression of sodium transporters and aquaporin-2 in hypothyroid rats. Am J Physiol Renal Physiol. 2003;284(5):F1097–F1104. doi: 10.1152/ajprenal.00368.2002. [DOI] [PubMed] [Google Scholar]
- 30.Reiche J, et al. SORLA/SORL1 functionally interacts with SPAK to control renal activation of Na(+)-K(+)-Cl(-) cotransporter 2. Mol Cell Biol. 2010;30(12):3027–3037. doi: 10.1128/MCB.01560-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang CL, et al. WNK1 and WNK4 modulate CFTR activity. Biochem Biophys Res Comm. 2007;353(3):535–540. doi: 10.1016/j.bbrc.2006.11.151. [DOI] [PubMed] [Google Scholar]
- 32.Piechotta K, Lu J, Delpire E. Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1). J Biol Chem. 2002;277(52):50812–50819. doi: 10.1074/jbc.M208108200. [DOI] [PubMed] [Google Scholar]
- 33.Zagorska A, et al. Regulation of activity and localization of the WNK1 protein kinase by hyperosmotic stress. J Cell Biol. 2007;176(1):89–100. doi: 10.1083/jcb.200605093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Castaneda-Bueno M, et al. Activation of the renal Na+:Cl- cotransporter by angiotensin II is a WNK4-dependent process. Proc Natl Acad Sci U S A. 2012;109(20):7929–7934. doi: 10.1073/pnas.1200947109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoorn EJ, et al. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med. 2011;17(10):1304–1309. doi: 10.1038/nm.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gamba G, et al. Primary structure and functional expression of a cDNA encoding the thiazide-sensitive, electroneutral sodium-chloride cotransporter. Proc Natl Acad Sci U S A. 1993;90(7):2749–2753. doi: 10.1073/pnas.90.7.2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sabath E, et al. Pathophysiology of functional mutations of the thiazide-sensitive Na-Cl cotransporter in Gitelman disease. Am J Physiol Renal Physiol. 2004;287(2):F195–F203. doi: 10.1152/ajprenal.00044.2004. [DOI] [PubMed] [Google Scholar]
- 38.San-Cristobal P, Ponce-Coria J, Vazquez N, Bobadilla NA, Gamba G. WNK3 and WNK4 amino-terminal domain defines their effect on the renal Na+-Cl– cotransporter. Am J Physiol Renal Physiol. 2008;295(4):F1199–F1206. doi: 10.1152/ajprenal.90396.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saritas T, et al. SPAK differentially mediates vascopressin effects on sodium cotransporters. J Am Soc Nephrol. 2013;24(3):407–418. doi: 10.1681/ASN.2012040404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Connexin 37 is localized in renal epithelia responds to changes in dietary salt intake Am J Physiol Renal Physiol. 2010;298(1):F216–F223. doi: 10.1152/ajprenal.00295.2009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.