

Abstract
Adipose tissue is a complex, multicellular organ that profoundly influences the function of nearly all other organ systems through its diverse metabolite and adipokine secretome. Adipocytes are the primary cell type of adipose tissue and play a key role in maintaining energy homeostasis. The efficiency with which adipose tissue responds to whole-body energetic demands reflects the ability of adipocytes to adapt to an altered nutrient environment, and has profound systemic implications. Deciphering adipocyte cell biology is an important component of understanding how the aberrant physiology of expanding adipose tissue contributes to the metabolic dysregulation associated with obesity.
Fat—properly defined as adipose tissue—is a biological caloric reservoir that expands in response to overnutrition and releases lipids in response to energy deficit. Adipocytes represent the primary cell type of adipose tissue and are responsible for storing excess calories as triglycerides in their cellular lipid droplets without the common lipotoxicity experienced by other cells under these conditions (Konige et al., 2014). This unparalleled capacity for lipid storage and release upon systemic metabolic demand links the cell biology of the adipocyte and adipose tissue physiology to whole body metabolism (Fig. 1).
Figure 1.

Benign and unhealthy adipose tissue. The overall health of adipose tissue can be visualized histologically. Benign murine subcutaneous adipose tissue (left; trichrome staining) is rich in unilocular white adipocytes with sparse ECM. On high-fat feeding (right), adipose tissue experiences hypoxia and inflammation, resulting in a dense, fibrous ECM. These histological tissue features are characteristic of insulin-resistant adipocytes.
Adipocytes exist in a spectrum of subtypes, identified by color, from white to brown (Box 1). White adipocytes, which are the focus of this review, constitute the classical fat cell and represent the majority of cells in visceral and subcutaneous adipose depots—the depots most noted for expansion with obesity. Brown adipocytes encompass smaller brown fat depots that play a role in thermogenesis in most mammalian species. Changes in adipocyte metabolism and nutrient handling are the basis for much of the pathophysiology associated with the metabolic syndrome, a condition encompassing multiple metabolic abnormalities including obesity, dyslipidemia (elevated circulating lipids), hyperglycemia (elevated blood glucose), hyperinsulinemia (elevated circulating insulin), and insulin resistance (Deng and Scherer, 2010; Sun et al., 2011). Adipocytes are also highly active secretory cells, producing proteins and hormones that function far beyond metabolism. An array of adipocyte-secreted factors contribute to immunity, inflammation, vasculogenesis, and matrix remodeling (Sun et al., 2011). Subcutaneous adipose tissue also serves as an immune barrier (Box 2) and secretes molecules, such as complement factors, that play a role in innate immunity (MacLaren et al., 2008; Caspar-Bauguil et al., 2009). Individual factors in the adipocyte secretome, termed adipokines (such as leptin and adiponectin), have a broad range of system-wide actions. Leptin, for example, signals in the hypothalamus to mediate food intake and adiponectin induces insulin sensitivity through action in the liver and skeletal muscle. Adipokines also correlate with multiple pathological states aside from the metabolic syndrome, such as cancer and osteoporosis (for review see Blüher, 2014; Deng and Scherer, 2010; Xia et al., 2014). Increased focus on the biogenesis of adipokines has identified basic mechanisms within the secretory machinery of the adipocyte that regulate the translation and release of factors from the cell (Box 3). The metabolic and redox states of the individual adipocyte dictate the composition of this secretome, and changes in the adipose tissue adipokine secretion profile occur with altered metabolic demand and fat expansion or reduction (Cao, 2014). Local adipocyte physiology is thus communicated systemically with potentially beneficial or detrimental effects.
Box 1. The adipocyte color spectrum.
Classical white adipocytes appear white due to their lipid content. In contrast, the high abundance of cytochromes in mitochondria-rich brown adipocytes gives the tissue a “brown” appearance. Additional differences between white and brown adipocytes reflect their unique progenitor populations. Recent studies have identified vascular pericyte stem cells and muscle stem cells as white and brown adipocyte progenitors, respectively (Seale et al., 2008; Tang et al., 2008; Gupta et al., 2012). Although this review focuses on the traditionally underappreciated white adipocyte lipid metabolism, brown adipocytes are metabolic powerhouses. Brown adipose fatty acid uptake and metabolism are critical for maintaining normal circulating lipid levels (Feldmann et al., 2009; Asterholm et al., 2014). Expression of mitochondrial UCP1 protein is requisite in brown adipocyte identity (Nedergaard and Cannon, 2013). However, a “brown” adipose tissue profile, and “browning” of white adipocytes with increased UCP1 expression occurs under numerous adrenergic, thermogenic, and protein/hormonal stimuli (for review see Nedergaard and Cannon, 2014). White adipose depots thus appear to be a collection of white and “beige/brite” cells (exhibiting white and brown characteristics) capable of dynamic changes in their thermogenic profile. Whether “beige/brite” cells are derived from a common white or unique beige progenitor pool continues to be debated, though recent studies support a separate origin for these cells (Gburcik et al., 2012; Waldén et al., 2012; Wu et al., 2012; Long et al., 2014).
Box 2. Did you know?
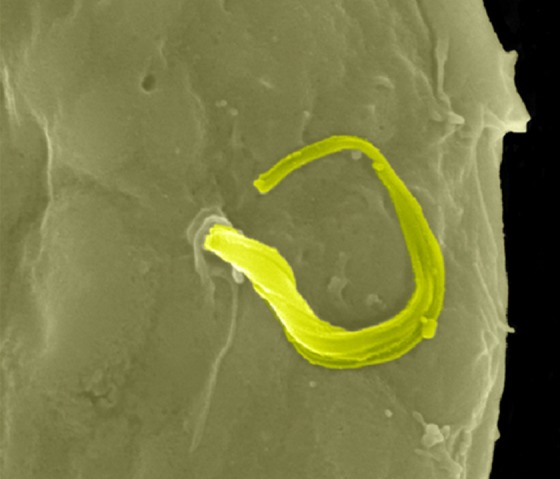
Chagas disease is endemic in Latin America, where the protozoan parasite Trypanosoma cruzi is transmitted by the triatomine bug. In the initial steps of infection, T. cruzi enters subcutaneous adipose tissue, a mechanism readily confirmed in infected 3T3-L1 adipocyte culture (Combs et al., 2005; Nagajyothi et al., 2008). Adipose tissue can host T. cruzi for decades, protecting the infected from Chagas disease progression to cardiovascular death (Ferreira et al., 2011; Nagajyothi et al., 2012). Interestingly, Chagas disease increases the incidence of hyperglycemia in human correlation studies and, experimentally, hyperglycemic mice harbor increased parasitic loads and exhibit increased mortality upon T. cruzi infection (Amole et al., 1985; Tanowitz et al., 1988). Conversely, T. cruzi infection into wild-type mice induced hypoglycemia through pancreatic inflammation (Combs et al., 2005; Nagajyothi et al., 2013). Altered glucose homeostasis in Chagas disease is thus due at least in part to a direct infection of adipocytes; how adipokines or the unique lipid biology of adipocytes promote T. cruzi infection remains to be fully understood (Nagajyothi et al., 2010, 2011). The figure shows a scanning electron micrograph of a single worm (highlighted in green) exiting a 3T3-L1 adipocyte. This research was originally published in The Journal of Biological Chemistry (Combs et al., 2005). Image © American Society for Biochemistry and Molecular Biology.
Box 3. Does the adipocyte have a triggered exocytic pathway?
Despite the resemblance between Glut4 vesicular trafficking and a triggered exocytic pathway analogous to neurotransmitter release, to date there is very little direct evidence that such a classical triggered exocytic pathway even exists in adipocytes. However, upon closer inspection, there are examples that suggest the adipocyte has the ability to rapidly respond through triggered exocytic events. For example, β3-adrenergic stimulation of murine adipose tissue rapidly triggers the release of insulin from the pancreatic-β cell within 2–5 min (Grujic et al., 1997). It is at this time not resolved whether this includes the release of a soluble factor from the adipocyte or whether the rapid kinetics are caused by neuronally mediated effects in the tissue. We know that the adipocyte has devised “alternative” approaches to triggered rapid release of proteins. For both lipoprotein lipase and adiponectin, significant intracellular stores exist in the late ER or early Golgi compartments (Roh et al., 2001; Wang et al., 2007) that could undergo a triggered release. For example, FGF21 rapidly triggers a burst of release of adiponectin from the adipocyte. This reserve shortens the response time; transcriptional activation, translation, and translocation into the ER followed by assembly of higher-order complexes would take adiponectin 10–45 min to reach the plasma membrane (Holland et al., 2013).
The adipocyte in the maintenance of systemic energy homeostasis
Adipose tissue is remarkably flexible in terms of energy storage and release. Responding to hormonal and energetic cues, it serves as source of energy-rich fatty acids during times of negative energy balance, reducing its lipid store and releasing fatty acids to target tissues in need of energy. In contrast, adipocyte lipid uptake, esterification, and storage in the form of triglyceride within the lipid droplet allows for expansion of adipose tissue, a beneficial, adaptive response to overnutrition that can prevent ectopic lipid deposition and lipotoxicity in other cell types. Once thought to be an inert storage site for lipid, we now know that the lipid droplet is an active player in maintaining systemic energy homeostasis. The large, unilocular lipid droplet occupies the majority of the adipocyte, placing the droplet’s borders within close proximity of the ER and mitochondria, where triglycerides are esterified and hydrolyzed, respectively.
Adipose tissue lipolysis and mitochondrial fatty acid oxidation.
Triglyceride stored within the lipid droplet is hydrolyzed to fatty acids and released to fuel peripheral tissues upon metabolic demand (Fig. 2). The lipid droplet surface contains structural proteins and metabolic enzymes specific to the adipocyte that respond to hormonal stimulation of lipolysis by increasing expression and through recruitment of additional proteins required for lipolysis (Brasaemle et al., 2004). The second messenger cAMP activates lipolytic enzymes via stimulation of the cAMP-dependent kinase, also referred to as protein kinase A (PKA), and thus plays a crucial role in the regulation of lipolysis (Zechner et al., 2005). β-Adrenergic (β-AR) signaling stimulates lipolysis through multiple signaling pathways, each of which is dependent on the phosphorylation of the lipid droplet membrane protein perilipin 1 (Plin1). Originally identified as a scaffolding protein, Plin1 is highly enriched in white adipocytes. As a mediator of protein–protein interactions during lipolysis, Plin1 plays a central role in regulating the hydrolysis of triglyceride molecules (Dalen et al., 2004; Takahashi et al., 2013). In fact, adipocytes isolated from Plin1-null mice display constitutively active lipolysis that is nonresponsive to isoproterenol, a β-AR agonist (Tansey et al., 2001). In the basal or anabolic state, lipolysis is inhibited by insulin and Plin1 is bound to comparative gene identification-58 (CGI-58), the coactivator of adipose triglyceride lipase (ATGL), the rate-limiting enzyme of lipolysis. Upon β-AR signaling and PKA activation, Plin1 is phosphorylated by PKA, which stimulates the release of CGI-58, activating ATGL that moves to the lipid droplet surface to hydrolyze the first fatty acid from the triglyceride (Lass et al., 2011). Also phosphorylated by PKA, hormone-sensitive lipase (HSL) binds Plin1 and hydrolyzes the resulting diacylglycerol to a monoacylglycerol, which is subsequently hydrolyzed, producing a third fatty acid and glycerol molecule. Fatty acids and glycerol are released from adipose tissue for systemic utilization.
Figure 2.

Expansion and reduction of the adipocyte. Sources of fatty acids for triglyceride synthesis within the adipocyte include exogenous NEFAs from the diet and those synthesized via de novo lipogenesis in the hepatocyte and adipocyte. NEFAs can enter the adipocyte via FATPs. In the presence of glucose, insulin binds to its receptor, stimulating the translocation of GLUT-4 transporters from the cytosol to the cell membrane. Increased flux through glycolysis leads to excess citrate generated in the TCA cycle, which is diverted into the cytosol for fatty acid synthesis. To synthesize triglyceride, three NEFAs are esterified to a glycerol backbone, and the triglyceride is processed through the ER for storage within the lipid droplet. In the expansive state, lipolysis is inhibited by insulin. PLIN1 is bound to CGI-58, the co-activator of ATGL, the rate-limiting enzyme of lipolysis. Through insulin-stimulated activation of phosphodiesterase 3B (PDE3B), which controls intracellular cAMP pools by converting cAMP to the inactive 5′ AMP, insulin prevents the phosphorylation of ATGL and HSL. Insulin also acts on the adipocyte via glucose flux to lactate to inhibit lipolysis indirectly via a lactate/GPR81-dependent inhibition of cAMP. In the lipolytic state, triglycerides are hydrolyzed to release NEFAs, and the size of the lipid droplet is reduced. Via β-AR signaling, Plin1 and HSL are phosphorylated by PKA, stimulating the release of CGI-58. ATGL moves to the lipid droplet surface to hydrolyze the first fatty acid from the triglyceride. Phosphorylated HSL binds Plin1 and hydrolyzes the resulting diacylglycerol to a monoacylglycerol, which is subsequently hydrolyzed by monoacylglycerol lipase (MGL), producing a third fatty acid and glycerol molecule. NEFAs are either exported to other tissues to undergo β-oxidation or re-esterification, or undergo β-oxidation within the mitochondrial matrix of adipocyte. Glycerol is transported to the liver, where it can be converted to glycolytic intermediates or phosphorylated to G3P for triglyceride synthesis (see text for details).
The metabolism of fatty acids to acetyl-coA for subsequent generation of energy (ATP and NADH) occurs primarily in the mitochondrial matrix of all oxidative tissues, but has been underappreciated as a significant part of white adipocyte metabolism. White adipocytes undergo mitochondrial expansion and exhibit increased β-oxidative capacity during differentiation, as indicated by increased expression of PGC1α, a regulator of mitochondrial biogenesis (Wilson-Fritch et al., 2004; Hao et al., 2010; Lu et al., 2010). Before entering the mitochondria for β-oxidation, fatty acids are activated to form fatty acyl-CoAs and are subsequently transported through the outer mitochondrial membrane via carnitine palmitoyl transferase-1 (CPT-1), the rate-limiting step of β-oxidation. Importantly, the activity of CPT-1 is inhibited by malonyl-CoA, an intermediate substrate of de novo lipogenesis. This prevents the oxidation of fatty acids when the cell is in a lipogenic state. Adipocytes lose oxidative capacity and insulin sensitivity in the obese and diabetic state (Choo et al., 2006; Keller and Attie, 2010), and targeting the oxidative metabolism of the adipocyte has demonstrated efficacy in treating type 2 diabetes. For example, peroxisome proliferator-activated receptor γ (PPARγ), a master transcriptional regulator of the adipogenic gene program, is the target of insulin-sensitizing thiazolidinedione drugs. Agonism of PPARγ has been shown to increase adipocyte oxidative potential and mitochondrial biogenesis (Wilson-Fritch et al., 2004; Bolten et al., 2007). Furthermore, transgenic mice lacking PGC1α, the coactivator of PPARγ, demonstrate reduced oxidative capacity and insulin resistance (Kleiner et al., 2012).
Insulin action on the adipocyte
Glucose transporter type 4 (GLUT-4)–mediated glucose transport.
Insulin signaling plays an important regulatory role in adipocyte metabolism by promoting the synthesis and storage of macronutrients and inhibiting their catabolism. In the fed state, high levels of circulating insulin bind to its receptor on adipocytes and signal the translocation of GLUT-4 transporters from the cytosol to the cell membrane, thereby allowing enhanced flux of glucose into the cell (Kahn and Cushman, 1985). Insulin affects GLUT-4 trafficking in three ways: increased exocytosis, decreased endocytosis, and increased GLUT-4 recycling at the cell surface (Stöckli et al., 2011). Insulin-stimulated GLUT-4 appears to be the predominant glucose transporter in the adipocyte despite the constitutive presence of GLUT-1 transporters on the adipocyte membrane. In fact, adipose tissue–specific deletion of GLUT-4 induces hepatic and skeletal muscle insulin resistance in mice (Abel et al., 2001), demonstrating the crucial role that adipocyte GLUT-4–mediated glucose uptake plays in maintaining systemic glucose homeostasis. Genetic or pharmacologic manipulations of the insulin receptor signaling cascade, such as targeting AMPK (Yamaguchi et al., 2005) or the use of thiazolidinedione drugs (Wu et al., 1998), alters the efficacy of insulin signaling and GLUT-4 expression and localization, exerting profound effects on adipocyte metabolism. As such, glucose is more than just a source of ATP to the adipocyte. Dihydroxyacetone phosphate, generated during glycolysis, is converted to glycerol-3-phosphate (G3P), and serves as the backbone for triglyceride synthesis and storage (Fig. 2). A study examining glucose and triglyceride turnover in adipose tissue during fasting in humans determined that 20–25% of the glucose taken up into adipocytes is subsequently used for triglyceride synthesis (Frayn and Humphreys, 2012). Although free glycerol is generated during lipolysis, adipocytes lack glycerol kinase, the enzyme that phosphorylates glycerol to G3P. Hence, glucose uptake and the reaction generating G3P are critical to adipocyte lipid packaging (Guan et al., 2002; Tan et al., 2003). For example, mice with adipocyte-specific deletion of insulin receptor are resistant to diet-induced and age-related obesity. Reduced adipocyte glucose uptake and triglyceride storage contribute to this phenotype (Blüher et al., 2002). Furthermore, increased adipocyte GLUT4 expression in transgenic mice enhances glucose uptake and leads to adipocyte hyperplasia via enhanced fatty acid esterification (Shepherd et al., 1993; Herman et al., 2012). When cellular glucose levels are low, however, adipocytes can use noncarbohydrate precursors such as lactate or amino acids to generate G3P via glyceroneogenesis (Forest et al., 2003).
Insulin and the regulation of lipolysis.
Insulin also acts on the adipocyte to suppress lipolysis during the fed state. This is accomplished in several ways. Through Akt-mediated phosphorylation and activation of phosphodiesterase 3B (PDE3B), which controls intracellular cAMP pools, insulin exerts its antilipolytic effects by inactivating cAMP, preventing the phosphorylation, and thus activation, of ATGL and HSL (Choi et al., 2010; Fig. 2). More recently, it has been demonstrated that insulin also suppresses lipolysis indirectly as a result of GLUT-4–mediated increased glucose flux through glycolysis. Glucose is metabolized to pyruvate, which then fluxes through the TCA cycle. However, a portion of this pyruvate is metabolized to lactate. Lactate exits the adipocyte and mediates insulin-dependent inhibition of lipolysis through an autocrine lactate loop. Lactate binds to the G protein–coupled receptor GPR81 to inhibit the formation of cAMP, and the downstream activation of PKA (Fig. 2; Ahmed et al., 2010).
Overnutrition and adipose tissue expansion
In response to overnutrition, excess energy stores partition into the adipocyte. Energy harvested in fatty acids either directly from the diet or via de novo lipogenesis in hepatocytes or adipocytes is esterified into triglyceride and stored within the lipid droplet, yielding expansive adipose tissue. Nonesterified fatty acids (NEFAs) are taken up from the extracellular pool through several fatty acid binding and transport proteins. CD36 and several members of the fatty acid transport protein (FATP) family are highly expressed in adipocytes and are essential for fatty acid sequestration. Mice with global deletion of CD36, for example, take up only 30–40% of labeled fatty acids compared with control littermates (Coburn et al., 2000). Adipocytes with reduced FATP1 levels exhibit a 25% reduction in fatty acid uptake in vitro (Lobo et al., 2007), and insulin-stimulated long chain fatty acid uptake is absent from FATP1-null adipocytes (Wu et al., 2006).
Upon transport into the adipocyte, NEFAs are esterified to a glycerol backbone, thus forming the triglyceride for storage within the lipid droplet of the adipocyte. Enzymes that catalyze the esterification of fatty acids to triglyceride are regulated through PPARγ (Festuccia et al., 2009), and genetically manipulating the expression of these enzymes leads to significantly impaired lipid accumulation and reduced adipogenic gene expression in 3T3-L1 adipocytes (Shan et al., 2010). Humans and rodents with impaired enzymatic activity for fatty acid esterification are lipodystrophic, highlighting the importance of this process to retain metabolically flexible adipose tissue expansion and function (Agarwal and Garg, 2003; Vergnes et al., 2006; Agarwal et al., 2011)
Lipid droplet expansion.
The fundamental mechanisms that regulate adipocyte lipid droplet formation and expansion are conserved across mammalian cells (Sturley and Hussain, 2012). The lipid monolayer membrane derives from the ER membrane. The mechanisms by which membrane-associated proteins target the newly formed droplet and how the droplet membrane mechanically functions are questions that are still being elucidated (Digel et al., 2010). However, the lipid droplet–associated proteome has been investigated in detail, and adipocyte-specific proteins, such as the previously discussed Plin1, are crucial for the metabolic flexibility of adipocyte lipid storage and metabolism (Brasaemle et al., 2004; Digel et al., 2010). Fat-specific protein (FSP) 27/Cidec (Fsp27) plays an important role in lipid droplet formation (Puri et al., 2007). A recent report found that Plin1 serves as an Fsp27 activator, increasing Fsp27-mediated lipid exchange, lipid transfer, and droplet growth (Sun et al., 2013a). This dual role of Plin1 in the regulation of adipose tissue lipolysis and lipid droplet expansion highlights the importance of lipid droplet proteins in the maintenance and the regulation of adipocyte metabolism. Indeed, mice with adipocyte-specific deletion of the Fsp27 gene have impaired lipid storage capacity, as indicated by small adipocyte size, resistance to weight gain upon high fat feeding, hepatic steatosis (fat accumulation in the liver), dyslipidemia, and systemic insulin resistance (Tanaka et al., 2015). This study supports the notion that expansion of the lipid droplet, and thus adipose tissue as a whole, can be protective against the metabolic abnormalities associated with excess caloric intake. Additional studies have demonstrated that the ability of adipocytes to expand the lipid droplet while maintaining insulin sensitivity conveys a protective, adaptive role (Kusminski et al., 2012). An area of great interest, and much debate, is adipocyte hyperplasia, or adipogenesis, in addition to hypertrophy as a mechanism for increased lipid sequestration and fat expansion (Box 4). As such, these states of increased fat mass with preserved metabolic fitness have been referred to as “healthy” expansion of adipose tissue.
Box 4. Hypertrophy versus hyperplasia.
Expanding fat mass requires either increased adipocyte size (hypertrophy) or increased adipocyte number (hyperplasia). Mechanisms that regulate adipocyte lipid storage permit hypertrophy with increased nutrient load. However, large, hypertrophic adipocytes face limits of expansion based on multiple factors, including hypoxia and differential matrix mechanics (Halberg et al., 2009) that result in dysfunctional adipocytes. Genetic mouse models to test these expansion limits by targeting HIF1α targets (Sun et al., 2013a), reducing ECM deposition (Khan et al., 2009), or protecting adipocytes from fatty acid oxidation (Kusminski et al., 2012) have been successful in demonstrating “healthy” hypertrophy. Alternatively, adipocyte hyperplasia may present a mechanism for healthy fat storage capacity (Zeve et al., 2009). Mature adipocytes are terminally differentiated cells. However, adipocyte precursors have been identified in adipose tissue that differentiate into fully mature white adipocytes under metabolic stimulation or PPARγ activation, both in vitro and in the mouse (Rodeheffer et al., 2008; Tang et al., 2008; Gupta et al., 2012). Although the relative turnover rate of mature adipocytes or induction of adipogenesis in the adult may be low (Spalding et al., 2008), an increasing genetic toolkit for lineage tracing experiments will permit advanced studies of the conditions and depots most primed for healthy expansion through hyperplasia (Lee et al., 2012; Wang et al., 2013).
Consequences of fat expansion
“Healthy” expansion of adipose tissue and the prevention of ectopic lipid deposition.
The most commonly known metabolic consequence of obesity is insulin resistance. This manifests itself as hyperinsulinemia in the presence of hyperglycemia and dyslipidemia. In systemic insulin resistance, the absence of these downstream effects of insulin signaling permits unregulated adipose tissue lipolysis, resulting in the ectopic deposition of lipid in nonadipose tissues, further exacerbating systemic insulin resistance. In fact, such dysregulated adipocyte lipolysis is implicated as the primary source of NEFA flux to the liver, which promotes nonalcoholic fatty liver disease (NAFLD) and hepatic insulin resistance, leading to an impairment of insulin actions to inhibit gluconeogenesis (Samuel et al., 2004).
Recent studies have shown that obesity-associated insulin resistance, hepatic steatosis, fibrosis, and systemic inflammation can be prevented by manipulating energy metabolism in the adipocyte to drive lipid droplet expansion. Adipose tissue overexpression of phosphoenolpyruvate carboxykinase (PEPCK), the rate-limiting enzyme in glyceroneogenesis (Forest et al., 2003), leads to obesity without associated insulin resistance in the mouse by increasing glycerol-3-phosphate substrate for re-esterification, thereby reducing NEFA release and increasing adipocyte triglyceride storage (Franckhauser et al., 2002; Tordjman et al., 2003). Eliminating PPARγ binding sites in the PEPCK gene leads to reduced PEPCK levels in adipocytes and was associated with profound insulin resistance, demonstrating that adipocyte glycerogenesis is protective (Millward et al., 2010). Silencing mitochondrial PEPCK lowered circulating glucose and triglyceride, as well as adipose tissue mass in the rat (Stark et al., 2014). Similarly, a reduction in adipocyte mitochondrial membrane potential through overexpressing the mitochondrial protein mitoNEET led to reduced fatty acid oxidation, increased PEPCK expression, increased adipocyte triglyceride storage, and insulin sensitivity in obese mice (Kusminski et al., 2012). Other disruptions in adipocyte mitochondrial function have also led to adipocyte triglyceride accumulation (Vankoningsloo et al., 2006; Wang et al., 2010). Despite the advantages of an expanding adipocyte, overwhelming the cell with lipid is a challenge to the adipocyte characterized by reactive oxygen species (ROS) generation and leads to ER stress and increased lipid “spillover” (Koves et al., 2008; Gao et al., 2010; Kusminski and Scherer, 2012).
This “lipid spillover” to other cell types, leading to ectopic lipid deposition, is partially causative for the collective pathologies that encompass the metabolic syndrome (Unger and Scherer, 2010). Clinically, ectopic lipids are most obviously concurrent with obesity in the diagnosis of NAFLD. A subset of patients, however, experience the metabolic syndrome through acquired or genetic lipodystrophies in which adipocytes fail to properly store lipids. Mutations in genes governing proteins that regulate adipogenesis, lipid metabolism, and lipid droplet formation, such as caveolin-1 (Cav1; Box 5), have all been identified as causing familial lipodystrophies (Agarwal and Garg, 2003; Reue and Dwyer, 2009; Garg, 2011). Interestingly, a common cause of acquired lipodystrophy was early generation antiretroviral protease inhibitors used to combat HIV infection (Garg, 2011; Pérez-Matute et al., 2013). The precise underlying mechanism of this type of lipoatrophy is unclear, but effects on adipocyte mitochondrial function may contribute (Apostolova et al., 2011; Pérez-Matute et al., 2013).
Box 5. Caveolae and adipocyte biology.
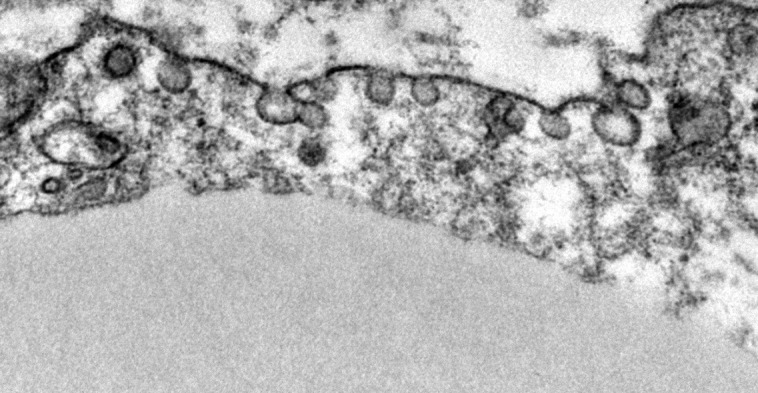
Adipocyte plasma membranes are rich in caveolae, occupying up to 30% of the membrane surface (Ding et al., 2014). These microdomain structures are up-regulated in the plasma membrane of adipocytes concurrent with adipogenesis (Scherer et al., 1994). Cavin-1 and Cav1, which are both required for caveolae formation, are enriched in adipocytes and critical for a host of its metabolic functions (Razani et al., 2002; Ding et al., 2014). Adipocyte Glut4 translocation and insulin–glucose responsiveness are in part caveolae dependent (Shigematsu et al., 2003; Yuan et al., 2007), and Cavin-1– and Cav1-null mice demonstrate insulin resistance with reduced adipocyte glucose uptake (Cohen et al., 2003; Liu et al., 2008). Fatty acid binding and transport proteins, such as CD36, localize to plasma membrane caveolae (Ring et al., 2006; Le Lay et al., 2009), and loss of caveolae reduces adipocyte fatty acid uptake (Liu et al., 2008) and lipolysis (Cohen et al., 2004; Ding et al., 2014). Caveolae are up-regulated with fasting to enhance fatty acid trafficking, and Cav1-null cells are more susceptible to lipotoxicity (Meshulam et al., 2011). Failure to up-regulate adipocyte caveolin-1 in a mouse model of type I diabetes resulted in severe dyslipidemia and rapid adipose wasting and death (Ye et al., 2014). Cav1-null adipocytes also exhibit increased ROS (Asterholm et al., 2012) and reduced mitochondrial biogenesis (Ding et al., 2014). In addition to nutrient handling, caveolae are also sites of membrane receptor clustering, e.g., for insulin, β-AR, and adiponectin signaling (Pilch and Liu, 2011; Wang et al., 2014). Adipocytes may, therefore, serve as a model for better understanding the many roles of caveolae that are highly relevant across other areas of cell biology. The image is a transmission electron micrograph of caveolae at the adipocyte plasma membrane.
ER stress and the unfolded protein response (UPR).
The ER plays important roles in protein and lipid synthesis, and its dysfunction/stress has been linked to perturbed cellular metabolism and local and systemic insulin resistance (Koves et al., 2008; Lim et al., 2009). Communication between the ER and mitochondria likely links ER stress and mitochondrial function (or dysfunction) in driving an increase in the cell’s UPR, increased ROS levels, and cellular insulin resistance (Kusminski and Scherer, 2012). Induction of ER stress activates PKA and results in increased lipolysis through Plin1 phosphorylation (Deng et al., 2012), a potential mechanism for increased NEFA release and systemic lipotoxicity. Conversely, ER stress may enhance lipid droplet formation in an attempt to further sequester lipid (Hapala et al., 2011). ER stress also reduces adipocyte insulin sensitivity (Xu et al., 2010) and regulates adiponectin oligomerization: increasing stress reduces high-molecular-weight forms of adiponectin (Huh et al., 2012). Reducing ROS reverses this response (Furukawa et al., 2004). As the relative abundance of adiponectin high-molecular-weight oligomers is better correlated with overall systemic insulin sensitivity than total adiponectin levels, this suggests a potential pathway by which adipocyte metabolic stress is communicated to other insulin-sensitive tissues.
Some degree of ER membrane–mediated UPR signaling may be necessary for adipogenesis and healthy function, but appears to be pathway dependent. Three distinct UPR pathways have been described. The PERK–eIF2a pathway leads to translational inhibition of adipogenesis, whereas the IREα and ATF6 pathways each appear to promote differentiation and lipid accumulation (Lowe et al., 2012; Han et al., 2013). Several interesting adipocyte functions have been recently attributed to IREα signaling of the UPR through Xbp1s. Although the Xbp1 pathway promotes adipogenesis (Cho et al., 2013), the posttranslational splicing of Xbp1 to Xbp1s specifically rescues adipogenesis in Xbp1 knockout cells (Sha et al., 2009). Xbp1s is also up-regulated in metabolically challenged adipocytes and in mammary adipose tissue during lactation (Gregor et al., 2013). Xbp1s expression in adipocytes increased insulin sensitivity in a transgenic mouse model (Sha et al., 2014). We recently uncovered an important role for Xbp1s as a stimulator of lipolysis in a variety of metabolic states (unpublished data). Thus, it is likely that Xbp1s function in the adipocytes stretches beyond glucose metabolism.
Adipocyte ceramide sequestration.
Ceramides are a class of sphingolipids strongly implicated in insulin resistance. Ceramides are formed predominantly from long-chain fatty acyl-CoA through de novo synthesis during times of intracellular fatty acid excess (Holland and Summers, 2008). Much like ectopic lipid deposition, increased cellular ceramides have been linked to cellular stress responses and apoptosis (Holland et al., 2011a). The analysis of ceramide metabolism is complicated by a high flux rate through upstream and downstream enzymes. Whether adipocyte-specific ceramide accumulation may contribute to the anti-diabetic effects of healthy adipose tissue still requires further investigation.
The protective effects exerted by adipocytes through lipid handling makes them ideal for studying the pathophysiology of aberrant ceramide production. Human adipose tissue ceramide species concentrations as well as those of their regulatory enzymes are significantly altered in individuals with the metabolic syndrome (Blachnio-Zabielska et al., 2012; Błachnio-Zabielska et al., 2012). Ceramides directly induce cellular insulin “resistance” in adipocytes by limiting the effects of downstream insulin signaling, such as GLUT4 translocation (Summers et al., 1998). Suppressing ceramide production or depleting its levels prevents or even enhances insulin sensitivity during fatty acid challenges (Holland et al., 2007; Lahiri et al., 2009; Bikman et al., 2012; Mitsutake et al., 2012). One study suggested that ceramide content was reduced with adipocyte differentiation (Choi et al., 2011). This could simply reflect the ability of adipocytes under heightened insulin signaling to esterify and sequester fatty acids, thus limiting sphingolipid biosynthesis (Hage Hassan et al., 2014). Ceramide-driven insulin resistance may be dependent on the particular chain length species that are present, either due to dietary exposure or relative expression levels of chain-specific ceramide synthases or ceramidases (Barbarroja et al., 2014; Turpin et al., 2014). Adipose tissue C16:0 ceramide, for example, strongly induces systemic insulin resistance (Turpin et al., 2014). The cytoprotective and insulin-sensitizing effects of adiponectin have been linked to its induction of ceramidase activity and ceramide conversion to sphingosine-1-phosphate (Holland et al., 2011b; Holland and Scherer, 2013). Adiponectin receptor agonism may thus demonstrate potent antidiabetic effects (Okada-Iwabu et al., 2013). Future work manipulating sphingolipid enzymes specifically in adipocytes will further define the role adipocytes play in regulating systemic ceramide levels.
Adipocyte cytoskeletal remodeling, matrix communication, and adipose fibrosis.
Another consequence of unhealthy adipose tissue expansion in obesity is the rapid induction of a dense extracellular matrix (Alligier et al., 2012; Sun et al., 2013b) with adipocytes expressing a pro-fibrotic transcriptome (Henegar et al., 2008). Genetic mouse models with metabolically dysfunctional adipocytes often exhibit increased adipose ECM density (Davis et al., 2013; Ding et al., 2014). The mechanical interactions of adipocytes with this fibrous matrix have been identified as potential cues that drive adipocyte dysfunction. In tissue culture systems, adipocyte precursors and mature adipocytes both exhibit marked responses to mechanical stimulation (Levy et al., 2012; Shoham et al., 2012, 2014). Culturing human adipocytes on dense, fibrous matrices leads to increased β1-integrin–dependent pro-inflammatory gene expression and cytoskeletal strain-activated transcriptional responses (Pellegrinelli et al., 2014). In pre-adipocytes, cytoskeletal remodeling of actin fibers prevents the nuclear translocation of MLK1 and allows PPARγ-mediated differentiation (Nobusue et al., 2014). The coalescing mechanism of large lipid droplet formation is microtubule dependent (Ariotti et al., 2012), and interfering with cytoskeletal remodeling disrupts lipid droplet maintenance and dynamics during lipolysis (Orlicky et al., 2013). The dynamic size changes of adipocytes with expansion and reduction are thus critically dependent on the mechanical environment.
In addition, extracellular matrix remodeling is an important step in adipose expansion. Interactions with the matrix—and the matrix composition itself—regulate adipocyte health. Mice heterozygous for MMP14 exhibit a reduced capacity to remodel the matrix of expanding adipose tissue and thus have limited fat expansion (Chun et al., 2010), though this effect was not apparent in MMP-9 knockout mice (Van Hul et al., 2010). An inverse genetic model with specifically increased MMP activity has yet to be metabolically characterized; however, mice lacking TIMP-2 have reduced native MMP inhibition and exhibit expanded adipose tissue (Jaworski et al., 2011). A similar effect was noted in mice lacking collagen 6 (Col6), a matrix collagen highly enriched in adipose tissue. These mice gained more weight, and had increased adipose depot size and massively hypertrophic adipocytes, but were metabolically healthier than their littermates with reduced inflammation (Khan et al., 2009). Tissues from Col6 knockout mice have been used ex vivo to further demonstrate the intimate role of adipose tissue mechanical stability on adipocyte metabolic and cellular health (Lackey et al., 2014). Although adipose tissue fibrosis is unquestionably of profound physiological and clinical importance (Sun et al., 2013b), a recent study integrating ECM architecture and subclinical inflammation revealed a more complex relationship. Complete inhibition of local adipocyte inflammatory responses was associated with severe metabolic inflexibility and insulin resistance. We found that a minimal level of inflammation in the adipocyte microenvironment was required for extracellular matrix remodeling during expansion and that a failure to do so resulted in dysfunctional adipose tissue (Wernstedt Asterholm et al., 2014).
Hypoxia in fat expansion.
As the adipocyte expands, adipose tissue interstitial oxygen tension decreases. Reduced oxygen tension has been directly measured in obese fat depots in mouse models and human subjects (Ye et al., 2007; Pasarica et al., 2009). HIF1α is a transcription factor that serves as an oxygen sensor in many cell types. Under normoxic conditions, HIF1α is rapidly degraded. However, hypoxic conditions stabilize HIF1α and permit up-regulation of genes containing HIF response elements as well as a distinct shift in the expression of a specific group of miRNAs, termed hypoxamirs (Nallamshetty et al., 2013). HIF1α protein is highly enriched in expanding adipocytes due to the need for increased adipose tissue vascularization. Because of the decreased oxygen availability in hypoxic conditions, glycolytic metabolic programs prevail under such conditions (Halberg et al., 2009; Regazzetti et al., 2010). In fact, local increases in fatty acids can uncouple the mitochondria and further increase oxygen consumption (Lee et al., 2014). Though HIF1α’s downstream targets should elicit a tissue response to improve oxygen supply, tissue vascularization effects have a complicated relationship with adipocyte health that is potentially both inhibitory or supportive, depending on the physiological state of the adipocyte (Sun et al., 2012). Direct inhibition of HIF1α improves adipose tissue health, further suggesting that other HIF1α responsive genes are more important for dysfunction than those that are specifically angiogenic (Sun et al., 2013a). Overexpression of HIF1α in the adipocyte proved to be more pro-fibrotic and pro-inflammatory than pro-angiogenic per se (Halberg et al., 2009). Adipocyte-specific deletion of HIF1α limited high-fat diet–induced adipose tissue inflammation and insulin resistance, and the tissue was equally vascularized as wild-type controls (Lee et al., 2014). A recent report echoed these effects by linking the protective signaling of estrogen receptor α on limiting adipose hypoxia and fibrosis with HIF1α ubiquitination and deactivation (Kim et al., 2014). In fact, the degree to which hypoxia prevails in adipose tissue may be directly proportional to adipocyte size. Large adipocytes (>100 µm) approximate the diffusional limits of oxygen or physiological capillary spacing. Leptin secretion increases with adipocyte size, and leptin is a direct HIF1α transcriptional target (Sun et al., 2013a). As a result, systemic sensing of available adipose storage capacity can be measured by how much leptin is produced. Thus, local adipocyte hypoxia plays an essential signaling role for assessing systemic energy reserves and is translated into altered leptin levels in circulation.
Concluding remarks
The adipocyte remains a mysterious cell full of surprises. Uniquely positioned to take up, store, and release free fatty acids depending on systemic nutritional status, it can cope with high concentrations of lipids without experiencing the lipotoxicity that essentially every other mammalian cell type is subject to. Once prompted to release fatty acids upon demand, the adipocyte also produces and releases several anti-lipotoxic adipokines (Unger et al., 2013) to protect other tissues from the cytotoxic effects of high concentrations of fatty acids. The adipocyte is therefore a major producer of secreted products that regulate a wide range of physiological systems. In light of its unique position as a gate-keeper of high-energy reserves, the adipocyte is highly sensitive to hormonal cues (such as insulin), nutritional substrate-driven responses (such as glucose and lipids), and central signals conveyed through sympathetic innervation into the tissue. We unfortunately have tended to underestimate the function of the adipocyte as part of a mere storage organ. We now increasingly appreciate it as an endocrine cell that is a key component in the maintenance of systemic energy homeostasis.
Acknowledgments
The authors were supported by the National Institutes of Health (grants R01-DK55758, R01-DK099110, and P01-DK088761 to P.E. Scherer) as well as a grant from the Cancer Prevention and Research Institute of Texas (CPRIT RP140412). J.H. Stern was supported by T32-DK007307. J.M. Rutkowski was supported by the American Heart Association (12SDG12050287). Fig. 2 was illustrated by Richard Howdy, a certified medical illustrator, at Visually Medical.
The authors declare no competing financial interests.
Footnotes
Abbreviations used in this paper:
- ATGL
- adipose triglyceride lipase
- β-Ar
- β-Adrenergic
- CGI-58
- comparative gene identification-58
- FATP
- fatty acid transport protein
- G3P
- glycerol-3-phosphate
- Glut-4
- glucose transporter type 4
- HSL
- hormone-sensitive lipase
- NEFA
- nonesterified fatty acid
- PEPCK
- phosphoenolpyruvate carboxykinase
- Plin1
- perilipin 1
- PPARγ
- peroxisome proliferator-activated receptor γ
- ROS
- reactive oxygen species
- UPR
- unfolded protein response
References
- Abel E.D., Peroni O., Kim J.K., Kim Y.B., Boss O., Hadro E., Minnemann T., Shulman G.I., and Kahn B.B.. 2001. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature. 409:729–733 10.1038/35055575 [DOI] [PubMed] [Google Scholar]
- Agarwal A.K., and Garg A.. 2003. Congenital generalized lipodystrophy: significance of triglyceride biosynthetic pathways. Trends Endocrinol. Metab. 14:214–221 10.1016/S1043-2760(03)00078-X [DOI] [PubMed] [Google Scholar]
- Agarwal A.K., Sukumaran S., Cortés V.A., Tunison K., Mizrachi D., Sankella S., Gerard R.D., Horton J.D., and Garg A.. 2011. Human 1-acylglycerol-3-phosphate O-acyltransferase isoforms 1 and 2: biochemical characterization and inability to rescue hepatic steatosis in Agpat2-/- gene lipodystrophic mice. J. Biol. Chem. 286:37676–37691 10.1074/jbc.M111.250449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed K., Tunaru S., Tang C., Müller M., Gille A., Sassmann A., Hanson J., and Offermanns S.. 2010. An autocrine lactate loop mediates insulin-dependent inhibition of lipolysis through GPR81. Cell Metab. 11:311–319 10.1016/j.cmet.2010.02.012 [DOI] [PubMed] [Google Scholar]
- Alligier M., Meugnier E., Debard C., Lambert-Porcheron S., Chanseaume E., Sothier M., Loizon E., Hssain A.A., Brozek J., Scoazec J.Y., et al. 2012. Subcutaneous adipose tissue remodeling during the initial phase of weight gain induced by overfeeding in humans. J. Clin. Endocrinol. Metab. 97:E183–E192 10.1210/jc.2011-2314 [DOI] [PubMed] [Google Scholar]
- Amole B.O., Wittner M., Hewlett D., and Tanowitz H.B.. 1985. Trypanosoma brucei: infection in murine diabetes. Exp. Parasitol. 60:342–347 10.1016/0014-4894(85)90040-2 [DOI] [PubMed] [Google Scholar]
- Apostolova N., Blas-García A., and Esplugues J.V.. 2011. Mitochondrial toxicity in HAART: an overview of in vitro evidence. Curr. Pharm. Des. 17:2130–2144 10.2174/138161211796904731 [DOI] [PubMed] [Google Scholar]
- Ariotti N., Murphy S., Hamilton N.A., Wu L., Green K., Schieber N.L., Li P., Martin S., and Parton R.G.. 2012. Postlipolytic insulin-dependent remodeling of micro lipid droplets in adipocytes. Mol. Biol. Cell. 23:1826–1837 10.1091/mbc.E11-10-0847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asterholm I.W., Mundy D.I., Weng J., Anderson R.G., and Scherer P.E.. 2012. Altered mitochondrial function and metabolic inflexibility associated with loss of caveolin-1. Cell Metab. 15:171–185 10.1016/j.cmet.2012.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asterholm I.W., Rutkowski J.M., Fujikawa T., Cho Y.R., Fukuda M., Tao C., Wang Z.V., Gupta R.K., Elmquist J.K., and Scherer P.E.. 2014. Elevated resistin levels induce central leptin resistance and increased atherosclerotic progression in mice. Diabetologia. 57:1209–1218 10.1007/s00125-014-3210-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbarroja N., Rodriguez-Cuenca S., Nygren H., Camargo A., Pirraco A., Relat J., Cuadrado I., Pellegrinelli V., Medina-Gomez G., Lopez-Pedrera C., et al. 2014. Increased dihydroceramide/ceramide ratio mediated by defective expression of degs1 impairs adipocyte differentiation and function. Diabetes. 10.2337/db14-0359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikman B.T., Guan Y., Shui G., Siddique M.M., Holland W.L., Kim J.Y., Fabriàs G., Wenk M.R., and Summers S.A.. 2012. Fenretinide prevents lipid-induced insulin resistance by blocking ceramide biosynthesis. J. Biol. Chem. 287:17426–17437 10.1074/jbc.M112.359950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blachnio-Zabielska A.U., Koutsari C., Tchkonia T., and Jensen M.D.. 2012. Sphingolipid content of human adipose tissue: relationship to adiponectin and insulin resistance. Obesity (Silver Spring). 20:2341–2347 10.1038/oby.2012.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Błachnio-Zabielska A.U., Pułka M., Baranowski M., Nikołajuk A., Zabielski P., Górska M., and Górski J.. 2012. Ceramide metabolism is affected by obesity and diabetes in human adipose tissue. J. Cell. Physiol. 227:550–557 10.1002/jcp.22745 [DOI] [PubMed] [Google Scholar]
- Blüher M.2014. Adipokines - removing road blocks to obesity and diabetes therapy. Mol. Metab. 3:230–240 10.1016/j.molmet.2014.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blüher M., Michael M.D., Peroni O.D., Ueki K., Carter N., Kahn B.B., and Kahn C.R.. 2002. Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Dev. Cell. 3:25–38 10.1016/S1534-5807(02)00199-5 [DOI] [PubMed] [Google Scholar]
- Bolten C.W., Blanner P.M., McDonald W.G., Staten N.R., Mazzarella R.A., Arhancet G.B., Meier M.F., Weiss D.J., Sullivan P.M., Hromockyj A.E., et al. 2007. Insulin sensitizing pharmacology of thiazolidinediones correlates with mitochondrial gene expression rather than activation of PPAR gamma. Gene Regul. Syst. Bio. 1:73–82. [PMC free article] [PubMed] [Google Scholar]
- Brasaemle D.L., Dolios G., Shapiro L., and Wang R.. 2004. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytes. J. Biol. Chem. 279:46835–46842 10.1074/jbc.M409340200 [DOI] [PubMed] [Google Scholar]
- Cao H.2014. Adipocytokines in obesity and metabolic disease. J. Endocrinol. 220:T47–T59 10.1530/JOE-13-0339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspar-Bauguil S., Cousin B., Bour S., Casteilla L., Penicaud L., and Carpéné C.. 2009. Adipose tissue lymphocytes: types and roles. J. Physiol. Biochem. 65:423–436 (published erratum appears in J. Physiol. Biochem. 2011. 67:497) 10.1007/BF03185938 [DOI] [PubMed] [Google Scholar]
- Cho Y.M., Kim D.H., Kwak S.N., Jeong S.W., and Kwon O.J.. 2013. X-box binding protein 1 enhances adipogenic differentiation of 3T3-L1 cells through the downregulation of Wnt10b expression. FEBS Lett. 587:1644–1649 10.1016/j.febslet.2013.04.005 [DOI] [PubMed] [Google Scholar]
- Choi S.M., Tucker D.F., Gross D.N., Easton R.M., DiPilato L.M., Dean A.S., Monks B.R., and Birnbaum M.J.. 2010. Insulin regulates adipocyte lipolysis via an Akt-independent signaling pathway. Mol. Cell. Biol. 30:5009–5020 10.1128/MCB.00797-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K.M., Lee Y.S., Choi M.H., Sin D.M., Lee S., Ji S.Y., Lee M.K., Lee Y.M., Yun Y.P., Hong J.T., and Yoo H.S.. 2011. Inverse relationship between adipocyte differentiation and ceramide level in 3T3-L1 cells. Biol. Pharm. Bull. 34:912–916 10.1248/bpb.34.912 [DOI] [PubMed] [Google Scholar]
- Choo H.J., Kim J.H., Kwon O.B., Lee C.S., Mun J.Y., Han S.S., Yoon Y.S., Yoon G., Choi K.M., and Ko Y.G.. 2006. Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia. 49:784–791 10.1007/s00125-006-0170-2 [DOI] [PubMed] [Google Scholar]
- Chun T.H., Inoue M., Morisaki H., Yamanaka I., Miyamoto Y., Okamura T., Sato-Kusubata K., and Weiss S.J.. 2010. Genetic link between obesity and MMP14-dependent adipogenic collagen turnover. Diabetes. 59:2484–2494 10.2337/db10-0073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coburn C.T., Knapp F.F. Jr, Febbraio M., Beets A.L., Silverstein R.L., and Abumrad N.A.. 2000. Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J. Biol. Chem. 275:32523–32529 10.1074/jbc.M003826200 [DOI] [PubMed] [Google Scholar]
- Cohen A.W., Razani B., Wang X.B., Combs T.P., Williams T.M., Scherer P.E., and Lisanti M.P.. 2003. Caveolin-1-deficient mice show insulin resistance and defective insulin receptor protein expression in adipose tissue. Am. J. Physiol. Cell Physiol. 285:C222–C235 10.1152/ajpcell.00006.2003 [DOI] [PubMed] [Google Scholar]
- Cohen A.W., Razani B., Schubert W., Williams T.M., Wang X.B., Iyengar P., Brasaemle D.L., Scherer P.E., and Lisanti M.P.. 2004. Role of caveolin-1 in the modulation of lipolysis and lipid droplet formation. Diabetes. 53:1261–1270 10.2337/diabetes.53.5.1261 [DOI] [PubMed] [Google Scholar]
- Combs T.P., Nagajyothi S., Mukherjee S., de Almeida C.J., Jelicks L.A., Schubert W., Lin Y., Jayabalan D.S., Zhao D., Braunstein V.L., et al. 2005. The adipocyte as an important target cell for Trypanosoma cruzi infection. J. Biol. Chem. 280:24085–24094 10.1074/jbc.M412802200 [DOI] [PubMed] [Google Scholar]
- Dalen K.T., Schoonjans K., Ulven S.M., Weedon-Fekjaer M.S., Bentzen T.G., Koutnikova H., Auwerx J., and Nebb H.I.. 2004. Adipose tissue expression of the lipid droplet-associating proteins S3-12 and perilipin is controlled by peroxisome proliferator-activated receptor-gamma. Diabetes. 53:1243–1252 10.2337/diabetes.53.5.1243 [DOI] [PubMed] [Google Scholar]
- Davis K.E., D Neinast M., Sun K., M Skiles W., D Bills J., A Zehr J., Zeve D., D Hahner L., W Cox D., M Gent L., et al. 2013. The sexually dimorphic role of adipose and adipocyte estrogen receptors in modulating adipose tissue expansion, inflammation, and fibrosis. Mol Metab. 2:227–242 10.1016/j.molmet.2013.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y., and Scherer P.E.. 2010. Adipokines as novel biomarkers and regulators of the metabolic syndrome. Ann. NY Acad. Sci. 1212:E1–E19 10.1111/j.1749-6632.2010.05875.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J., Liu S., Zou L., Xu C., Geng B., and Xu G.. 2012. Lipolysis response to endoplasmic reticulum stress in adipose cells. J. Biol. Chem. 287:6240–6249 10.1074/jbc.M111.299115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digel M., Ehehalt R., and Füllekrug J.. 2010. Lipid droplets lighting up: insights from live microscopy. FEBS Lett. 584:2168–2175 10.1016/j.febslet.2010.03.035 [DOI] [PubMed] [Google Scholar]
- Ding S.Y., Lee M.J., Summer R., Liu L., Fried S.K., and Pilch P.F.. 2014. Pleiotropic effects of cavin-1 deficiency on lipid metabolism. J. Biol. Chem. 289:8473–8483 10.1074/jbc.M113.546242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann H.M., Golozoubova V., Cannon B., and Nedergaard J.. 2009. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 9:203–209 10.1016/j.cmet.2008.12.014 [DOI] [PubMed] [Google Scholar]
- Ferreira A.V., Segatto M., Menezes Z., Macedo A.M., Gelape C., de Oliveira Andrade L., Nagajyothi F., Scherer P.E., Teixeira M.M., and Tanowitz H.B.. 2011. Evidence for Trypanosoma cruzi in adipose tissue in human chronic Chagas disease. Microbes Infect. 13:1002–1005 10.1016/j.micinf.2011.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festuccia W.T., Blanchard P.G., Turcotte V., Laplante M., Sariahmetoglu M., Brindley D.N., Richard D., and Deshaies Y.. 2009. The PPARγ agonist rosiglitazone enhances rat brown adipose tissue lipogenesis from glucose without altering glucose uptake. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296:R1327–R1335 10.1152/ajpregu.91012.2008 [DOI] [PubMed] [Google Scholar]
- Forest C., Tordjman J., Glorian M., Duplus E., Chauvet G., Quette J., Beale E.G., and Antoine B.. 2003. Fatty acid recycling in adipocytes: a role for glyceroneogenesis and phosphoenolpyruvate carboxykinase. Biochem. Soc. Trans. 31:1125–1129 10.1042/BST0311125 [DOI] [PubMed] [Google Scholar]
- Franckhauser S., Muñoz S., Pujol A., Casellas A., Riu E., Otaegui P., Su B., and Bosch F.. 2002. Increased fatty acid re-esterification by PEPCK overexpression in adipose tissue leads to obesity without insulin resistance. Diabetes. 51:624–630 10.2337/diabetes.51.3.624 [DOI] [PubMed] [Google Scholar]
- Frayn K.N., and Humphreys S.M.. 2012. Metabolic characteristics of human subcutaneous abdominal adipose tissue after overnight fast. Am. J. Physiol. Endocrinol. Metab. 302:E468–E475 10.1152/ajpendo.00527.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa S., Fujita T., Shimabukuro M., Iwaki M., Yamada Y., Nakajima Y., Nakayama O., Makishima M., Matsuda M., and Shimomura I.. 2004. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Invest. 114:1752–1761 10.1172/JCI21625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C.L., Zhu C., Zhao Y.P., Chen X.H., Ji C.B., Zhang C.M., Zhu J.G., Xia Z.K., Tong M.L., and Guo X.R.. 2010. Mitochondrial dysfunction is induced by high levels of glucose and free fatty acids in 3T3-L1 adipocytes. Mol. Cell. Endocrinol. 320:25–33 10.1016/j.mce.2010.01.039 [DOI] [PubMed] [Google Scholar]
- Garg A.2011. Clinical review#: Lipodystrophies: genetic and acquired body fat disorders. J. Clin. Endocrinol. Metab. 96:3313–3325 10.1210/jc.2011-1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gburcik V., Cawthorn W.P., Nedergaard J., Timmons J.A., and Cannon B.. 2012. An essential role for Tbx15 in the differentiation of brown and “brite” but not white adipocytes. Am. J. Physiol. Endocrinol. Metab. 303:E1053–E1060 10.1152/ajpendo.00104.2012 [DOI] [PubMed] [Google Scholar]
- Gregor M.F., Misch E.S., Yang L., Hummasti S., Inouye K.E., Lee A.H., Bierie B., and Hotamisligil G.S.. 2013. The role of adipocyte XBP1 in metabolic regulation during lactation. Cell Reports. 3:1430–1439 10.1016/j.celrep.2013.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grujic D., Susulic V.S., Harper M.E., Himms-Hagen J., Cunningham B.A., Corkey B.E., and Lowell B.B.. 1997. β3-adrenergic receptors on white and brown adipocytes mediate β3-selective agonist-induced effects on energy expenditure, insulin secretion, and food intake. A study using transgenic and gene knockout mice. J. Biol. Chem. 272:17686–17693 10.1074/jbc.272.28.17686 [DOI] [PubMed] [Google Scholar]
- Guan H.P., Li Y., Jensen M.V., Newgard C.B., Steppan C.M., and Lazar M.A.. 2002. A futile metabolic cycle activated in adipocytes by antidiabetic agents. Nat. Med. 8:1122–1128 10.1038/nm780 [DOI] [PubMed] [Google Scholar]
- Gupta R.K., Mepani R.J., Kleiner S., Lo J.C., Khandekar M.J., Cohen P., Frontini A., Bhowmick D.C., Ye L., Cinti S., and Spiegelman B.M.. 2012. Zfp423 expression identifies committed preadipocytes and localizes to adipose endothelial and perivascular cells. Cell Metab. 15:230–239 10.1016/j.cmet.2012.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hage Hassan R., Bourron O., and Hajduch E.. 2014. Defect of insulin signal in peripheral tissues: Important role of ceramide. World J Diabetes. 5:244–257 10.4239/wjd.v5.i3.244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halberg N., Khan T., Trujillo M.E., Wernstedt-Asterholm I., Attie A.D., Sherwani S., Wang Z.V., Landskroner-Eiger S., Dineen S., Magalang U.J., et al. 2009. Hypoxia-inducible factor 1α induces fibrosis and insulin resistance in white adipose tissue. Mol. Cell. Biol. 29:4467–4483 10.1128/MCB.00192-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J., Murthy R., Wood B., Song B., Wang S., Sun B., Malhi H., and Kaufman R.J.. 2013. ER stress signalling through eIF2α and CHOP, but not IRE1α, attenuates adipogenesis in mice. Diabetologia. 56:911–924 10.1007/s00125-012-2809-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Q., Hansen J.B., Petersen R.K., Hallenborg P., Jørgensen C., Cinti S., Larsen P.J., Steffensen K.R., Wang H., Collins S., et al. 2010. ADD1/SREBP1c activates the PGC1-α promoter in brown adipocytes. Biochim. Biophys. Acta. 1801:421–429 10.1016/j.bbalip.2009.11.008 [DOI] [PubMed] [Google Scholar]
- Hapala I., Marza E., and Ferreira T.. 2011. Is fat so bad? Modulation of endoplasmic reticulum stress by lipid droplet formation. Biol. Cell. 103:271–285 10.1042/BC20100144 [DOI] [PubMed] [Google Scholar]
- Henegar C., Tordjman J., Achard V., Lacasa D., Cremer I., Guerre-Millo M., Poitou C., Basdevant A., Stich V., Viguerie N., et al. 2008. Adipose tissue transcriptomic signature highlights the pathological relevance of extracellular matrix in human obesity. Genome Biol. 9:R14 10.1186/gb-2008-9-1-r14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman M.A., Peroni O.D., Villoria J., Schön M.R., Abumrad N.A., Blüher M., Klein S., and Kahn B.B.. 2012. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature. 484:333–338 10.1038/nature10986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland W.L., and Scherer P.E.. 2013. Cell Biology. Ronning after the adiponectin receptors. Science. 342:1460–1461 10.1126/science.1249077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland W.L., and Summers S.A.. 2008. Sphingolipids, insulin resistance, and metabolic disease: new insights from in vivo manipulation of sphingolipid metabolism. Endocr. Rev. 29:381–402 10.1210/er.2007-0025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland W.L., Brozinick J.T., Wang L.P., Hawkins E.D., Sargent K.M., Liu Y., Narra K., Hoehn K.L., Knotts T.A., Siesky A., et al. 2007. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 5:167–179 10.1016/j.cmet.2007.01.002 [DOI] [PubMed] [Google Scholar]
- Holland W.L., Bikman B.T., Wang L.P., Yuguang G., Sargent K.M., Bulchand S., Knotts T.A., Shui G., Clegg D.J., Wenk M.R., et al. 2011a. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J. Clin. Invest. 121:1858–1870 10.1172/JCI43378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland W.L., Miller R.A., Wang Z.V., Sun K., Barth B.M., Bui H.H., Davis K.E., Bikman B.T., Halberg N., Rutkowski J.M., et al. 2011b. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat. Med. 17:55–63 10.1038/nm.2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland W.L., Adams A.C., Brozinick J.T., Bui H.H., Miyauchi Y., Kusminski C.M., Bauer S.M., Wade M., Singhal E., Cheng C.C., et al. 2013. An FGF21-adiponectin-ceramide axis controls energy expenditure and insulin action in mice. Cell Metab. 17:790–797 10.1016/j.cmet.2013.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh J.Y., Seo E.Y., Lee H.B., and Ha H.. 2012. Glucose-based peritoneal dialysis solution suppresses adiponectin synthesis through oxidative stress in an experimental model of peritoneal dialysis. Perit. Dial. Int. 32:20–28 10.3747/pdi.2009.00228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski D.M., Sideleva O., Stradecki H.M., Langlois G.D., Habibovic A., Satish B., Tharp W.G., Lausier J., Larock K., Jetton T.L., et al. 2011. Sexually dimorphic diet-induced insulin resistance in obese tissue inhibitor of metalloproteinase-2 (TIMP-2)-deficient mice. Endocrinology. 152:1300–1313 10.1210/en.2010-1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn B.B., and Cushman S.W.. 1985. Subcellular translocation of glucose transporters: role in insulin action and its perturbation in altered metabolic states. Diabetes Metab. Rev. 1:203–227 10.1002/dmr.5610010301 [DOI] [PubMed] [Google Scholar]
- Keller M.P., and Attie A.D.. 2010. Physiological insights gained from gene expression analysis in obesity and diabetes. Annu. Rev. Nutr. 30:341–364 10.1146/annurev.nutr.012809.104747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan T., Muise E.S., Iyengar P., Wang Z.V., Chandalia M., Abate N., Zhang B.B., Bonaldo P., Chua S., and Scherer P.E.. 2009. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol. Cell. Biol. 29:1575–1591 10.1128/MCB.01300-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M., Neinast M.D., Frank A.P., Sun K., Park J., Zehr J.A., Vishvanath L., Morselli E., Amelotte M., Palmer B.F., et al. 2014. ERα upregulates Phd3 to ameliorate HIF-1 induced fibrosis and inflammation in adipose tissue. Mol Metab. 3:642–651 10.1016/j.molmet.2014.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner S., Mepani R.J., Laznik D., Ye L., Jurczak M.J., Jornayvaz F.R., Estall J.L., Chatterjee Bhowmick D., Shulman G.I., and Spiegelman B.M.. 2012. Development of insulin resistance in mice lacking PGC-1α in adipose tissues. Proc. Natl. Acad. Sci. USA. 109:9635–9640 10.1073/pnas.1207287109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konige M., Wang H., and Sztalryd C.. 2014. Role of adipose specific lipid droplet proteins in maintaining whole body energy homeostasis. Biochim. Biophys. Acta. 1842:393–401 10.1016/j.bbadis.2013.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koves T.R., Ussher J.R., Noland R.C., Slentz D., Mosedale M., Ilkayeva O., Bain J., Stevens R., Dyck J.R., Newgard C.B., et al. 2008. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 7:45–56 10.1016/j.cmet.2007.10.013 [DOI] [PubMed] [Google Scholar]
- Kusminski C.M., and Scherer P.E.. 2012. Mitochondrial dysfunction in white adipose tissue. Trends Endocrinol. Metab. 23:435–443 10.1016/j.tem.2012.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusminski C.M., Holland W.L., Sun K., Park J., Spurgin S.B., Lin Y., Askew G.R., Simcox J.A., McClain D.A., Li C., and Scherer P.E.. 2012. MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat. Med. 18:1539–1549 10.1038/nm.2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackey D.E., Burk D.H., Ali M.R., Mostaedi R., Smith W.H., Park J., Scherer P.E., Seay S.A., McCoin C.S., Bonaldo P., and Adams S.H.. 2014. Contributions of adipose tissue architectural and tensile properties toward defining healthy and unhealthy obesity. Am. J. Physiol. Endocrinol. Metab. 306:E233–E246 10.1152/ajpendo.00476.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri S., Park H., Laviad E.L., Lu X., Bittman R., and Futerman A.H.. 2009. Ceramide synthesis is modulated by the sphingosine analog FTY720 via a mixture of uncompetitive and noncompetitive inhibition in an Acyl-CoA chain length-dependent manner. J. Biol. Chem. 284:16090–16098 10.1074/jbc.M807438200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lass A., Zimmermann R., Oberer M., and Zechner R.. 2011. Lipolysis - a highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog. Lipid Res. 50:14–27 10.1016/j.plipres.2010.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Lay S., Blouin C.M., Hajduch E., and Dugail I.. 2009. Filling up adipocytes with lipids. Lessons from caveolin-1 deficiency. Biochim. Biophys. Acta. 1791:514–518 10.1016/j.bbalip.2008.10.008 [DOI] [PubMed] [Google Scholar]
- Lee Y.H., Petkova A.P., Mottillo E.P., and Granneman J.G.. 2012. In vivo identification of bipotential adipocyte progenitors recruited by β3-adrenoceptor activation and high-fat feeding. Cell Metab. 15:480–491 10.1016/j.cmet.2012.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.S., Kim J.W., Osborne O., Oh Y., Sasik R., Schenk S., Chen A., Chung H., Murphy A., Watkins S.M., et al. 2014. Increased adipocyte O2 consumption triggers HIF-1α, causing inflammation and insulin resistance in obesity. Cell. 157:1339–1352 10.1016/j.cell.2014.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy A., Enzer S., Shoham N., Zaretsky U., and Gefen A.. 2012. Large, but not small sustained tensile strains stimulate adipogenesis in culture. Ann. Biomed. Eng. 40:1052–1060 10.1007/s10439-011-0496-x [DOI] [PubMed] [Google Scholar]
- Lim J.H., Lee H.J., Ho Jung M., and Song J.. 2009. Coupling mitochondrial dysfunction to endoplasmic reticulum stress response: a molecular mechanism leading to hepatic insulin resistance. Cell. Signal. 21:169–177 10.1016/j.cellsig.2008.10.004 [DOI] [PubMed] [Google Scholar]
- Liu L., Brown D., McKee M., Lebrasseur N.K., Yang D., Albrecht K.H., Ravid K., and Pilch P.F.. 2008. Deletion of Cavin/PTRF causes global loss of caveolae, dyslipidemia, and glucose intolerance. Cell Metab. 8:310–317 10.1016/j.cmet.2008.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo S., Wiczer B.M., Smith A.J., Hall A.M., and Bernlohr D.A.. 2007. Fatty acid metabolism in adipocytes: functional analysis of fatty acid transport proteins 1 and 4. J. Lipid Res. 48:609–620 10.1194/jlr.M600441-JLR200 [DOI] [PubMed] [Google Scholar]
- Long J.Z., Svensson K.J., Tsai L., Zeng X., Roh H.C., Kong X., Rao R.R., Lou J., Lokurkar I., Baur W., et al. 2014. A smooth muscle-like origin for beige adipocytes. Cell Metab. 19:810–820 10.1016/j.cmet.2014.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe C.E., Dennis R.J., Obi U., O’Rahilly S., and Rochford J.J.. 2012. Investigating the involvement of the ATF6α pathway of the unfolded protein response in adipogenesis. Int. J. Obes. (Lond). 36:1248–1251 10.1038/ijo.2011.233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R.H., Ji H., Chang Z.G., Su S.S., and Yang G.S.. 2010. Mitochondrial development and the influence of its dysfunction during rat adipocyte differentiation. Mol. Biol. Rep. 37:2173–2182 10.1007/s11033-009-9695-z [DOI] [PubMed] [Google Scholar]
- MacLaren R., Cui W., and Cianflone K.. 2008. Adipokines and the immune system: an adipocentric view. Adv. Exp. Med. Biol. 632:1–21 10.1007/978-0-387-78952-1_1 [DOI] [PubMed] [Google Scholar]
- Meshulam T., Breen M.R., Liu L., Parton R.G., and Pilch P.F.. 2011. Caveolins/caveolae protect adipocytes from fatty acid-mediated lipotoxicity. J. Lipid Res. 52:1526–1532 10.1194/jlr.M015628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millward C.A., Desantis D., Hsieh C.W., Heaney J.D., Pisano S., Olswang Y., Reshef L., Beidelschies M., Puchowicz M., and Croniger C.M.. 2010. Phosphoenolpyruvate carboxykinase (Pck1) helps regulate the triglyceride/fatty acid cycle and development of insulin resistance in mice. J. Lipid Res. 51:1452–1463 10.1194/jlr.M005363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsutake S., Date T., Yokota H., Sugiura M., Kohama T., and Igarashi Y.. 2012. Ceramide kinase deficiency improves diet-induced obesity and insulin resistance. FEBS Lett. 586:1300–1305 10.1016/j.febslet.2012.03.032 [DOI] [PubMed] [Google Scholar]
- Nagajyothi F., Desruisseaux M.S., Thiruvur N., Weiss L.M., Braunstein V.L., Albanese C., Teixeira M.M., de Almeida C.J., Lisanti M.P., Scherer P.E., and Tanowitz H.B.. 2008. Trypanosoma cruzi infection of cultured adipocytes results in an inflammatory phenotype. Obesity (Silver Spring). 16:1992–1997 10.1038/oby.2008.331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagajyothi F., Zhao D., Machado F.S., Weiss L.M., Schwartz G.J., Desruisseaux M.S., Zhao Y., Factor S.M., Huang H., Albanese C., et al. 2010. Crucial role of the central leptin receptor in murine Trypanosoma cruzi (Brazil strain) infection. J. Infect. Dis. 202:1104–1113 10.1086/656189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagajyothi F., Weiss L.M., Silver D.L., Desruisseaux M.S., Scherer P.E., Herz J., and Tanowitz H.B.. 2011. Trypanosoma cruzi utilizes the host low density lipoprotein receptor in invasion. PLoS Negl. Trop. Dis. 5:e953 10.1371/journal.pntd.0000953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagajyothi F., Machado F.S., Burleigh B.A., Jelicks L.A., Scherer P.E., Mukherjee S., Lisanti M.P., Weiss L.M., Garg N.J., and Tanowitz H.B.. 2012. Mechanisms of Trypanosoma cruzi persistence in Chagas disease. Cell. Microbiol. 14:634–643 10.1111/j.1462-5822.2012.01764.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagajyothi F., Kuliawat R., Kusminski C.M., Machado F.S., Desruisseaux M.S., Zhao D., Schwartz G.J., Huang H., Albanese C., Lisanti M.P., et al. 2013. Alterations in glucose homeostasis in a murine model of Chagas disease. Am. J. Pathol. 182:886–894 10.1016/j.ajpath.2012.11.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nallamshetty S., Chan S.Y., and Loscalzo J.. 2013. Hypoxia: a master regulator of microRNA biogenesis and activity. Free Radic. Biol. Med. 64:20–30 10.1016/j.freeradbiomed.2013.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard J., and Cannon B.. 2013. UCP1 mRNA does not produce heat. Biochim. Biophys. Acta. 1831:943–949 10.1016/j.bbalip.2013.01.009 [DOI] [PubMed] [Google Scholar]
- Nedergaard J., and Cannon B.. 2014. The browning of white adipose tissue: some burning issues. Cell Metab. 20:396–407 10.1016/j.cmet.2014.07.005 [DOI] [PubMed] [Google Scholar]
- Nobusue H., Onishi N., Shimizu T., Sugihara E., Oki Y., Sumikawa Y., Chiyoda T., Akashi K., Saya H., and Kano K.. 2014. Regulation of MKL1 via actin cytoskeleton dynamics drives adipocyte differentiation. Nat. Commun. 5:3368 10.1038/ncomms4368 [DOI] [PubMed] [Google Scholar]
- Okada-Iwabu M., Yamauchi T., Iwabu M., Honma T., Hamagami K., Matsuda K., Yamaguchi M., Tanabe H., Kimura-Someya T., Shirouzu M., et al. 2013. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature. 503:493–499 10.1038/nature12656 [DOI] [PubMed] [Google Scholar]
- Orlicky D.J., Monks J., Stefanski A.L., and McManaman J.L.. 2013. Dynamics and molecular determinants of cytoplasmic lipid droplet clustering and dispersion. PLoS ONE. 8:e66837 10.1371/journal.pone.0066837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasarica M., Sereda O.R., Redman L.M., Albarado D.C., Hymel D.T., Roan L.E., Rood J.C., Burk D.H., and Smith S.R.. 2009. Reduced adipose tissue oxygenation in human obesity: evidence for rarefaction, macrophage chemotaxis, and inflammation without an angiogenic response. Diabetes. 58:718–725 10.2337/db08-1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrinelli V., Heuvingh J., du Roure O., Rouault C., Devulder A., Klein C., Lacasa M., Clément E., Lacasa D., and Clément K.. 2014. Human adipocyte function is impacted by mechanical cues. J. Pathol. 233:183–195 10.1002/path.4347 [DOI] [PubMed] [Google Scholar]
- Pérez-Matute P., Pérez-Martínez L., Blanco J.R., and Oteo J.A.. 2013. Role of mitochondria in HIV infection and associated metabolic disorders: focus on nonalcoholic fatty liver disease and lipodystrophy syndrome. Oxid. Med. Cell. Longev. 2013:493413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilch P.F., and Liu L.. 2011. Fat caves: caveolae, lipid trafficking and lipid metabolism in adipocytes. Trends Endocrinol. Metab. 22:318–324 10.1016/j.tem.2011.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri V., Konda S., Ranjit S., Aouadi M., Chawla A., Chouinard M., Chakladar A., and Czech M.P.. 2007. Fat-specific protein 27, a novel lipid droplet protein that enhances triglyceride storage. J. Biol. Chem. 282:34213–34218 10.1074/jbc.M707404200 [DOI] [PubMed] [Google Scholar]
- Razani B., Combs T.P., Wang X.B., Frank P.G., Park D.S., Russell R.G., Li M., Tang B., Jelicks L.A., Scherer P.E., and Lisanti M.P.. 2002. Caveolin-1-deficient mice are lean, resistant to diet-induced obesity, and show hypertriglyceridemia with adipocyte abnormalities. J. Biol. Chem. 277:8635–8647 10.1074/jbc.M110970200 [DOI] [PubMed] [Google Scholar]
- Regazzetti C., Bost F., Le Marchand-Brustel Y., Tanti J.F., and Giorgetti-Peraldi S.. 2010. Insulin induces REDD1 expression through hypoxia-inducible factor 1 activation in adipocytes. J. Biol. Chem. 285:5157–5164 10.1074/jbc.M109.047688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reue K., and Dwyer J.R.. 2009. Lipin proteins and metabolic homeostasis. J. Lipid Res. 50:S109–S114 10.1194/jlr.R800052-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring A., Le Lay S., Pohl J., Verkade P., and Stremmel W.. 2006. Caveolin-1 is required for fatty acid translocase (FAT/CD36) localization and function at the plasma membrane of mouse embryonic fibroblasts. Biochim. Biophys. Acta. 1761:416–423 10.1016/j.bbalip.2006.03.016 [DOI] [PubMed] [Google Scholar]
- Rodeheffer M.S., Birsoy K., and Friedman J.M.. 2008. Identification of white adipocyte progenitor cells in vivo. Cell. 135:240–249 10.1016/j.cell.2008.09.036 [DOI] [PubMed] [Google Scholar]
- Roh C., Roduit R., Thorens B., Fried S., and Kandror K.V.. 2001. Lipoprotein lipase and leptin are accumulated in different secretory compartments in rat adipocytes. J. Biol. Chem. 276:35990–35994 10.1074/jbc.M102791200 [DOI] [PubMed] [Google Scholar]
- Samuel V.T., Liu Z.X., Qu X., Elder B.D., Bilz S., Befroy D., Romanelli A.J., and Shulman G.I.. 2004. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 279:32345–32353 10.1074/jbc.M313478200 [DOI] [PubMed] [Google Scholar]
- Scherer P.E., Lisanti M.P., Baldini G., Sargiacomo M., Mastick C.C., and Lodish H.F.. 1994. Induction of caveolin during adipogenesis and association of GLUT4 with caveolin-rich vesicles. J. Cell Biol. 127:1233–1243 10.1083/jcb.127.5.1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P., Bjork B., Yang W., Kajimura S., Chin S., Kuang S., Scimè A., Devarakonda S., Conroe H.M., Erdjument-Bromage H., et al. 2008. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 454:961–967 10.1038/nature07182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha H., He Y., Chen H., Wang C., Zenno A., Shi H., Yang X., Zhang X., and Qi L.. 2009. The IRE1α-XBP1 pathway of the unfolded protein response is required for adipogenesis. Cell Metab. 9:556–564 10.1016/j.cmet.2009.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha H., Yang L., Liu M., Xia S., Liu Y., Liu F., Kersten S., and Qi L.. 2014. Adipocyte spliced form of X-box-binding protein 1 promotes adiponectin multimerization and systemic glucose homeostasis. Diabetes. 63:867–879 10.2337/db13-1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan D., Li J.L., Wu L., Li D., Hurov J., Tobin J.F., Gimeno R.E., and Cao J.. 2010. GPAT3 and GPAT4 are regulated by insulin-stimulated phosphorylation and play distinct roles in adipogenesis. J. Lipid Res. 51:1971–1981 10.1194/jlr.M006304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd P.R., Gnudi L., Tozzo E., Yang H., Leach F., and Kahn B.B.. 1993. Adipose cell hyperplasia and enhanced glucose disposal in transgenic mice overexpressing GLUT4 selectively in adipose tissue. J. Biol. Chem. 268:22243–22246. [PubMed] [Google Scholar]
- Shigematsu S., Watson R.T., Khan A.H., and Pessin J.E.. 2003. The adipocyte plasma membrane caveolin functional/structural organization is necessary for the efficient endocytosis of GLUT4. J. Biol. Chem. 278:10683–10690 10.1074/jbc.M208563200 [DOI] [PubMed] [Google Scholar]
- Shoham N., Gottlieb R., Sharabani-Yosef O., Zaretsky U., Benayahu D., and Gefen A.. 2012. Static mechanical stretching accelerates lipid production in 3T3-L1 adipocytes by activating the MEK signaling pathway. Am. J. Physiol. Cell Physiol. 302:C429–C441 10.1152/ajpcell.00167.2011 [DOI] [PubMed] [Google Scholar]
- Shoham N., Girshovitz P., Katzengold R., Shaked N.T., Benayahu D., and Gefen A.. 2014. Adipocyte stiffness increases with accumulation of lipid droplets. Biophys. J. 106:1421–1431 10.1016/j.bpj.2014.01.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalding K.L., Arner E., Westermark P.O., Bernard S., Buchholz B.A., Bergmann O., Blomqvist L., Hoffstedt J., Näslund E., Britton T., et al. 2008. Dynamics of fat cell turnover in humans. Nature. 453:783–787 10.1038/nature06902 [DOI] [PubMed] [Google Scholar]
- Stark R., Guebre-Egziabher F., Zhao X., Feriod C., Dong J., Alves T.C., Ioja S., Pongratz R.L., Bhanot S., Roden M., et al. 2014. A role for mitochondrial phosphoenolpyruvate carboxykinase (PEPCK-M) in the regulation of hepatic gluconeogenesis. J. Biol. Chem. 289:7257–7263 10.1074/jbc.C113.544759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöckli J., Fazakerley D.J., and James D.E.. 2011. GLUT4 exocytosis. J. Cell Sci. 124:4147–4159 10.1242/jcs.097063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturley S.L., and Hussain M.M.. 2012. Lipid droplet formation on opposing sides of the endoplasmic reticulum. J. Lipid Res. 53:1800–1810 10.1194/jlr.R028290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers S.A., Garza L.A., Zhou H., and Birnbaum M.J.. 1998. Regulation of insulin-stimulated glucose transporter GLUT4 translocation and Akt kinase activity by ceramide. Mol. Cell. Biol. 18:5457–5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K., Kusminski C.M., and Scherer P.E.. 2011. Adipose tissue remodeling and obesity. J. Clin. Invest. 121:2094–2101 10.1172/JCI45887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K., Wernstedt Asterholm I., Kusminski C.M., Bueno A.C., Wang Z.V., Pollard J.W., Brekken R.A., and Scherer P.E.. 2012. Dichotomous effects of VEGF-A on adipose tissue dysfunction. Proc. Natl. Acad. Sci. USA. 109:5874–5879 10.1073/pnas.1200447109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K., Halberg N., Khan M., Magalang U.J., and Scherer P.E.. 2013a. Selective inhibition of hypoxia-inducible factor 1α ameliorates adipose tissue dysfunction. Mol. Cell. Biol. 33:904–917 10.1128/MCB.00951-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K., Tordjman J., Clément K., and Scherer P.E.. 2013b. Fibrosis and adipose tissue dysfunction. Cell Metab. 18:470–477 10.1016/j.cmet.2013.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y., Shinoda A., Furuya N., Harada E., Arimura N., Ichi I., Fujiwara Y., Inoue J., and Sato R.. 2013. Perilipin-mediated lipid droplet formation in adipocytes promotes sterol regulatory element-binding protein-1 processing and triacylglyceride accumulation. PLoS ONE. 8:e64605 10.1371/journal.pone.0064605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan G.D., Debard C., Tiraby C., Humphreys S.M., Frayn K.N., Langin D., Vidal H., and Karpe F.. 2003. A “futile cycle” induced by thiazolidinediones in human adipose tissue? Nat. Med. 9:811–812 10.1038/nm0703-811 [DOI] [PubMed] [Google Scholar]
- Tanaka N., Takahashi S., Matsubara T., Jiang C., Sakamoto W., Chanturiya T., Teng R., Gavrilova O., and Gonzalez F.J.. 2015. Adipocyte-specific disruption of fat-specific protein 27 causes hepatosteatosis and insulin resistance in high-fat diet-fed mice. J. Biol. Chem. 290:3092–3105 10.1074/jbc.M114.605980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W., Zeve D., Suh J.M., Bosnakovski D., Kyba M., Hammer R.E., Tallquist M.D., and Graff J.M.. 2008. White fat progenitor cells reside in the adipose vasculature. Science. 322:583–586 10.1126/science.1156232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanowitz H.B., Amole B., Hewlett D., and Wittner M.. 1988. Trypanosoma cruzi infection in diabetic mice. Trans. R. Soc. Trop. Med. Hyg. 82:90–93 10.1016/0035-9203(88)90272-6 [DOI] [PubMed] [Google Scholar]
- Tansey J.T., Sztalryd C., Gruia-Gray J., Roush D.L., Zee J.V., Gavrilova O., Reitman M.L., Deng C.X., Li C., Kimmel A.R., and Londos C.. 2001. Perilipin ablation results in a lean mouse with aberrant adipocyte lipolysis, enhanced leptin production, and resistance to diet-induced obesity. Proc. Natl. Acad. Sci. USA. 98:6494–6499 10.1073/pnas.101042998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tordjman J., Chauvet G., Quette J., Beale E.G., Forest C., and Antoine B.. 2003. Thiazolidinediones block fatty acid release by inducing glyceroneogenesis in fat cells. J. Biol. Chem. 278:18785–18790 10.1074/jbc.M206999200 [DOI] [PubMed] [Google Scholar]
- Turpin S.M., Nicholls H.T., Willmes D.M., Mourier A., Brodesser S., Wunderlich C.M., Mauer J., Xu E., Hammerschmidt P., Brönneke H.S., et al. 2014. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. 20:678–686 10.1016/j.cmet.2014.08.002 [DOI] [PubMed] [Google Scholar]
- Unger R.H., and Scherer P.E.. 2010. Gluttony, sloth and the metabolic syndrome: a roadmap to lipotoxicity. Trends Endocrinol. Metab. 21:345–352 10.1016/j.tem.2010.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger R.H., Scherer P.E., and Holland W.L.. 2013. Dichotomous roles of leptin and adiponectin as enforcers against lipotoxicity during feast and famine. Mol. Biol. Cell. 24:3011–3015 10.1091/mbc.E12-10-0774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hul M., Piccard H., and Lijnen H.R.. 2010. Gelatinase B (MMP-9) deficiency does not affect murine adipose tissue development. Thromb. Haemost. 104:165–171 10.1160/TH09-10-0739 [DOI] [PubMed] [Google Scholar]
- Vankoningsloo S., De Pauw A., Houbion A., Tejerina S., Demazy C., de Longueville F., Bertholet V., Renard P., Remacle J., Holvoet P., et al. 2006. CREB activation induced by mitochondrial dysfunction triggers triglyceride accumulation in 3T3-L1 preadipocytes. J. Cell Sci. 119:1266–1282 10.1242/jcs.02848 [DOI] [PubMed] [Google Scholar]
- Vergnes L., Beigneux A.P., Davis R., Watkins S.M., Young S.G., and Reue K.. 2006. Agpat6 deficiency causes subdermal lipodystrophy and resistance to obesity. J. Lipid Res. 47:745–754 10.1194/jlr.M500553-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldén T.B., Hansen I.R., Timmons J.A., Cannon B., and Nedergaard J.. 2012. Recruited vs. nonrecruited molecular signatures of brown, “brite,” and white adipose tissues. Am. J. Physiol. Endocrinol. Metab. 302:E19–E31 10.1152/ajpendo.00249.2011 [DOI] [PubMed] [Google Scholar]
- Wang Z.V., Schraw T.D., Kim J.Y., Khan T., Rajala M.W., Follenzi A., and Scherer P.E.. 2007. Secretion of the adipocyte-specific secretory protein adiponectin critically depends on thiol-mediated protein retention. Mol. Cell. Biol. 27:3716–3731 10.1128/MCB.00931-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.H., Wang C.C., and Wei Y.H.. 2010. Mitochondrial dysfunction in insulin insensitivity: implication of mitochondrial role in type 2 diabetes. Ann. NY Acad. Sci. 1201:157–165 10.1111/j.1749-6632.2010.05625.x [DOI] [PubMed] [Google Scholar]
- Wang Q.A., Tao C., Gupta R.K., and Scherer P.E.. 2013. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat. Med. 19:1338–1344 10.1038/nm.3324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Wang X., Lau W.B., Yuan Y., Booth D., Li J.J., Scalia R., Preston K., Gao E., Koch W., and Ma X.L.. 2014. Adiponectin inhibits tumor necrosis factor-α-induced vascular inflammatory response via caveolin-mediated ceramidase recruitment and activation. Circ. Res. 114:792–805 10.1161/CIRCRESAHA.114.302439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernstedt Asterholm I., Tao C., Morley T.S., Wang Q.A., Delgado-Lopez F., Wang Z.V., and Scherer P.E.. 2014. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 20:103–118 10.1016/j.cmet.2014.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson-Fritch L., Nicoloro S., Chouinard M., Lazar M.A., Chui P.C., Leszyk J., Straubhaar J., Czech M.P., and Corvera S.. 2004. Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. J. Clin. Invest. 114:1281–1289 10.1172/JCI200421752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z., Xie Y., Morrison R.F., Bucher N.L., and Farmer S.R.. 1998. PPARγ induces the insulin-dependent glucose transporter GLUT4 in the absence of C/EBPα during the conversion of 3T3 fibroblasts into adipocytes. J. Clin. Invest. 101:22–32 10.1172/JCI1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q., Ortegon A.M., Tsang B., Doege H., Feingold K.R., and Stahl A.. 2006. FATP1 is an insulin-sensitive fatty acid transporter involved in diet-induced obesity. Mol. Cell. Biol. 26:3455–3467 10.1128/MCB.26.9.3455-3467.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J., Boström P., Sparks L.M., Ye L., Choi J.H., Giang A.H., Khandekar M., Virtanen K.A., Nuutila P., Schaart G., et al. 2012. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 150:366–376 10.1016/j.cell.2012.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J.Y., Morley T.S., and Scherer P.E.. 2014. The adipokine/ceramide axis: key aspects of insulin sensitization. Biochimie. 96:130–139 10.1016/j.biochi.2013.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L., Spinas G.A., and Niessen M.. 2010. ER stress in adipocytes inhibits insulin signaling, represses lipolysis, and alters the secretion of adipokines without inhibiting glucose transport. Horm. Metab. Res. 42:643–651 10.1055/s-0030-1255034 [DOI] [PubMed] [Google Scholar]
- Yamaguchi S., Katahira H., Ozawa S., Nakamichi Y., Tanaka T., Shimoyama T., Takahashi K., Yoshimoto K., Imaizumi M.O., Nagamatsu S., and Ishida H.. 2005. Activators of AMP-activated protein kinase enhance GLUT4 translocation and its glucose transport activity in 3T3-L1 adipocytes. Am. J. Physiol. Endocrinol. Metab. 289:E643–E649 10.1152/ajpendo.00456.2004 [DOI] [PubMed] [Google Scholar]
- Ye J., Gao Z., Yin J., and He Q.. 2007. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am. J. Physiol. Endocrinol. Metab. 293:E1118–E1128 10.1152/ajpendo.00435.2007 [DOI] [PubMed] [Google Scholar]
- Ye R., Holland W.L., Gordillo R., Wang M., Wang Q.A., Shao M., Morley T.S., Gupta R.K., Stahl A., and Scherer P.E.. 2014. Adiponectin is essential for lipid homeostasis and survival under insulin deficiency and promotes β-cell regeneration. eLife. 3 10.7554/eLife.03851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan T., Hong S., Yao Y., and Liao K.. 2007. Glut-4 is translocated to both caveolae and non-caveolar lipid rafts, but is partially internalized through caveolae in insulin-stimulated adipocytes. Cell Res. 17:772–782 10.1038/cr.2007.73 [DOI] [PubMed] [Google Scholar]
- Zechner R., Strauss J.G., Haemmerle G., Lass A., and Zimmermann R.. 2005. Lipolysis: pathway under construction. Curr. Opin. Lipidol. 16:333–340 10.1097/01.mol.0000169354.20395.1c [DOI] [PubMed] [Google Scholar]
- Zeve D., Tang W., and Graff J.. 2009. Fighting fat with fat: the expanding field of adipose stem cells. Cell Stem Cell. 5:472–481 10.1016/j.stem.2009.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]