

Abstract
Rapid nitric oxide (NO) production via endothelial NO synthase (eNOS) activation represents a major signaling pathway for the cardiovascular protective effects of estrogens; however, the pathways after NO biosynthesis that estrogens use to function remain largely unknown. Covalent adduction of a NO moiety to cysteines, termed S-nitrosylation (SNO), has emerged as a key route for NO to directly regulate protein function. Cofilin-1 (CFL1) is a small actin-binding protein essential for actin dynamics and cytoskeleton remodeling. Despite being identified as a major SNO protein in endothelial cells, whether SNO regulates CFL-1 function is unknown. We hypothesized that estradiol-17β (E2β) stimulates SNO of CFL1 via eNOS-derived NO and that E2β-induced SNO-CFL1 mediates cytoskeleton remodeling in endothelial cells. Point mutation studies determined Cys80 as the primary SNO site among the 4 cysteines (Cys39/80/139/147) in CFL1. Substitutions of Cys80 with Ala or Ser were used to prepare the SNO-mimetic/deficient (C80A/S) CFL1 mutants. Recombinant wild-type (wt) and mutant CFL1 proteins were prepared; their actin-severing activity was determined by real-time fluorescence imaging analysis. The activity of C80A CFL1 was enhanced to that of the constitutively active S3/A CFL1, whereas the other mutants had no effects. C80A/S mutations lowered Ser3 phosphorylation. Treatment with E2β increased filamentous (F)-actin and filopodium formation in endothelial cells, which were significantly reduced in cells overexpressing wt-CFL. Overexpression of C80A, but not C80S, CFL1 decreased basal F-actin and further suppressed E2β-induced F-actin and filopodium formation compared with wt-CFL1 overexpression. Thus, SNOCys80 of cofilin-1 via eNOS-derived NO provides a novel pathway for mediating estrogen-induced endothelial cell cytoskeleton remodeling.
The cardiovascular protective role of estrogens has been deduced for decades from a lower incidence in cardiovascular diseases in premenopausal women compared with age-matched men (1). Endothelial cells, expressing estrogen receptors (ERs; ie, ERα and ERβ), are direct targets of estrogen action (2, 3). Estrogens stimulate endothelial cell cytoskeleton remodeling and angiogenesis, which is important for endothelial repair in postmenopausal women receiving estrogen replacement therapy (4, 5). Nitric oxide (NO) derived from endothelial NO synthase (eNOS) is a determinant for vascular health, contributing significantly to the vasoprotective effects of estrogens (6, 7). However, the mechanisms by which NO regulates the endothelial effects of estrogens after biosynthesis remain largely unresolved.
Generation of the second messenger cyclic is the best-defined NO signaling (8); however, many NO bioactivities are cyclic GMP independent. NO can directly modulate protein function at the level of posttranslation (9). Covalent adduction of a NO moiety to reactive cysteines is termed S-nitrosylation (SNO). SNO has emerged as a redox-based posttranslational protein modification, which is now recognized as a prototypic redox-based signaling mechanism in cell regulation (10). The importance of SNO has been compared with O-phosphorylation (11) because SNO can regulate the entire proteome, thus participating in numerous biological pathways. For example, SNO of β-catenin by eNOS-derived NO promotes vascular endothelial growth factor (VEGF)-induced endothelial cell permeability (12), showing that SNO of specific proteins regulates endothelial cell function directly.
Cofilin-1 (CFL1) is an important actin-binding protein essential for cytoskeleton remodeling by regulating actin dynamics and plasticity (13). It severs and depolymerizes filamentous (F)-actin to yield the barbed ends for polymerization and actin monomers for generating new actin filaments. The activity of CFL1 is well known to be reversibly regulated by Ser3 phosphorylation, with phosphorylated form to be inactive (14). CFL1 is also known to be redox sensitive. Oxidative stress such as local inflammation renders CFL1 incapable of enhancing actin depolymerization (15). We have recently identified CFL1 as a major SNO target highly responsive to estrogen stimulation in endothelial cells via specific ER- and eNOS-NO-dependent mechanisms (16, 17). Furthermore, we have recently shown that SNO of CFL1 on Cys80/139 is important for mediating endothelial cell migration upon VEGF stimulation (18). However, the mechanisms by which SNO regulates CFL1 functions remain incompletely understood, and it is unknown whether SNO of CFL1 plays a role in the estrogen regulation of endothelial cell cytoskeleton remodeling.
The objective of this study was to determine the mechanism by which SNO regulates CFL1 function for testing a hypothesis that SNO of CFL1 via eNOS-derived NO mediates estrogen-induced endothelial cytoskeleton remodeling. We have shown that estradiol-17β (E2β) rapidly stimulates SNO of CFL1 via eNOS-derived NO in endothelial cells and identified Cys80 as a major site for SNO. SNO on Cys80 results in increased actin severing activity of CFL1, which in turn regulates endothelial actin dynamics and cytoskeleton remodeling upon estrogen stimulation. Hence, we have demonstrated that SNO of CFL1 is a novel mechanism for controlling endothelial cytoskeleton remodeling.
Materials and Methods
Chemicals and antibodies
Anti-Flag and fluorescein isothiocyanate (FITC)-labeled anti-Flag antibodies, BSA, HEPES, N-nitro-L-arginine-methylester (L-NAME), and all other chemicals unless specified, were from Sigma. S-nitrosoglutathione (GSNO) was from Cayman. Anti-CFL1 antibody was from Abcam. Anti-β-actin monoclonal antibody was from Ambion. Anti-eNOS antibody was from Santa Cruz Biotechnology, Inc. Prolong Gold antifade reagent with 4′,6-diamidino-2-phenylindole, M199 was purchased from Invitrogen.
Cell culture, biotin switch, avindin capture, and immunoblotting
Human umbilical vein endothelial cells (HUVECs) were isolated from healthy term placentas by collagenase digestion as described (16). Cords were collected from University of California Irvine Medical Center Hospital (Irvine, California) and approved by the institutional review boards. HUVECs were cultured in endothelial cell medium containing 5% fetal bovine serum and supplemented with 1% antibiotics and 1% endothelial cell growth supplement (ScienCell) and used within five passages. When cells were grown to approximately 80% confluence, the medium was replaced with M199 containing 0.1% BSA and 25 mM HEPES overnight. After 1 hour of equilibration with fresh M199–0.1% BSA-25 mM HEPES, the cells were treated with E2β for various time periods with/without the NOS inhibitor L-NAME; drugs were added 1 hour before E2β treatment.
SNO proteins in cell lysate were labeled by the biotin switch technique (BST) as described previously (16, 17, 19). The biotinylated SNO proteins were captured by NeutrAvidin protein coated beads (Thermo Scientific) at 4°C overnight. The avidin-captured SNO proteins were eluted from the beads with 1× sodium dodecyl sulfate sample buffer containing 100 mM 2-mercaptoethenol at 37°C for 20 minutes and used for immunoblotting with specific antibodies exactly as previously described (16, 17, 19, 20).
Small interfering RNA (siRNA) knockdown of eNOS
Endogenous eNOS was knockdown by specific siRNAs as previously described (17). The eNOS-specific (5′-GAGTTACAAGATCCGCTTCdTdT-3′) and a control (5′-AUUGUAUGCG AUCGCAGACdTdT-3′) siRNAs were synthesized by Dharmacon. Transfection of siRNA was performed with RNAiMAX reagent (Invitrogen) following the manufacturer's protocol.
Site-directed mutagenesis and cell transfection
The plasmid CFL1-pCMV (from OriGene Technologies) was used to prepare mutants of CFL1 by site-directed mutagenesis (21) and confirmed by sequencing. All the recombinant proteins have a Flag-tag at the N terminus. Cell transfection was achieved with GeneJet transfection reagent (SignaGen Laboratories) following the manufacturer's instructions.
Preparation of recombinant CFL1 and its mutant proteins
The pCMV plasmids containing CFL1 or its mutants were transfected into 293T cells. The 293T cells were grown in DMEM medium (Invitrogen) containing 10% fetal bovine serum for 48 hours. The cells were harvested and sonicated in Tris-buffered saline (50 mM Tris-HCl, pH 7.4; 150 mM NaCl) containing 1 mM EDTA, 1% Triton X-100, and protease inhibitors. Anti-Flag M2 affinity beads (Sigma) were added into protein lysates (20 μL resin slurry per 10 mg protein), and incubated at 4°C for 2 hours. The recombinant Flag-tagged CFL1 and its mutants were pulled down by 3× Flag peptide according to the manufacturer's instructions. All recombinant proteins displayed as a single band on SDS-PAGE, with an estimated molecular mass of approximately 26 kDa.
Actin filament-severing assay
The activity of recombinant CFL1 and its mutant proteins was measured by the ability of severing in vitro formed actin filament (F-actin) using fluorescence microscopy as described previously (22). Briefly, a nitrocellulose-coated flow cell was coated with antibiotin antibodies (Sigma) in ISAP buffer (20 mM Tris-HCl, pH 7.2; 5 mM EGTA; 2 mM MgCl2; 50 mM KCl; 1 mM ATP; 1 mM dithiothreitol) by perfusion for 5 minutes, followed by incubation with ISAP buffer containing 0.5 mg/mL BSA for 5 minutes. Rhodamine-labeled actin (0.4 μM), biotin-labeled actin (0.2 μM), and unlabeled actin (1.4 μM) were purchased from Cytoskeleton Inc and copolymerized at room temperature for 2 hours in ISAP buffer. The in vitro-formed, fluorescently labeled F-actin was injected into the antibiotin antibody-coated flow cell with antibleaching buffer (ISAP buffer containing 5 mg/mL BSA, 0.036 mg/mL catalase, 0.02 mg/mL glucose oxidase, 6 mg/mL glucose, 100 mM dithiothreitol, 1 mM ATP) for 5 minutes. Unbound F-actin was washed off with antibleaching buffer. The assay was initiated by the addition of recombinant CFL1 proteins (1 μM). F-actin deploymerization was observed under an inverted Leica fluorescence microscopy. Images were captured by a Hamamatsu charge-coupled device camera using SimplePCI software (Hamamatsu) for measuring the length and number of actin filaments by using ImageJ software (National Institutes of Health, Bethesda, Maryland). Actin filaments from five images of each treatment at different incubation times were measured and subpopulated by length greater than 1 μm or 1μm or less. An increased number of short actin filaments (length ≤ 1 μm) and a decreased number of long actin filament (length > 1 μm) indicate F-actin depolymerization.
Immunofluorescence staining of F-actin and Flag-CFL1
HUVECs grown on coverslips were transfected with Flag-tagged wt-CFL1 or its mutant plasmids. After treatment with E2β, the cells were fixed for immunofluorescence staining as described previously (23), with tetramethylrhodamine isothiocyanate (TRITC)-labeled antiphalloidin to visualize F-actin (red) and with an FITC-labeled anti-Flag antibody (Sigma-Aldrich) to visualize Flag-CFL1 (green). Relative immunofluorescence intensities were quantified by using SimplePCI software (Hamamatsu).
Filopodium formation
HUVEC was seeded on gelatin-coated glass coverslips and grew to ∼70% confluence. Following serum starvation overnight, cells were incubated with or without 10 nM E2β for 16 hours. After treatment, cells were fixed with 4% formaldehyde in Ca2+/Mg2+-free PBS and washed with Ca2+/Mg2+-free PBS containing 0.1% Triton X-100. The fixed cells were then stained with TRITC-phalloidin to visualize F-actin under a Leica fluorescence microscopy for image acquisition by a Hamamatsu high-resolution charge-coupled device camera using the SimplePCI image analysis software (Hamamatsu). Protrusions that contain actin with a width of 0.6–1.2 μm and an average length between 6 and 15 μm were defined as filopodia (24). The average filopodium number of 30 cells was calculated for statistic comparison among different groups.
Experimental replication and statistical analysis
All experiments were repeated at least three times using HUVECs derived from different placentas. Data were presented as mean ± SEM. Statistical analysis was performed by a one-way ANOVA, followed by a Student-Newman-Keuls test for multiple comparisons using SigmaStat 3.5 (Systat Software Inc). A significant difference was accepted as P < .05.
Results
E2β rapidly stimulates SNO of CFL1 in HUVECs via an eNOS-dependent pathway
To access the effects of E2β on CFL1 SNO in HUVECs, the cells were treated with 10 nM E2β for various times, and the cellular CFL1 and SNO-CFL1 levels were determined. After treatment with E2β, the level of SNO-CFL1 increased discernibly within 15 minutes, maximized at 30 minutes, and then returned to baseline at 60 minutes (Figure 1A). E2β failed to stimulate SNO-CFL1 in HUVECs pretreated with the NOS inhibitor L-NAME, suggesting a NOS-dependent pathway. When eNOS was knocked down by using eNOS-specific siRNAs, the E2β-stimulated SNO of CFL1 was blocked (Figure 1B). As a control, treatment with scramble siRNAs did not affect E2β-stimulated SNO of CFL1. Thus, our findings demonstrate that E2β stimulation of SNO on CFL1 is mediated by eNOS-derived NO.
Figure 1.

E2β stimulates dynamic S-nitrosylation in CFL1 in HUVECs via eNOS-derived NO. A, Subconfluent HUVECs were treated by 10 nM of E2β with or without 1 mM of L-NAME for the indicated time periods. Total protein extracts were harvested and subjected to biotin switch and avidin capture. The biotin-labeled SNO proteins were analyzed by immunoblotting with anti-CFL1 antibody. *, P < .05 vs untreated control. B, HUVECs were transfected with or without scrambled or eNOS-specific siRNAs, followed by treatment with or without E2β for 30 minutes. SNO-CFL1 was measured. Representative blots of SNO-CFL1, total CFL1, eNOS, and β-actin of a typical experiment are shown. Graphs summarize data (mean ± SEM) from three independent experiments using cells from different fetuses. Data were normalized and expressed fold to control. Bars with different letters differ from each other significantly; paired comparisons of importance were indicated with an asterisk (P < .05).
Cys80 is the major SNO site in CFL1
Identification of specific SNO-site(s) in target proteins is required for functional analysis of SNO because SNO occurs only on certain specific cysteine residues (9). CFL1 contains four cysteines (at positions 39, 80, 139, and 147). To identify the SNO site(s) of CFL1, we introduced a single-point mutation into CFL1 on each of its cysteines (Cys39/80/139/147) by replacement of Ala or Ser. The constructed mutants were named as C39A/S, C80A/S, C139A/S, and C147A/S, respectively. A constitutively active CFL1 mutant S3A (replacing Ser3 with Ala) (25) was also constructed for the comparisons.
The wild-type (wt)-CFL1 and its mutants were overexpressed in 293T cells. The cell lysates were incubated with or without 1 mM GSNO, an NO donor and potent SNO inducer (26), at 37°C for 30 minutes. After GSNO removal with Amicon centrifugal filters (Millipore), SNO-modified cysteines in proteins were labeled with biotin-HPDP by the BST. Total biotin-labeled SNO proteins were captured by avidin-coated beads for determining SNO-CFL1 by immunoblotting with an anti-CFL1 antibody. Basal SNO of the CFL1s were barely detectable in all samples; treatment with 1 mM of GSNO dramatically increased SNO levels of wt-CFL1 and all the mutants except the C80A/S (Figure 2A). Compared with wt-CFL1 and other mutants, SNO of C80A or C80S CFL1 mutants was barely detectable with or without GSNO treatment. Consistently, when overexpressed in HUVECs, the basal SNO level of C80ACFL1 was much lower compared with wt-CFL1 and C139A, which was not changed by E2β stimulation (Supplemental Figure 1). These data show that a single-point mutation on Cys80 results in SNO deficiency, indicating that Cys80 is the major SNO site in CFL1. Interestingly, the GSNO- and E2β-stimulated SNO level of S3A-CFL1 were much lower compared with that of wt-CFL1 (Figure 2B and Supplemental Figure 1), implying that SNO also affects Ser3 phosphorylation in CFL1.
Figure 2.

Cysteine80 is the major S-nitrosylation site in CFL1. Flag-tagged wt-CFL1 and the mutants were overexpressed in 293T cells. Whole-cell lysates were collected and treated with 1 mM of GSNO at 37°C for 30 minutes. The biotin-labeled total SNO proteins from equal amounts of proteins were avidin captured for analyzing SNO-Flag-CFL1 and total Flag-CFL1 by immunoblotting with an anti-Flag antibody. Total Flag-CFL1 or β-actin was used as a loading control. Blots shown represent one of three similar transfection experiments.
Ser3 phosphorylation of CFL1 is affected by E2β treatment and SNO in HUVECs
We then tested whether E2β and SNO affect Ser3 phosphorylation of CFL1 because the function of CFL1 is known to be regulated by Ser3 phosphorylation, with the phosphorylated form to be inactive (27). Treatment with E2β (10 nM) stimulated CFL1 phosphorylation in a time-dependent manner. After treatment with E2β, the levels of phosphorylated CFL1 decreased within 2 minutes and decreased between 5 and 15 minutes and then returned to baseline in 1 hour (Figure 3A). This finding shows that E2β regulates Ser3 phosphorylation of CFL1, which may further regulate the activity of CFL1 in endothelial cells. Contrary to SNO, Ser3 phosphorylation of CFL1 was not mediated by NO because neither L-NAME nor eNOS siRNA eliminated its response to E2β (Figure 3B).
Figure 3.

Effects of E2β and S-nitrosylation on CFL1 phosphorylation. A, Subconfluent HUVECs were treated with 10 nM of E2β for the indicated time periods. Total protein extracts were harvested and analyzed by immunoblotting with antibodies against pSer3-CFL1 and CFL1. Representative blots of pSer3-CFL1 and total CFL1 of a typical experiment are shown. Graphs summarize data (mean ± SEM) from three independent experiments using cells from different fetuses. Data were normalized and expressed fold to control. *, P < .05 vs untreated control. B, HUVECs were pretreated with or without NOS inhibitor, L-NAME, and transfected with or without scrambled or eNOS-specific siRNAs, followed by treatment with or without E2β for 30 minutes. Representative blots of pSer3-CFL1, total CFL1, and eNOS of a typical experiment are shown. C, Flag-tagged CFL1 and the mutants were overexpressed in HUVECs, respectively. Cellular proteins were harvested for immunoblotting with antibodies against pSer3-CFL1 and CFL1. Total β-actin was used as a loading control. Representative blots of pSer3-CFL1, pSer3-Cfl1flag, and total CFL1 of a typical experiment are shown. Data (mean ± SEM) were summarized in the lower graph from three independent experiments using cells from different fetuses. Bars with different letters differ from each other significantly (P < .05); paired comparisons of importance were indicated with an asterisk (P < .05).
To test whether SNO on CFL1 affects its phosphorylation, we transfected wt-CFL1 and its mutants into HUVECs and determined the effects of point mutation of each cysteine in CFL1 on its phosphorylation. Similar to endogenous CFL1, wt- and C139A-CFL1 were readily phosphorylated when transfected in HUVECs. Consistently, the constitutively active S3A mutant was not phosphorylated when overexpressed in HUVECs. In these cells, levels of endogenous CFL1 and its phosphorylation response to E2β were not affected by transfection. Basal Ser3 phosphorylation levels of the C80A/S mutants were markedly lower than that of wt-CFL1 (Figure 3C). Treatment with E2β resulted in dephosphorylation of wt- and C139A-CFL1; whereas phosphorylation of C80A-CFL1 was unaffected by E2β stimulation (Supplemental Figure 1). Our data therefore show that SNO on Cys80 affects Ser3 phosphorylation of CFL1. In addition, the basal Ser3 phosphorylation levels of C39A/S mutants were also markedly decreased when overexpressed in HUVECs; however, this site seemed not to be important for SNO of CFL1 (Figure 2). Thus, our data show that Cys80 is an important site for SNO of CFL1, and modification on Cys80 dramatically alters the Ser3 phosphorylation of CFL1.
SNO enhances the F-actin-severing activity of CFL1 in vitro
Because SNO increases local hydrophobicity, substitution with relatively hydrophobic residues exerts a SNO-mimetic effect (28), Cys→Ala mutation on SNO sites has been used as a tool for characterizing SNO-mimetic effect. On the other hand, substitution with hydrophilic residues, Cys→Ser, exerts a SNO-deficient effect (28–32). We then compared the activities of recombinant wt-, and all of the Cys→Ala/Ser mutant CFL1 proteins by using an in vitro actin filament-severing assay (18, 22). In this assay, the constitutively active CFL1 mutant S3A was used as the positive control. As shown in Figure 4, when no recombinant CFL1 proteins were added, both length and number of the fluorescently labeled F-actin fibers were unchanged over time. Incubation with wt-CFL1 protein significantly depolymerized F-actin as evidenced by reduced number and length of F-actin. As a positive control, the S3A mutant showed the highest activity in severing F-actin. When incubation with the SNO-mimetic mutant C80A, the length of F-actin was significantly decreased with a markedly increased number of the shortened F-actin fibers compared with wt-CFL1; however, incubation with SNO-deficient C80S CFL1 did not result in any changes compared with that of wt-CFL1. Moreover, a point mutation of any other cysteines with either Ala or Ser did not alter the F-actin-severing activity of CFL1. These data show that the SNO-mimetic Cys80→Ala mutant is with increased severing activity, suggesting that Cys80 is the major functional site for SNO in CFL1. Thus, our findings show that SNO of CFL1 on Cys80 enhances its F-actin-severing activity. Of note, although mutation on Cys39 in CFL1 affected its Ser3 phosphorylation (Figure 3A), the F-actin-severing activities of C39A/S mutants were not significantly different from that of wt-CFL1. Therefore, Cys39 is not a key site for SNO to regulate CFL1 function in severing actin.
Figure 4.

SNO-mimetic/deficiency on in vitro actin-severing activity of CFL1. Rhodamine-labeled F-actin was prepolymerized and observed under fluorescence microscope with ×100 oil lens. The changes in both length and number of F-actin fibers were recorded over time with or without the addition of 1 μmol/L recombinant CFL1 or the mutants. Images of one representative experiment are shown for each group. The length and number of F-actin at 5 minutes of incubation were quantified. Data (mean ± SEM) were summarized in the lower graph from three independent experiments, in which F-actin greater than 1 μm were averaged. Bars with different letters differ from each other significantly (P < .05); paired comparisons of importance were indicated with an asterisk (P < .05). Scale bar, 5 μm.
SNO enhances CFL1 binding to actin in vivo
Because binding to actin is a critical step for CFL1 to elicit its actin-severing activity (33), we next measured the in vivo actin-binding ability of the recombinant CFL1 proteins in HUVECs. We overexpressed HUVEC with Flag-tagged wt-CFL1 and C80A/S mutants and then treated the cells with/without 10 nM E2β for 30 minutes. A coimmunoprecipitation assay was used to determine CFL1 binding to actin with the use of antiactin and anti-Flag antibodies.
In resting cells, the levels of actin-bound wt-CFL1 were barely detectable; treatment with E2β dramatically increased the levels of actin bound wt-CFL1 (Figure 5). In comparison with wt-CFL1, the levels of actin bound C80A-CFL1 (SNO mimetic CFL1 mutant) in nonstimulated cells was similar to that of the E2β-stimulated actin-bound wt-CFL1, and this was not altered by E2β treatment (Figure 5). The basal level of actin-bound C80S-CFL1 (SNO deficient mutant) was detectable but low and also was not changed by E2β treatment. As expected, Flag-tagged CFL1 was undetectable in the vector-transfected controls. These data suggest that SNO of CFL1 on Cys80 may have maximized its actin-binding capacity, and SNO seems to be a major mechanism responsible for increasing the actin-binding activity of CFL1 upon E2β stimulation.
Figure 5.

SNO-mimetic/deficiency on the actin binding activity of CFL1. Flag-tagged CFL1 and the mutants were overexpressed in HUVECs, respectively. After treatment with or without E2β, lysates were harvested for immunoprecipitation with antiflag antibody. Vector-transfected cells were used as controls. The immunoprecipitates were analyzed by immunoblotting with antibodies against actin and Flag, respectively. Representative blots of actin and CFL1 of a typical experiment are shown. Data (mean ± SEM) were summarized in the lower graph from three independent experiments using cells from different fetuses. Bars with different letters differ from each other significantly; paired comparisons of importance were indicated with an asterisk (P < .05). IB, immunoblotting; IP, immunoprecipitation; Vect, vector.
SNO-CFL1 decreases F-actin formation in HUVECs
To investigate the effects of SNO-CFL1 on E2β-induced F-actin formation in HUVECs, we overexpressed the Flag-tagged wt-CLF1 and its mutants in HUVECs, followed by treatment with or without 10 nM E2β for 16 hours. The Flag-tagged CFL1s and F-actin were labeled with red and green fluorescence in the cells, respectively. Nontransfected cells that were not labeled with green fluorescence were used as controls in each group. As shown in Figure 6, the basal levels of F-actin in the cells overexpressing wt-CFL1 were significantly lower than that in nontransfected control cells. Compared with wt-CFL1, the C80A mutant further decreased basal F-actin levels, whereas the C80S mutant was ineffective. Of note, C39A/S mutants had no effect on the basal F-actin levels, although it reduced Ser3 phosphorylation when overexpressed (Figure 3B). As expected, the constitutively active S3A mutant had the lowest level of basal F-actin among all groups. These results indicate that SNO on CFL1 decreased F-actin formation in endothelial cells.
Figure 6.
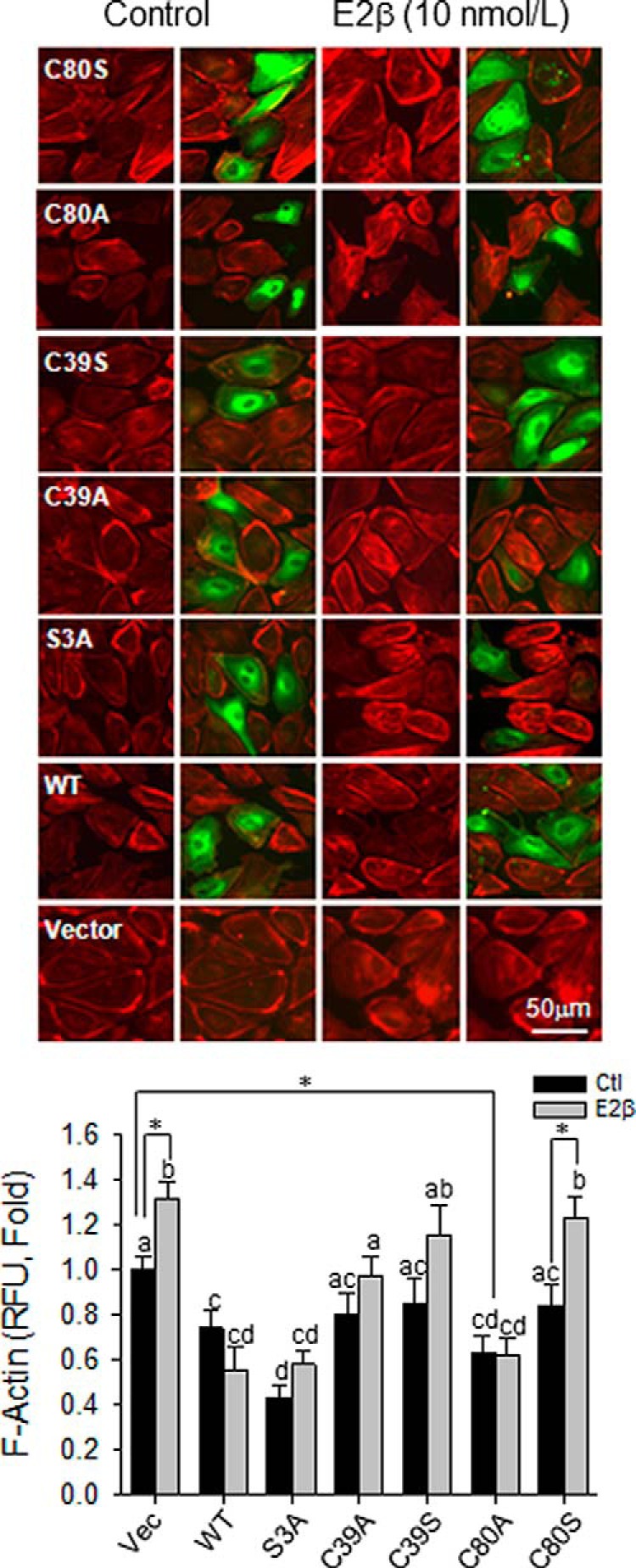
S-nitrosylation of CFL1 regulates F-actin formation upon estradiol-17β stimulation in HUVECs. Flag-tagged CFL1 and the mutants were overexpressed in HUVECs, respectively. After treated with or without 10 nM of E2β for 16 hours, the cells were fixed for labeling F-actin with TRITC-phalloidin (red) and Flag-CFL1 with FITC-conjugated (green) anti-Flag antibody. Double-fluorescence images (top panels) were captured for measuring the relative fluorescence intensities of F-actin by using SimplePCI software (Hamamatsu). Levels of F-actin (mean red fluorescence intensity) of 50 cells per group were obtained and summarized in the graph. Graph summarized data (mean ± SEM) from three independent experiments using cells from different fetuses. Bars with different letters differ from each other significantly (P < .05); paired comparisons of importance were indicated with an asterisk (P < .05). Scale bar, 50 μm.
E2β stimulated F-actin formation in nontransfected cells, and overexpression of wt-CFL1 inhibited this response. The F-actin level was unresponsive to E2β treatment in the cells overexpressed with S3A and C80A mutants; however, this was increased by E2β in cells overexpressed with C39S and C80S mutants (Figure 6). These data show that once the regulatory sites Cys80 and Ser3 are mutated with Ala, E2β is unable to regulate F-actin formation via CFL1, implicating that CFL1 regulates F-actin formation in endothelial cells via both SNO and phosphorylation.
SNO of CFL1 inhibits filopodium formation in endothelial cells
Actin remodeling results in filopodium formation at the leading edges of cells to initiate cell migration, which is an important component of angiogenesis (34). We then investigated whether SNO on CFL1 is involved in E2β-induced filopodium formation in HUVECs. The cells were overexpressed with wt-CFL1, S3A, C39A/S, or C80A/S mutants. After treatment with or without E2β for 16 hours, the cells were fixed and F-actin was fluorescently labeled and examined under a fluorescence microscope to visualize filopodia at the leading edges of the cells. Overexpression of wt-CFL1 significantly decreased the basal number of filopodium per cell in HUVECs. This number was further decreased in cells overexpressed with S3A or C80A mutants, whereas the C39A/S and C80S had limited effects in comparison with wt-CFL1. Treatment with E2β was not able to stimulate filopodium formation in HUVECs overexpressing wt-, S3A-, or C80A-CFL1; however, E2β significantly increased the number in the other groups (Figure 7). These findings show that once a single mutation of either C80A or S3A is made with Ala, E2β is unable to regulate filipodium formation via CFL1. Thus, both SNO and phosphorylation of CFL1 are involved in the regulation of filipodium formation by E2β in endothelial cells.
Figure 7.
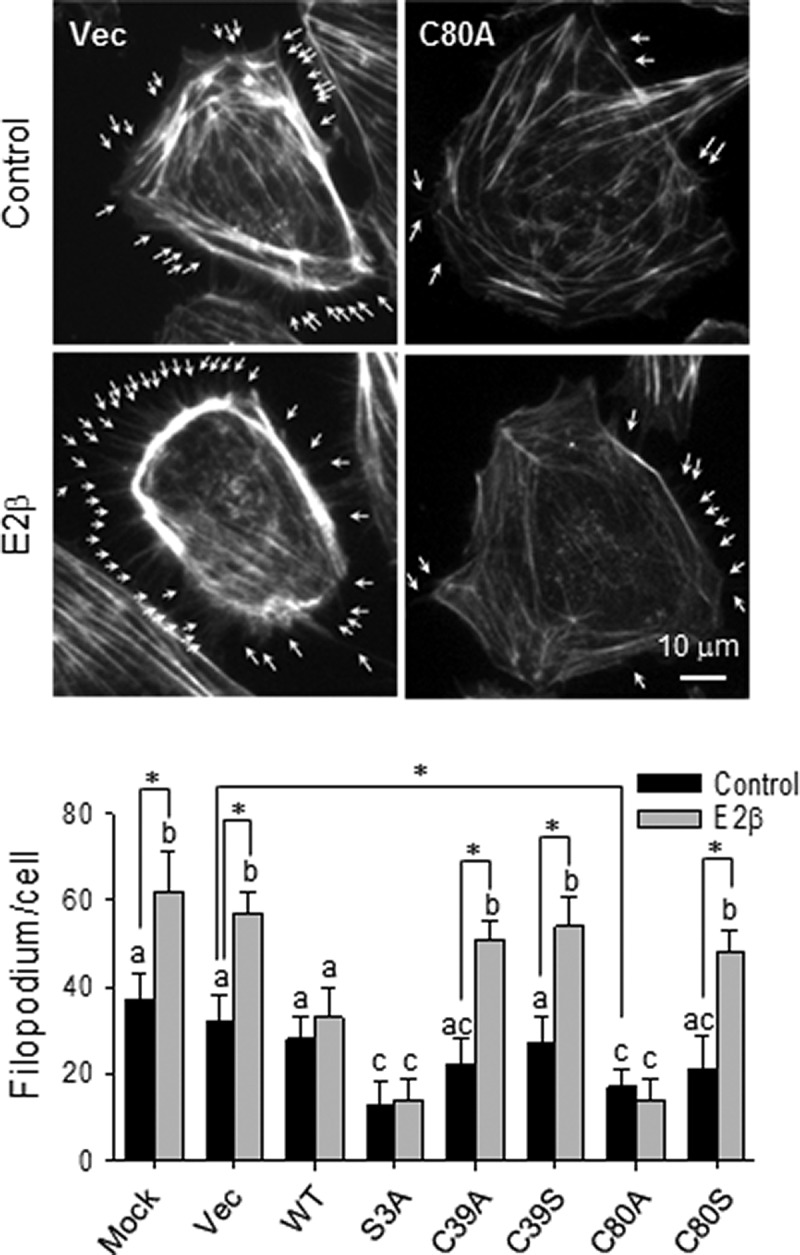
S-nitrosylation of CFL1 regulates filopodium formation upon estradiol-17β stimulation in HUVECs. Flag-tagged wt-CFL1 and the mutants were overexpressed in HUVECs. After treatment with 10 nM of E2β for 16 hours, the cells were fixed for labeling F-actin with TRITC-phalloidin. The average number of filopodium per cell of 30 cells in each group was quantified. Typical fluorescence images of vector/C80A transfected HUVECs with/without E2β treatment were shown; arrows indicate typical filipodia of the HUVECs. Scale bar, 10 μm. Graph summarized data (mean ± SEM) from three independent experiments using cells from different fetuses. Bars with different letters differ from each other significantly (P < .05); paired comparisons of importance were indicated with an asterisk (P < .05). Vec, vector.
Discussion
Estrogens stimulate the reendothelialization of damaged endothelium in animal models of postmenopause (35, 36) and are also proangiogenic in the cardiovascular system (37). Endothelial cytoskeleton reorganization is critical for the reendothelialization and angiogenesis (38) in which eNOS-derived NO has been shown to play a determinant role (23). However, the mechanisms by which estrogens stimulate endothelial cytoskeleton remodeling remain largely unknown. By using BST combined with proteomics approaches, we and other groups have recently identified CFL1 as a major SNO protein in endothelial cells (16, 17, 39, 40), whose function is to sever and depolymerize actin filaments during cytoskeleton remodeling (13). Thus, it is important to determine whether SNO regulates CFL1 function as related to endothelial actin dynamics and cytoskeleton remodeling. In this study, by using HUVECs as a cell model, we have shown the following: 1) that E2β rapidly stimulates CFL1 SNO via endogenous NO derived from eNOS; 2) that SNO increases the actin-severing activity of CFL1, which is linked to its dephosphorylation on Ser3; and 3) that SNO decreases basal and E2β-induced formation of F-actin and filopodia. Thus, our findings show that SNO on CFL1 via eNOS-derived NO provides a novel pathway for mediating estrogen stimulation of actin dynamics and cytoskeleton remodeling in endothelial cells.
Although SNO has been identified as a crucial mechanism by which NO regulates protein functions directly, the fragile S-NO bond can be accurately measured until BST was invented in 2001 (41); this method has now been accepted as the most reliable approach for SNO measurement (41–45). In this method, SNO groups are selectively reduced by ascorbate and then labeled with biotin, thus allowing nitrosoproteins to be readily displayed and affinity purified and identified. Similar to O-phosphorylation and other posttranslational modification, SNO occurs on only some certain cysteine residues with specific surrounding sequence (9). Thus, identification of the specific cysteines of SNO is a prerequisite for deciphering the functional consequences of SNO of a specific protein. Among the four cysteines in CFL1, Cys80 and Cys139 were suspected to be the SNO sites (39, 40), which we have recently confirmed to be important in mediating placental endothelial cell migration upon VEGF stimulation by double mutations on these sites (18). However, our current data demonstrate that Cys80 is the major site for SNO in CFL1 upon estrogen stimulation because of the following findings. First, in comparison with wt-CFL1, the point mutation of Cys80 dramatically decreases the GSNO- and E2β-induced SNO on CFL1 (Figure 2 and Supplemental Figure 1), whereas point mutation of Cys139 does not. Second, compared with wt-CFL1, the point mutation in Cys80 with Ala/Ser, but not Cys139, greatly reduces CFL1 phosphorylation when overexpressed in HUVECs (Figure 3 and Supplemental Figure 1). Third, C80A (SNO mimetic) mutant CFL1 results in enhanced actin-severing activity, reaching to levels comparable with that of the constitutively active S3A mutant CFL1, whereas the actin-severing activity of C139A mutant CFL1 is similar to that of wt-CFL1 (Figure 4). Fourth, point mutations with Ala/Ser on other cysteine sites have no effect on CFL1 function, as shown in Figure 4. Of note, treatment with GSNO induces a weak SNO response in the C80 mutant CFL1 in vitro, suggesting that the other three cysteines (Cys-39/139/147) in CFL1 may be able to be nitrosylated by exogenous NO from NO donors; however, they are not the dominant SNO sites in CFL1 because of their low GSNO-induced SNO levels compared with that of wt-CFL1.
In functional studies using SNO-mimetic or SNO-deficient CFL1 mutants, we have shown that SNO results in enhanced actin binding and severing activities of CFL1 due to the following observations. First, the SNO-mimetic C80A-CFL1 possesses greater capacity of depolymerizing in vitro-formed actin filaments than that of wt-CFL1, whereas the activity of SNO-deficient C80S-CFL1 seems to be comparable with that of wt-CFL1. Second, when overexpressed in endothelial cells, C80S-CFL1 has a greater actin-binding activity than that of wt- and C80S-CFL1. Third, the basal levels of F-actin in endothelial cells are significantly decreased by overexpression of wt-CFL1 and the SNO-mimetic C80A-CFL1 but not the SNO-deficient C80S-CFL1. In addition, E2β dramatically stimulates the binding of actin to wt-CFL1, but not C80A-CFL1, when they are overexpressed in endothelial cells. These data suggest that SNO may have maximized the actin binding and severing activity of CFL1.
It is noteworthy that E2β stimulation of both SNO and Ser3 phosphorylation of CFL1 is rapid and reversible in time-dependent manners, suggesting potential coregulation between the mechanisms by which SNOCys80 and PSer3 regulate the activity of CFL1. In fact, our point-mutation studies of CFL1 may have suggested the importance of coregulation of SNOCys80 and PSer3 in regulating CFL1 function. For example, C80A/S point mutations result in lower basal Ser3 phosphorylation relative to wt-CFL1. However, NO mediates only SNO, but not phosphorylation, of CFL1 because blockade of endogenous NO by L-NAME or eNOS siRNA inhibits only E2β stimulation of SNO, but not Ser3 phosphorylation, of CFL1 in in HUVECs (Figures 1B and 2B).
Although the underlying mechanisms are elusive, E2β stimulation of both SNO and Ser3 phosphorylation of CFL1 may be caused by conformational or structural changes in CFL1 as suggested in the CFL1 point-mutation studies. C80A/S point mutations result in a lower basal PSer3 level of CFL1 compared with wt-CFL1. Because CFL1 may form disulfide bridge(s) between cysteines, especially on Cys80 (Research Collaboratory for Structural Bioinformatics Protein Data Bank), our results indicate that SNO on Cys80 may alter the secondary structure by breaking inter/intramolecular disulfide bridge(s), thereby preventing CFL1 from phosphorylation by protein kinases and phosphatases (33). Consistently, it has been shown that the structure of CFL1 was dramatically changed when the Cys80 was mutated (46). Our S3A point-mutation studies also support this notion. S3A mutation causes phosphorylation deficiency and constitutive activation of CFL1 (14). However, GSNO and E2β can barely stimulate the SNO of CFL1 on this mutant, although all cysteines are intact (Figure 2 and Supplemental Figure 1). Of note, C39A/S point mutations also result in a low basal level of PSer3 compared with those of wt-CFL1, possibly related to structural/conformational changes like C80A/S as aforementioned. However, unlike C80A, C39A mutation does not change the activity of CFL1. Moreover, our proteomics studies show that C39 is not nitrosylated in endothelial cells upon stimulation with E2β (16, 17). Therefore, our findings demonstrate that CFL1 function is coregulated via SNOCys80 and PSer3.
Altogether, we have shown herein for the first time that E2β stimulates the dynamic SNOCys80 of CFL1 via eNOS-derived NO in endothelial cells, resulting in increased actin-severing activity of CFL1 for regulating endothelial cell actin dynamics and cytoskeleton remodeling as well as filopodium formation. This pathway cooperates with PSer3 to regulate CFL1 function in the control of actin dynamics and cytoskeleton remodeling in response to estrogen stimulation (Figure 8).
Figure 8.

SNO of CFL1 serves as a novel pathway after NO biosynthesis for mediating estrogen-induced cytoskeleton remodeling in endothelial cells. Estrogen stimulates dynamic SNOCys80 of CFL1 via eNOS-derived NO. Activation of this pathway cooperates with the CFL1 pathway via dephosphorylation on Ser3, resulting in increased actin-severing activity of CFL1 for regulating actin dynamics and cytoskeleton remodeling in endothelial cells.
Acknowledgments
This work was supported by National Institutes of Health Grants R21 HL98746, RO1 HL74947, and RO1 HL70562 (to D.B.C.) and American Heart Association Grant SDG13910006 (to H.H.Z.). T.J.L. is an American Heart Association Pre-doctoral Fellow (AHA 14PRE18570033).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- CFL1
- cofilin-1
- E2β
- estradiol-17β
- eNOS
- endothelial NO synthase
- ER
- estrogen receptor
- F
- filamentous
- FITC
- fluorescein isothiocyanate
- GSNO
- S-nitrosoglutathione
- HUVEC
- human umbilical vein endothelial cell
- ISAP
- buffer of Tris-HCl, EGTA, MgCl2, KCl, ATP, and dithiothreitol
- L-NAME
- N-nitro-L-arginine-methylester
- NO
- nitric oxide
- siRNA
- small interfering RNA
- SNO
- S-nitrosylation
- TRITC
- tetramethylrhodamine isothiocyanate
- VEGF
- vascular endothelial growth factor
- wt
- wild type.
References
- 1. Barrett-Connor E. Sex differences in coronary heart disease. Why are women so superior? The 1995 Ancel Keys Lecture. Circulation. 1997;95:252–264. [DOI] [PubMed] [Google Scholar]
- 2. Kim KH, Bender JR. Membrane-initiated actions of estrogen on the endothelium. Mol Cell Endocrinol. 2009;308:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Straub RH. The complex role of estrogens in inflammation. Endocr Rev. 2007;28:521–574. [DOI] [PubMed] [Google Scholar]
- 4. Simoncini T, Scorticati C, Mannella P, et al. Estrogen receptor α interacts with Gα13 to drive actin remodeling and endothelial cell migration via the RhoA/Rho kinase/moesin pathway. Mol Endocrinol. 2006;20:1756–1771. [DOI] [PubMed] [Google Scholar]
- 5. Kublickiene K, Fu XD, Svedas E, Landgren BM, Genazzani AR, Simoncini T. Effects in postmenopausal women of estradiol and medroxyprogesterone alone and combined on resistance artery function and endothelial morphology and movement. J Clin Endocrinol Metab. 2008;93:1874–1883. [DOI] [PubMed] [Google Scholar]
- 6. Chambliss KL, Shaul PW. Estrogen modulation of endothelial nitric oxide synthase. Endocr Rev. 2002;23:665–686. [DOI] [PubMed] [Google Scholar]
- 7. Chen DB, Bird IM, Zheng J, Magness RR. Membrane estrogen receptor-dependent extracellular signal-regulated kinase pathway mediates acute activation of endothelial nitric oxide synthase by estrogen in uterine artery endothelial cells. Endocrinology. 2004;145:113–125. [DOI] [PubMed] [Google Scholar]
- 8. Arnold WP, Mittal CK, Katsuki S, Murad F. Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proc Natl Acad Sci USA. 1977;74:3203–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. [DOI] [PubMed] [Google Scholar]
- 10. Stamler JS, Lamas S, Fang FC. Nitrosylation. the prototypic redox-based signaling mechanism. Cell. 2001;106:675–683. [DOI] [PubMed] [Google Scholar]
- 11. Lane P, Hao G, Gross SS. 2001 S-nitrosylation is emerging as a specific and fundamental posttranslational protein modification: head-to-head comparison with O-phosphorylation. Sci STKE. 2001:RE1. [DOI] [PubMed] [Google Scholar]
- 12. Thibeault S, Rautureau Y, Oubaha M, et al. S-nitrosylation of β-catenin by eNOS-derived NO promotes VEGF-induced endothelial cell permeability. Mol Cell. 2010;39:468–476. [DOI] [PubMed] [Google Scholar]
- 13. Theriot JA. Accelerating on a treadmill: ADF/cofilin promotes rapid actin filament turnover in the dynamic cytoskeleton. J Cell Biol. 1997;136:1165–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moriyama K, Iida K, Yahara I. Phosphorylation of Ser-3 of cofilin regulates its essential function on actin. Genes Cells. 1996;1:73–86. [DOI] [PubMed] [Google Scholar]
- 15. Klemke M, Wabnitz GH, Funke F, Funk B, Kirchgessner H, Samstag Y. Oxidation of cofilin mediates T cell hyporesponsiveness under oxidative stress conditions. Immunity. 2008;29:404–413. [DOI] [PubMed] [Google Scholar]
- 16. Zhang HH, Feng L, Livnat I, et al. Estradiol-17β stimulates specific receptor and endogenous nitric oxide-dependent dynamic endothelial protein S-nitrosylation: analysis of endothelial nitrosyl-proteome. Endocrinology. 2010;151:3874–3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang HH, Feng L, Wang W, Magness RR, Chen DB. Estrogen-responsive nitroso-proteome in uterine artery endothelial cells: role of endothelial nitric oxide synthase and estrogen receptor-β. J Cell Physiol. 2012;227:146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang HH, Wang W, Feng L, et al. S-nitrosylation of cofilin-1 serves as a novel pathway for VEGF-stimulated endothelial cell migration. J Cell Physiol. 2015;230:406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang HH, Wang YP, Chen DB. Analysis of nitroso-proteomes in normotensive and severe preeclamptic human placentas. Biol Reprod. 2011;84:966–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Satohisa S, Zhang HH, Feng L, Yang YY, Huang L, Chen DB. Endogenous NO upon estradiol-17β stimulation and NO donor differentially regulate mitochondrial S-nitrosylation in endothelial cells. Endocrinology. 2014;155:3005–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hemsley A, Arnheim N, Toney MD, Cortopassi G, Galas DJ. A simple method for site-directed mutagenesis using the polymerase chain reaction. Nucleic Acids Res. 1989;17:6545–6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chan AY, Bailly M, Zebda N, Segall JE, Condeelis JS. Role of cofilin in epidermal growth factor-stimulated actin polymerization and lamellipod protrusion. J Cell Biol. 2000;148:531–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liao WX, Feng L, Zheng J, Chen DB. Deciphering mechanisms controlling placental artery endothelial cell migration stimulated by vascular endothelial growth factor. Endocrinology. 2009;151:3432–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lim KB, Bu W, Goh WI, et al. The Cdc42 effector IRSp53 generates filopodia by coupling membrane protrusion with actin dynamics. J Biol Chem. 2008;283:20454–20472. [DOI] [PubMed] [Google Scholar]
- 25. Yang N, Higuchi O, Ohashi K, et al. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 1998;393:809–812. [DOI] [PubMed] [Google Scholar]
- 26. Foster MW, Stamler JS. New insights into protein S-nitrosylation. Mitochondria as a model system. J Biol Chem. 2004;279:25891–25897. [DOI] [PubMed] [Google Scholar]
- 27. Kobayashi M, Nishita M, Mishima T, Ohashi K, Mizuno K. MAPKAPK-2-mediated LIM-kinase activation is critical for VEGF-induced actin remodeling and cell migration. EMBO J. 2006;25:713–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ozawa K, Whalen EJ, Nelson CD, et al. S-nitrosylation of β-arrestin regulates beta-adrenergic receptor trafficking. Mol Cell. 2008;31:395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McMahon TJ, Exton Stone A, Bonaventura J, Singel DJ, Solomon Stamler J. Functional coupling of oxygen binding and vasoactivity in S-nitrosohemoglobin. J Biol Chem. 2000;275:16738–16745. [DOI] [PubMed] [Google Scholar]
- 30. McMahon TJ, Moon RE, Luschinger BP, et al. Nitric oxide in the human respiratory cycle. Nat Med. 2002;8:711–717. [DOI] [PubMed] [Google Scholar]
- 31. Hara MR, Agrawal N, Kim SF, et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005;7:665–674. [DOI] [PubMed] [Google Scholar]
- 32. Huang Y, Man HY, Sekine-Aizawa Y, et al. S-nitrosylation of N-ethylmaleimide sensitive factor mediates surface expression of AMPA receptors. Neuron. 2005;46:533–540. [DOI] [PubMed] [Google Scholar]
- 33. Condeelis J. How is actin polymerization nucleated in vivo? Trends Cell Biol. 2001;11:288–293. [DOI] [PubMed] [Google Scholar]
- 34. Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84:359–369. [DOI] [PubMed] [Google Scholar]
- 35. Brouchet L, Krust A, Dupont S, Chambon P, Bayard F, Arnal JF. Estradiol accelerates reendothelialization in mouse carotid artery through estrogen receptor-α but not estrogen receptor-β. Circulation. 2001;103:423–428. [DOI] [PubMed] [Google Scholar]
- 36. Krasinski K, Spyridopoulos I, Asahara T, van der Zee R, Isner JM, Losordo DW. Estradiol accelerates functional endothelial recovery after arterial injury. Circulation. 1997;95:1768–1772. [DOI] [PubMed] [Google Scholar]
- 37. Morales DE, McGowan KA, Grant DS, et al. Estrogen promotes angiogenic activity in human umbilical vein endothelial cells in vitro and in a murine model. Circulation. 1995;91:755–763. [DOI] [PubMed] [Google Scholar]
- 38. Lamalice L, Le Boeuf F, Huot J. Endothelial cell migration during angiogenesis. Circ Res. 2007;100:782–794. [DOI] [PubMed] [Google Scholar]
- 39. Han P, Chen C. Detergent-free biotin switch combined with liquid chromatography/tandem mass spectrometry in the analysis of S-nitrosylated proteins. Rapid Commun Mass Spectrom. 2008;22:1137–1145. [DOI] [PubMed] [Google Scholar]
- 40. Dall'Agnol M, Bernstein C, Bernstein H, Garewal H, Payne CM. Identification of S-nitrosylated proteins after chronic exposure of colon epithelial cells to deoxycholate. Proteomics. 2006;6:1654–1662. [DOI] [PubMed] [Google Scholar]
- 41. Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE. 2001;2001:PL1. [DOI] [PubMed] [Google Scholar]
- 42. Derakhshan B, Wille PC, Gross SS. Unbiased identification of cysteine S-nitrosylation sites on proteins. Nat Protoc. 2007;2:1685–1691. [DOI] [PubMed] [Google Scholar]
- 43. Wang X, Kettenhofen NJ, Shiva S, Hogg N, Gladwin MT. Copper dependence of the biotin switch assay: modified assay for measuring cellular and blood nitrosated proteins. Free Radic Biol Med. 2008;44:1362–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Foster MW, Forrester MT, Stamler JS. A protein microarray-based analysis of S-nitrosylation. Proc Natl Acad Sci USA. 2009;106:18948–18953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chouchani ET, Hurd TR, Nadtochiy SM, et al. Identification of S-nitrosated mitochondrial proteins by S-nitrosothiol difference in gel electrophoresis (SNO-DIGE): implications for the regulation of mitochondrial function by reversible S-nitrosation. Biochem J. 2010;430:49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schulte B, John I, Simon B, et al. A reducing milieu renders cofilin insensitive to phosphatidylinositol 4,5-bisphosphate (PIP2) inhibition. J Biol Chem. 2013;288:29430–29439. [DOI] [PMC free article] [PubMed] [Google Scholar]