

Abstract
REV-ERBα has emerged as an important target for regulation of circadian rhythm and its associated physiology. Herein, we report on the optimization of a series of REV-ERBα agonists based on GSK4112 (1) for potency, selectivity, and bioavailability. Potent REV-ERBα agonists 4, 10, 16, and 23 are detailed for their ability to suppress BMAL and IL-6 expression from human cells while also demonstrating excellent selectivity over LXRα. Amine 4 demonstrated in vivo bioavailability after either IV or oral dosing.
Introduction
The circadian clock aligns all the tissues in most organisms with the day and night cycle of our planet. Through a transcriptional mechanism, the clock controls many important biological pathways, such as metabolism, inflammation, and sleep-wake cycles.1-3 REV-ERBα is a nuclear receptor that has been demonstrated to, upon activation by heme, form a complex with co-factors that represses the transcription of target genes.4,5 REV-ERBα is at the heart of the circadian clock and is a mechanism by which the circadian clock gates inflammatory response and controls the metabolic state of the organism.6-9 Investigation into the function of REV-ERBα will provide further insights into circadian biology and may identify the receptor as a therapeutic target for a variety of diseases.
Pharmacological investigations into the biological role of the REV-ERBα have used the sub-optimal chemical probe GSK4112 (1) and, its analogs SR9011 (2) and SR9009 (3) (Figure 1).10,11 All three compounds are closely related by a common tertiary amine core and two of three identical substituents off the amine. Use of these compounds to interrogate REV-ERBα biology is complicated by high metabolic clearance rates that necessitate high dosing to achieve meaningful levels of exposure in vivo. In addition, the tertiary amine chemotype has known activity on the nuclear receptor LXRα, a potential liability for interpretation of results from cell-based and animal pharmacology.12-14 Herein we describe our optimization of the tertiary amine series of REV-ERBα agonists to address these liabilities and allow further exploration of the role of REV-ERBα in circadian control and inflammation.
Figure 1.

Reported REV-ERBα agonists 1-3.
Results and Discussion
To deliver a tool suitable for in vivo studies we evaluated the literature compounds 1-3 to assess opportunities for improvement. First we assessed the ability of the literature compounds 1-3 to bind to both REV-ERBα and LXRα. The tertiary amine 1 induced recruitment of co-factor NCOR peptide fragment to purified REV-ERBα protein, indicating that the compound binds to the protein and induces a conformational change (Table 1). Interestingly, neither compounds 2 or 3 increased recruitment of the NCOR peptide fragment to REV-ERBα in vitro. Amines 2 and 3 were reported to have other activities associated with REV-ERBα activation, so we investigated the compounds ability to activate the receptor by a different mechanism.11 To determine if these two compounds were recruiting a different set of peptides to the REV-ERBα protein, we performed a scan of co-factor derived peptide fragments recruited to REV-ERBα in the presence of amine 1 (See Supporting Information Figure 1). Aside from NCOR and SMRT, the only other tested peptide that showed significant interaction with the REV-ERBα was a fragment of PGC1-β. Full curve analysis of the ability of compounds 1-3 demonstrated that the compounds were dose-dependently able to induce recruitment of PGC1-β peptide to REV-ERBα (Figure 2). This result demonstrated that amines 2 and 3 do bind to the REV-ERBα protein to induce a conformational change and that the induced conformation is different from that induced by amine 1. These data raise the intriguing possibility that the different peptide recruitment profiles of the compounds may result in distinct biological activities. They also indicate that PGC1-β may have a yet uninvestigated role in REV-ERBα pharmacology. The investigation of these possibilities is beyond the scope of this paper.
Table 1.
REV-ERBα and LXRα activity of GSK4112 analogs.
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | cLogP | REV-ERBαEC50a | LXRα | BMAL1 | ||
|
| |||||||||
| IC50(μM) | Fold Sel. | Supp.b | Phase Delayc | ||||||
| 1 | tBuO2C- | 4-Cl-C6H4- | 5-Nitro-thiophen-2-yl | 5.2 | 0.50 | 5.0 | 10 | 6% | 0 |
| 2 | 1-Ethylcarboxylate-pyrrolidin-3-yl | 4-Cl-C6H4- | 5-Nitro-thiophen-2-yl | 4.8 | >50 | 6.3 | 0.13 | 20% | 1.3 |
| 3 | 1-N-Pentylcarboxamide-pyrrolidin-3-yl | 4-Cl-C6H4- | 5-Nitro-thiophen-2-yl | 5.3 | >50 | 13 | 0.25 | 14% | 1.3 |
| 4 | 4-Cl-2-Me-C6H3- | 4-Cl-C6H4- | 5-Nitro-thiophen-2-yl | 6.8 | 0.050 | 63 | 1,259 | 21% | 0 |
| 5 | Ph- | 4-Cl-C6H4- | 5-Nitro-thiophen-2-yl | 5.6 | 0.10 | >100 | >1,000 | NT | NT |
| 6 | 2-F-C6H4- | 4-Cl-C6H4- | 5-Nitro-thiophen-2-yl | 5.7 | 0.063 | 20 | 320 | 38% | 0 |
| 7 | iso-Butyl- | 4-Cl-C6H4- | 5-Nitro-thiophen-2-yl | 5.9 | 0.16 | 16 | 100 | 21% | 0 |
| 8 | 3-Pyridyl- | 4-Cl-C6H4- | 5-Nitro-thiophen-2-yl | 4.1 | 0.16 | 13 | 79 | NT | NT |
| 9 | 3-Pyridyl- | 4-Me-C6H4- | 5-Nitro-thiophen-2-yl | 3.9 | 0.16 | 40 | 250 | 23% | 0 |
| 10 | 3-Pyridyl- | 4-F-C6H4- | 5-Nitro-thiophen-2-yl | 3.5 | 0.16 | 40 | 250 | 14% | 0 |
| 11 | 3-Pyridyl- | 2-Me-C6H4- | 5-Nitro-thiophen-2-yl | 3.8 | 0.20 | 32 | 160 | 37% | 0 |
| 12 | 3-Pyridyl- | 3-Me-C6H4- | 5-Nitro-thiophen-2-yl | 3.9 | 0.20 | 25 | 130 | 41% | 0.8 |
| 13 | 3-Pyridyl- | 4-OMe-C6H4- | 5-Nitro-thiophen-2-yl | 3.3 | 0.20 | 32 | 160 | NT | NT |
| 14 | 3-Pyridyl- | 3-OMe-C6H4- | 5-Nitro-thiophen-2-yl | 3.3 | 0.40 | 10 | 25 | 42% | 0.5 |
| 15 | 3-Pyridyl- | 3-CF3-C6H4- | 5-Nitro-thiophen-2-yl | 4.3 | 0.40 | 16 | 40 | NT | NT |
| 16 | 3-Pyridyl- | 4-Cl-C6H4- | 5-Nitrile-thiophen-2-yl | 3.8 | 0.20 | 20 | 100 | 23% | 0 |
| 17 | 3-Pyridyl- | 4-Cl-C6H4- | 4-Br-5-Me-thiophen-2-yl | 5.7 | 0.25 | 32 | 130 | 16% | 0 |
| 18 | 3-Pyridyl- | 4-Cl-C6H4- | Benzo[b]thiophen-2-yl | 5.7 | 0.40 | 40 | 100 | 24% | 0 |
| 19 | 3-Pyridyl- | 4-Cl-C6H4- | 3-(1H-Pyrrol-1-yl)thiophen-2-yl | 5.5 | 0.63 | 40 | 63 | 30% | 0 |
| 20 | 3-Pyridyl- | 4-Cl-C6H4- | Thiazol-2-yl | 3.0 | >50 | 50 | <1 | NT | NT |
| 21 | 3-Pyridyl- | 4-Cl-C6H4- | 1H-Imidazol-2-yl | 2.5 | >50 | >100 | NT | NT | NT |
| 22 | 3-Pyridyl- | 4-F-C6H4- | 5-Nitrile-thiophen-2-yl | 3.2 | 0.20 | NT | NT | NT | NT |
| 23 | 3-Pyridyl- | 3,4-F2-C6H3- | 5-Nitrile-thiophen-2-yl | 3.3 | 0.20 | 16 | 79 | 37% | 0 |
| 24 | 3-Pyridyl- | 4-OMe-C6H4- | 5-Nitrile-thiophen-2-yl | 3.0 | 0.63 | >100 | >160 | NT | NT |
| 25 | 3-Pyridyl- | 2,5-F2-C6H3- | 5-Nitrile-thiophen-2-yl | 3.4 | 0.63 | 6.3 | 10 | 27% | 0 |
| 26 | 3-Pyridyl- | 2-F-C6H4- | 5-Nitrile-thiophen-2-yl | 3.3 | >50 | 32 | <1 | 6% | 0 |
All active compounds demonstrated a 30% – 60% increase in NCOR peptide recruitment.
Suppression of BMAL1 expression after 40 h of 20 μM compound treatment.
Delay of peak of second cycle in hours of 20 μM treated cells relative to DMSO treated cells.
Figure 2.

Effect of compounds 1-3 on PGC1-β peptide fragment recruitment to REV-ERBα.
Each of the compounds 1-3 was able to displace a radio ligand from the LXRα binding site, confirming the compounds bind to the LXRα at ~10 μM (Table 1). We investigated if the observed binding of the compounds to both REV-ERBα and LXRα induced related effects in a living cell. To investigate REV-ERBα cellular efficacy, we measured the ability of agonists 1 and 2 to inhibit IL-6 production from human THP-1 cells following LPS stimulation (Figure 3a). To assess the cellular activity of the compounds in an LXRα-dependent system, we compared the effects of the compounds 1 and 2 on expression of ABCA1 in THP-1 cells (Figure 3b). Both compounds were able to significantly inhibit LPS-stimulated IL-6 expression and up-regulate ABCA1 levels indicating significant cellular activity of each compound in both REV-ERBα and LXRα dependent phenotypes that correlated with the ability of the compounds to bind to target proteins. The original report on compound 2 described a lack of activity in an LXRα Gal4 reporter gene assay,11 but our results indicated that the compounds bound directly to LXRα and induced cellular affects through LXRα activation. As activation of LXRα has been reported to have diverse effects on multiple pharmacological pathways, including inflammation, cholesterol metabolism, and insulin resistance, a key goal for this project was to improve the selectivity profile for the series.12-14
Figure 3.

THP-1 cells were stimulated with LPS 10 μg/mL in the presence of DMSO, 1 (10 μM) or compound 2 (10 μM). After six hours the cells were lysed and the RNA analyzed using RT-qPCR. Both compounds significantly repressed IL-6 but increased ABCA1 transcript expression (**<0.01, T test). A representative experiment of three experiments is shown.
In addition to the observed off-target activity, compounds 1- 3 have relatively high cLogP values of > 5 that may contribute to off-target binding, particularly with nuclear receptors, and the high clearance observed in reported mouse DMPK studies.15
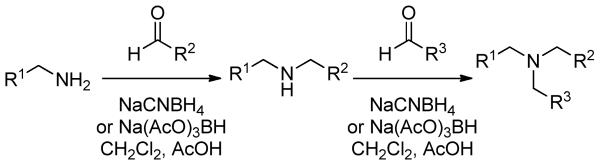
The goal of our synthetic efforts was therefore to provide an effective and readily accessible in vivo probe for REV-ERBα that could be used as an efficient tool to explore the biological profile of this fascinating receptor. To improve on the significant short falls of 1-3 it was crucial to increase selectivity over LXRα to allow clear and precise interpretations of subsequent data and secondarily reduce the lipophilicity of the compounds. Preparation of the target tertiary amines was through a series of reductive amination procedures (Scheme 1). The overall process could be carried out under ambient reaction conditions or accelerated through microwave irradiation. It also proved possible to carry out the entire reaction process through a one-pot two-step protocol allowing rapid access to range of tertiary amine scaffolds for evaluation. Pertinent data in this ligand development on a selection of the amines prepared are collected in Table 1.
Scheme 1. Synthesis of tertiary amine compounds reported.
Initial efforts focussed on replacing the tert-butyl ester arm (R1) of compound 1. Alternative substitution at this position provided multiple options for improved REV-ERBα activity and LXRα selectivity, but often at the cost of an increase in compound cLogP (Table 1), however, significant improvements in the overall compound profile proved possible. Replacement of the tert-butyl ester with both substituted benzyl (4-6) and alkyl (7) groups was not only well tolerated but led to stunning improvements in LXRα selectivity. Although many of these changes resulted in an increase in cLogP, it showed that LXRα activity could ultimately be removed from this tertiary amine series through simple structural modification. In order to address the lipophilicity issue a variety of heterocyclic substituents were examined in this position, the vast majority of which were not tolerated (data not shown), however, introduction of a 3-pyridyl group (e.g. 8) provided a modest increase in REV-ERBα activity (EC50 = 0.16), significant improvement in LXRα selectivity (79 fold), and importantly a meaningful lowering in cLogP (4.1).
Using compound 8 as the basis for continued exploration of scaffold modification we examined variation of the 4-chlorobenzyl arm (R2) of compound 1. In contrast to substitution of R1, both alkyl chains and heterocyclic substituents were not tolerated in this position with all changes resulting in compounds with no significant activity at REV-ERBα (data not shown). It was possible to replace this substituent with a variety substituted benzyl groups (e.g. 9-15) that provided multiple options that maintained REV-ERBα activity and concurrently improved LXRα selectivity and reduced cLogP. For example, introduction of a 4-fluorobenzyl unit (10) lowered the cLogP significantly (3.5) and improved selectivity over LXRα to >250 fold. Other changes at R2 such as the introduction of a 4-methoxybenzyl substituent (13) provided a similar and substantial improvement in overall compound profile.
In parallel to these modifications, an array of compounds was prepared replacing the nitro-substituent on the thiophene ring (R3). Interestingly, the majority of replacements for the nitrothiophene ring system with alternative polar 5-membered ring heterocycles as exemplified by compounds 20 and 21, resulted in complete loss of activity in the primary NCOR peptide recruitment assay. It did prove possible to introduce different substituents to the thiophene ring and maintain REV-ERBα potency and improve LXRα selectivity as exemplified by compounds 17 and 18, however, these changes often proved detrimental to the cLogP. One crucial exception to this trend was introduction of the 5-nitrile thiophene in compound 16, which not only maintained REV-ERBα selectivity (EC50 = 0.2) but also resulted in a significant drop in cLogP (3.8) and a slight improvement in LXRα selectivity (100 fold).
Combination of the optimal substituents discovered for R1, R2, and R3 resulted in the tertiary amines 22-26 which on evaluation revealed the key compound 23 which maintained potency in the NCOR peptide recruitment assay and also provided the desired LXRα selectivity profile and reduced lipophilicity.
Initially, to confirm the ability of the compounds to induce REV-ERBα-driven phenotypes in a cellular system, profiling for the ability to affect the circadian expression of BMAL in synchronized U2OS cells was examined. By stably expressing a BMAL promoter-driven luciferase reporter in U2OS cells, we were able to capture real time bioluminescent oscillations of synchronized cells. A single endpoint assay of BMAL-luciferase suppression by REV-ERBα agonists (including compound 1) has been described in the literature.16 We confirmed that comparable data (a statistically significant 15% suppression) can be generated with compound 1 through our in-house U2OS reporter assay. Further, we showed that the natural ligand for REV-ERBα (heme), produced a >50% suppression in the same assay (data not shown). A representative data set from the assay for amine 4 is shown in Figure 5. Additional data from the assay are also shown in Table 1 and Supporting Information Figure 2. As can be seen, the increase in potency for recruitment of the NCOR peptide promoted by compound 4 correlates with the suppression and shift of the BMAL oscillation curve in a dose-dependent manner.
Figure 5.

Oscillation of BMAL-Luc gene over time in synchronized U2OS cells with and without treatment with compound 4.
During the evolution of this SAR knowledge, compounds 4, 10, 16, and 23 (Figure 4) were selected for further profiling and evaluation of their suitability to be adopted as in vivo probes for REV-ERBα.
Figure 4.

Key compounds selected during study for further evaluation as potential probes.
To assess the cellular efficacy of REV-ERBα activation and selectivity over a LXRα driven pathway, amines 4, 10, 16, and 23 were profiled for their ability to inhibit LPS induction of IL-6 production and to upregulate expression of ABCA1 in human THP-1 cells. All compounds demonstrated a significant reduction of IL-6 secretion following treatment with 4, 10, 16, and 23 at 1 μM with no measurable effect on ABCA1 levels (Figure 6). Additionally, none of the compounds showed any toxicity in measurement of ATP levels in THP-1 cells (See Supporting Information Figure 3). These data indicated that each of these tertiary amines was able to potently activate REV-ERBα in cells, that suitable selectivity over LXRα had been achieved, and that no general toxicity was skewing the results.
Figure 6.

THP-1 cells were stimulated with LPS 10 mcg/mL in the presence of DMSO or compounds 4, 10, 16, 23 (1 μM), in triplicate. After six hours the cells were lysed and the RNA analyzed using RT-qPCR. All compounds repressed IL-6 without affecting ABCA1 transcript expression (*=p<0.05,**<0.01, One way ANOVA with post-hoc Dunnett’s).
The IV and oral DMPK profiles of compounds 3, 4, 10, 16, and 23 were evaluated in a 1 mg/Kg cassette dose experiment in C57Bl/6 mice. Four of the compounds demonstrated essentially identical profiles with short half-lives, high clearance, and low oral bioavailability. Compound 4 (GSK2945) was differentiated from the group with a longer half-life of 2.0 h and an oral bioavailability of 23%, despite the higher cLogP of the compound (Table 2 and Supporting Information Figure 4). The profile of amine 4 is suitable to achieve plasma concentrations around the IC50 of the compound with oral doses around 20-30 mg/Kg, a dosing regimen that is compatible with long term in vivo studies. The remaining four compounds (3, 10, 16, 23) would be suitable for acute time-of-day dosing by injection where short exposure of the compound at meaningful levels in desired. The specific advantage of 10, 16, and 23 over the previously reported tertiary amine 3 is the selectivity profile observed for REV-ERBα over LXRα.
Table 2.
Pharmokinetic Parameters.
| Dose Route (1 mg/Kg) | PK Parameter | 3 | 4 | 10 | 16 | 23 |
|---|---|---|---|---|---|---|
| IV | CL (L/h/Kg) | 2.7 | 0.4 | 2.8 | 2.8 | 2.6 |
| Vss (L/Kg) | 1.6 | 0.3 | 1.5 | 1.8 | 1.4 | |
| Terminal t1/2 (h) | 0.55 | 1.65 | 0.74 | 1.27 | 0.74 | |
| AUClast(h*ng/mL) | 365 | 2,538 | 354 | 361 | 375 | |
|
| ||||||
| Oral | Tmax (h) | 0.50 | 2.0 | 0.25 | 0.25 | 0.25 |
| Cmax (ng/mL) | 7.41 | 180 | 8.98 | 9.14 | 9.94 | |
| AUClast(h*ng/mL) | 7.93 | 594 | 8.39 | 10.2 | 9.39 | |
| F (%) | 2.2 | 23.4 | 2.4 | 3.5 | 3.0 | |
Conclusion
Herein we report on the discovery of REV-ERBα agonists from the GSK4112 series that demonstrated > 1,000 fold selectivity over LXRα and have improved DMPK properties. In addition, we have identified PGC1-β as a novel co-factor for REV-ERBα and have noted that reported ligands 1-3 recruit different co-factor peptides to REV-ERBα and may induce distinct pharmacology as a result. GSK2945 (4) is a potent agonist with a profile suitable for chronic in vivo dosing via both oral and IV routes. Other compounds (10, 16, and 23) are identified with a shorter half-life in rodents that could be used for acute time-of-day dosing studies. These compounds represent valuable additions to the pharmacological toolbox to investigate the biology of REV-ERBα without the complication of activity on LXRα.
Experimental Section
General
All chemical reagents were purchased and used as received. 1H NMR spectra were recorded on a Varian Unity-300 or Varian Unity Plus-400. Chemical shifts are expressed in parts per million (ppm, δ units). Coupling constants are in units of hertz (Hz). Splitting patterns describe apparent multiplicities and are designated as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), or br (broad).
Unless otherwise noted, the LCMS system used to determine purity was a UPLC analysis was conducted on a Waters Acquity system with BEH C18, 2 × 50 mm, 1.7 micron column at 40 °C 95% H2O, 5% MeCN to 99% MeCN in 1.1 minutes, holding at 100% MeCN for 40 seconds. Water contained 0.2% v/v formic acid. MeCN contained 0.15% v/v formic acid. The flow rate was 1 mL/min with 5 μL of solution injected. Mass spectra were recorded utilizing electrospray ionization (ESI) or atmospheric pressure chemical ionization (APCI) switching between positive and negative modes with DAD scanning from 210 to 350 nm. All compounds tested were of ≥95% purity.
Normal phase chromatography was accomplished using either Isco or Biotage equipment using pre-packed silica columns.
Reverse phase HPLC was accomplished using Agilent 110 series prep HPLC systems using C18 Phenomenex Luna, 75 × 30 mm, 5-micron column using the gradient described. The flow rate was 70 mL/min and the product was collected based on UV detection at 220 or 254 nm.
N-(4-Chlorobenzyl)-1-(5-nitrothiophen-2-yl)methanamine hydrochloride salt (27)
4-Chlorobenzylamine (5.41 g, 38.2 mmol) and 5-carboxaldehyde-2-nitrothiophene (2.00 g, 12.7 mmol) were dissolved in the anhydrous THF (80 mL) and acetic acid (20 mL) to give a clear solution. The reaction was stirred at RT for 1 h. Sodium triacetoxyborohydride (3.24 g, 15.3 mmol) was added, and the reaction was stirred at RT for 1 h. The reaction was concentrated to an oil and dissolved in 250 mL EtOAc. The organic solution was washed 2 × 100 mL NaOH and 1 × 100 mL brine, dried with magnesium sulfate, and concentrated to give a red oil. The oil was purified via normal phase silica gel chromatography (2-10% MeOH in CH2Cl2 with 0.5% DIEA) to give a red oil. The oil was taken up in ether and washed 3 × 50 mL 1N NaOH and dried. 4N HCl in dioxane (6 mL) was added to the ether to give a white solid. The solid was collected by filtration to give the title compound (2.40 g, 7.4 mmol, 58%) as a white solid. 1H NMR (400 MHz, chloroform-d) δ 10.16 (br. s., 2H), 8.11 (d, J = 4.31 Hz, 1H), 7.56-7.69 (m, 2H), 7.38-7.56 (m, 3H), 4.47 (br. s., 2H), 4.18 (br. s., 2H). NMR (DEPTQ-135) (101 MHz, CHLOROFORM-d) δ 155.2, 150.5, 137.9, 133.1, 129.5, 128.9, 128.7, 123.5, 52.4, 48.0. HRMS (APCI+) C12H10O2N2Cl1S1H+ requires m/z 283.0303, found 283.0306. LCMS: tR = 0.53 min, 100%. MS m/z = 283 (M + H)+.
General Procedure for reductive aminations Method A (used for compounds 5-16 and 25-26). N-(4-chloro-2-methylbenzyl)-N-(4-chlorobenzyl)-1-(5-nitrothiophen-2-yl)methanamine hydrochloride salt (4)
MP-Sodium cyanoborohydride (200 mg, 400 μmol) was weighed into a 5 mL microwave reaction vessel. Compound 27 (56 mg, 200 μmol) and 4-chloro-2-methylbenzaldehyde (34 mg, 220 μmol) were dissolved in CH2Cl2 (2 mL), ethanol (2 mL), and acetic acid (0.8 mL) then added to the reaction vessel. The reaction was heated to 120 °C for 10 min using microwave irradiation. The crude reaction was filtered, concentrated to an oil, dissolved in DMSO, and purified by reverse phase prep-HPLC (20%-100% MeCN in water with 0.5% TFA) to give an oil. The oil was dissolved in CH2Cl2 and treated with 300 mg of MP-carbonate resin and filtered. The filtrate was treated with 2 mL of 1N HCl in MeOH and concentrated to give the title compound (27 mg, 59.0 μmol, 28%) as a white solid. 1H NMR (400 MHz, methanol-d4) δ 7.81 (d, J = 4.02 Hz, 1H), 7.29-7.41 (m, 5H), 7.10-7.19 (m, 2H), 6.95 (d, J = 4.02 Hz, 1H), 3.74 (s, 2H), 3.60 (s, 2H), 3.58 (s, 2H), 2.26 (s, 3H). 13C NMR (101 MHz, methanol-d4) δ 155.3, 152.0, 140.9, 138.5, 136.5, 134.4, 134.2, 132.5, 131.9, 131.3, 130.1, 129.7, 127.0, 126.4, 59.0, 56.9, 53.9, 19.6. LCMS: tR = 1.32 min, 100%. MS m/z = 422 (M + H)+. HRMS calcd. for C20H18 35Cl2N2O2S (M + H)+ 421.0539, found 421.0544.
N-Benzyl-N-(4-chlorobenzyl)-1-(5-nitrothiophen-2-yl)methanamine (5)
General Method A was used to give the title compound (27 mg, 59 μmol, 28%) as a white solid using 4-chloro-2-methylbenzaldehyde and N-(4-chlorobenzyl)-1-(5-nitrothiophen-2-yl)methanamine hydrochloride salt. 1H NMR (400 MHz, chloroform-d) δ 7.79 (d, J = 4.10 Hz, 1H), 7.28-7.50 (m, 10H), 6.91 (br. s., 1H), 3.78 (br. s., 2H), 3.66 (d, J = 9.63 Hz, 4H). LCMS: tR = 1.21 min, 100%. MS m/z = 373 (M + H)+. HRMS calcd. for C19H1835ClN2O2S (M + H)+ 373.0778, found 373.0780.
N-(4-Chlorobenzyl)-N-(2-fluorobenzyl)-1-(5-nitrothiophen-2-yl)methanamine hydrochloride salt (6)
General Method A was used to give the title compound (20 mg, 47.0 μmol, 22%) as a white solid using 2-fluorobenzaldehyde and N-(4-chlorobenzyl)-1-(5-nitrothiophen-2-yl)methanamine hydrochloride salt. 1H NMR (400 MHz, methanol-d4) δ 7.82 (d, J = 4.27 Hz, 1H), 7.51 (t, J = 7.03 Hz, 1H), 7.38-7.44 (m, 2H), 7.24-7.36 (m, 3H), 7.12-7.22 (m, 1H), 7.07 (t, J = 9.29 Hz, 1H), 6.98 (d, 1H), 3.79 (s, 2H), 3.72 (s, 2H), 3.64 (s, 2H). 13C NMR (101 MHz, methanol-d4) δ 155.6, 138.8, 134.3, 132.5, 131.5, 130.6, 130.6, 130.1, 129.7, 126.3, 126.1, 125.5, 116.6, 116.4, 58.6, 53.9, 51.9. LCMS: tR = 1.25 min, 100%. MS m/z = 391 (M + H)+. HRMS calcd. for C19H1735ClFN2O2S (M + H)+ 391.0683, found 391.0684.
N-(4-Chlorobenzyl)-3-methyl-N-((5-nitrothiophen-2-yl)methyl)butan-1-amine hydrochloride salt (7)
General Method A was used to give the title compound (13 mg, 36.0 μmol, 18%) as a white solid using 3-methylbutanal and N-(4-chlorobenzyl)-1-(5-nitrothiophen-2-yl)methanamine hydrochloride salt. 1H NMR (400 MHz, chloroform-d) δ 8.11 (br. s., 1H), 7.36-7.79 (m, 5H), 4.14-4.96 (m, 3H), 3.01-3.17 (m, 1H), 2.91 (br. s., 1H), 1.30-1.91 (m, 3H), 1.20 (t, J = 7.24 Hz, 1H), 0.51-0.98 (m, 6H). 13C NMR (101 MHz, METHANOL-d4) d 163.0, 155.6, 138.8, 134.3, 132.5, 132.5, 131.5, 130.6, 130.6, 130.1, 129.7, 126.1, 125.5, 116.5, 58.6, 53.9, 51.9. LCMS: tR = 1.13 min, 100%. MS m/z = 353 (M + H)+. HRMS calcd. for C17H2235ClN2O2S (M + H)+ 353.1091, found 353.1091.
N-(4-Chlorobenzyl)-1-(5-nitrothiophen-2-yl)-N-(pyridin-3-ylmethyl)methanamine hydrochloride salt (8)
General Method A was used to give the title compound (27 mg, 59 μmol, 28%) as a white solid using 3-pyridylcarbaldehyde and N-(4-chlorobenzyl)-1-(5-nitrothiophen-2-yl)methanamine hydrochloride salt. 1H NMR (400 MHz, chloroform-d) δ 8.77 (d, J = 1.37 Hz, 1H), 8.67 (dd, J = 1.17, 5.27 Hz, 1H), 8.25 (d, J = 7.81 Hz, 1H), 7.80 (d, J = 4.10 Hz, 1H), 7.71 (dd, J = 5.37, 7.91 Hz, 1H), 7.28-7.37 (m, 4H), 6.92 (d, J = 4.10 Hz, 1H), 3.80 (s, 2H), 3.77 (s, 2H), 3.65 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 154.3, 149.6, 146.6, 145.8, 137.0, 135.6, 132.0, 132.0, 130.5, 130.3, 128.5, 125.9, 125.0, 56.6, 54.2, 52.2. LCMS: tR = 0.81 min, 95%. MS m/z = 374 (M + H)+. HRMS calcd. for C18H1735ClN2O2S (M + H)+ 374.0730, found 374.0731.
1-(5-Nitrothiophen-2-yl)-N-(pyridin-3-ylmethyl)methanamine (28)
The 3-aminomethylpyridine (1.50 g, 13.87 mmol) and 5-carboxaldehyde-2-nitrothiophene (2.00 g, 12.73 mmol) were dissolved in the anhydrous THF (40 mL) and acetic acid (10.00 mL) to give a clear solution. The reaction was stirred at room temperature for 30 min. Sodium triacetoxyborohydride (3.24 g, 15.27 mmol) was added, and the reaction was stirred for 1 h at RT. The reaction was concentrated to an oil which was purified via normal phase silica gel chromatography (2-10% MeOH in CH2Cl2) to give the title compound (1.40 g, 4.53 mmol, 36%) as a brown oil. 1H NMR (400 MHz, chloroform-d) δ 8.55 (s, 1H), 8.37-8.51 (m, 1H), 8.01 (d, J = 4.11 Hz, 1H), 7.78 (d, J = 7.83 Hz, 1H), 7.37 (dd, J = 4.80, 7.54 Hz, 1H), 7.10 (d, J = 4.31 Hz, 1H), 3.95 (s, 2H), 3.79 (s, 2H). 13C NMR (DEPTQ-135) (101 MHz, CHLOROFORM-d) δ 154.6, 150.6, 149.7, 149.0, 135.9, 134.8, 128.9, 123.6, 123.6, 50.5, 48.1. HRMS (APCI+) C11H10O2N3S1H+ requires m/z 250.0645, found 250.0641. LCMS: tR = 0.27 min, 100%. MS m/z = 250 (M + H)+.
N-(4-Methylbenzyl)-1-(5-nitrothiophen-2-yl)-N-(pyridin-3-ylmethyl)methanamine hydrochloride salt (9)
General Method A was used to give the title compound (35 mg, 75 μmol, 32%) as a white solid using 4-methylbenzaldehyde and 1-(5-nitrothiophen-2-yl)-N-(pyridin-3-ylmethyl)methanamine. 1H NMR (400 MHz, methanol-d4) δ 8.71 (s, 1H), 8.62 (d, J = 5.48 Hz, 1H), 8.43 (d, J = 7.83 Hz, 1H), 7.77-7.90 (m, 2H), 7.25 (d, J = 7.63 Hz, 2H), 7.12 (d, J = 7.63 Hz, 2H), 7.02 (d, 1H), 3.88 (s, 2H), 3.83 (s, 2H), 3.66 (s, 2H), 2.27 (s, 3H). LCMS: tR = 0.82 min, 100%. MS m/z = 354 (M + H)+. 13C NMR (101 MHz, DMSO-d6) δ 154.7, 151.8, 149.8, 146.6, 145.6, 136.6, 134.7, 130.3, 130.0, 129.3, 129.1, 128.7, 125.8, 57.0, 54.1, 52.1, 20.7. HRMS calcd. for C19H20N3O2S (M + H)+ 354.1276, found 354.1278.
N-(4-Fluorobenzyl)-1-(5-nitrothiophen-2-yl)-N-(pyridin-3-ylmethyl)methanamine hydrochloride salt (10)
General Method A was used to give the title compound (27 mg, 57 μmol, 24%) as a white solid using 4-fluorobenzaldehyde and 1-(5-nitrothiophen-2-yl)-N-(pyridin-3-ylmethyl)methanamine. 1H NMR (400 MHz, methanol-d4) δ 8.55 (d, 1H), 8.40-8.46 (m, 1H), 7.91 (d, J = 7.78 Hz, 1H), 7.83 (d, J = 4.02 Hz, 1H), 7.42 (dd, J = 5.40, 8.16 Hz, 3H), 7.07 (t, J = 8.78 Hz, 2H), 7.00 (d, J = 4.02 Hz, 1H), 3.81 (s, 2H), 3.70 (s, 2H), 3.65 (s, 2H). 13C NMR (101 MHz, methanol-d4) δ 165.0, 162.6, 155.2, 150.6, 149.3, 138.9, 136.6, 135.6, 131.9, 130.2, 126.3, 125.4, 116.4, 58.5, 56.3, 53.9. LCMS: tR = 0.77 min, 100%. MS m/z = 358 (M + H)+. HRMS calcd. for C17H17FN3O2S (M + H)+ 358.1026, found 358.1028.
N-(2-Methylbenzyl)-1-(5-nitrothiophen-2-yl)-N-(pyridin-3-ylmethyl)methanamine hydrochloride salt (11)
General Method A was used to give the title compound (31 mg, 66 μmol, 28%) as a white solid using 2-methylbenzaldehyde and 1-(5-nitrothiophen-2-yl)-N-(pyridin-3-ylmethyl)methanamine. 1H NMR (400 MHz, methanol-d4) δ 8.61 (s, 1H), 8.57 (d, J = 5.29 Hz, 1H), 8.35 (d, J = 8.03 Hz, 1H), 7.74-7.87 (m, 2H), 7.29-7.37 (m, 1H), 7.04-7.15 (m, 3H), 7.02 (d, 1H), 3.92 (s, 2H), 3.85 (s, 2H), 3.73 (s, 2H), 2.30 (s, 3H). LCMS: tR = 0.79 min, 100%. MS m/z = 354 (M + H)+. HRMS calcd. for C19H20N3O2S (M + H)+ 354.1276, found 354.1278.
N-(3-Methylbenzyl)-1-(5-nitrothiophen-2-yl)-N-(pyridin-3-ylmethyl)methanamine hydrochloride salt (12)
General Method A was used to give the title compound (34 mg, 73 μmol, 31%) as a white solid using 3-methylbenzaldehyde and 1-(5-nitrothiophen-2-yl)-N-(pyridin-3-ylmethyl)methanamine. 1H NMR (400 MHz, chloroform-d) δ 8.71 (s, 1H), 8.61 (d, J = 5.48 Hz, 1H), 8.44 (d, J = 7.83 Hz, 1H), 7.81-7.91 (m, 2H), 7.13-7.23 (m, 3H), 6.96-7.09 (m, 2H), 3.89 (s, 2H), 3.85 (s, 2H), 3.67 (s, 2H), 2.30 (s, 3H). 13C NMR (101 MHz, methanol-d4) δ 155.5, 151.0, 150.6, 149.2, 139.5, 139.2, 138.8, 136.7, 130.8, 130.2, 129.7, 129.4, 127.1, 126.2, 125.4, 59.4, 56.4, 54.0, 21.6. LCMS: tR = 0.81 min, 100%. MS m/z = 354 (M + H)+. HRMS calcd. for C19H20N3O2S (M + H)+ 354.1271, found 354.1271.
N-(4-Methoxybenzyl)-1-(5-nitrothiophen-2-yl)-N-(pyridin-3-ylmethyl)methanamine hydrochloride salt (13)
General Method A was used to give the title compound (24 mg, 49 μmol, 21%) as a white solid using 4-methoxybenzaldehyde and 1-(5-nitrothiophen-2-yl)-N-(pyridin-3-ylmethyl)methanamine. 1H NMR (400 MHz, methanol-d4) δ 8.55 (s, 1H), 8.38-8.51 (m, 2H), 7.90 (d, J = 7.78 Hz, 1H), 7.83 (d, J = 4.27 Hz, 1H), 7.42 (dd, J = 5.02, 7.78 Hz, 1H), 7.31 (d, J = 8.53 Hz, 2H), 6.98 (d, J = 4.02 Hz, 1H), 6.90 (d, J = 8.53 Hz, 2H), 3.79 (s, 2H), 3.77 (s, 3H), 3.68 (s, 2H), 3.59 (s, 2H). 13C NMR (101 MHz, methanol-d4) δ 160.8, 155.7, 150.6, 149.2, 138.9, 136.8, 131.4, 131.3, 130.2, 126.1, 125.4, 115.3, 115.1, 58.7, 56.2, 55.9, 53.8. LCMS: tR = 0.75 min, 100%. MS m/z = 370 (M + H)+. HRMS calcd. for C19H20N3O3S (M + H)+ 370.1225, found 370.1229.
N-(3-Methoxybenzyl)-1-(5-nitrothiophen-2-yl)-N-(pyridin-3-ylmethyl)methanamine hydrochloride salt (14)
General Method A was used to give the title compound (36 mg, 75 μmol, 32%) as a white solid using 3-methoxybenzaldehyde and 1-(5-nitrothiophen-2-yl)-N-(pyridin-3-ylmethyl)methanamine. 1H NMR (400 MHz, chloroform-d) δ 8.72 (s, 1H), 8.62 (d, J = 5.29 Hz, 1H), 8.44 (d, J = 7.83 Hz, 1H), 7.79-7.91 (m, 2H), 7.22 (t, J = 7.83 Hz, 1H), 7.02 (d, J = 4.11 Hz, 1H), 6.88-6.99 (m, 2H), 6.68-6.84 (m, 1H), 3.90 (s, 2H), 3.86 (s, 2H), 3.77 (s, 3H), 3.68 (s, 2H). 13C NMR (101 MHz, methanol-d4) δ 161.5, 155.4, 152.0, 150.6, 149.2, 141.2, 138.8, 136.6, 130.8, 130.2, 126.2, 125.4, 122.2, 115.4, 114.2, 59.3, 56.4, 55.8, 54.0. LCMS: tR = 0.75 min, 100%. MS m/z = 370 (M + H)+. HRMS calcd. for C19H20N3O3S (M + H)+ 370.1225, found 370.1220.
N-(3-Trifluromethylbenzyl)-1-(5-nitrothiophen-2-yl)-N-(pyridin-3-ylmethyl)methanamine hydrochloride salt (15)
General Method A was used to give the title compound (21 mg, 40 μmol, 17%) as a white solid using 3-trifluoromethylbenzaldehyde and 1-(5-nitrothiophen-2-yl)-N-(pyridin-3-ylmethyl)methanamine. 1H NMR (400 MHz, methanol-d4) δ 8.70 (s, 1H), 8.60 (d, J = 5.09 Hz, 1H), 8.35 (br. s., 1H), 7.74-7.86 (m, 2H), 7.62-7.73 (m, 2H), 7.47-7.58 (m, 2H), 7.02 (d, J = 4.11 Hz, 1H), 3.90 (s, 2H), 3.86 (s, 2H), 3.80 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 154.2, 149.6, 147.1, 146.1, 139.7, 138.9, 135.2, 132.7, 130.3, 129.6, 129.3, 129.0, 127.9, 126.0, 124.8, 124.6, 56.9, 54.6, 52.5. 13C NMR (101 MHz, DMSO-d6) d 149.6, 147.1, 139.7, 139.4, 135.2, 133.3, 132.6, 130.3, 130.2, 129.6, 129.3, 129.0, 127.9, 126.0, 124.8, 125.6, 56.9, 54.6, 52.5. LCMS: tR = 0.84 min, 100%. MS m/z = 408 (M + H)+. HRMS calcd. for C19H17F3N3O2S (M + H)+ 408.0994, found 408.0997.
5-(((Pyridin-3-ylmethyl)amino)methyl)thiophene-2-carbonitrile (29)
3-Aminomethylpyridine (1.51 g, 14.0 mmol) and 5-formyl-2-thiophenecarbonitrile (1.60 g, 11.7 mmol) were dissolved in the anhydrous THF (40 mL) and acetic acid (10 mL) to give a clear solution. The reaction was stirred at room temperature for 30 min. Sodium triacetoxyborohydride (2.97 g, 14 mmol) was added, and the reaction was stirred at RT for 1 h. The reaction was concentrated to an oil which was purified via normal phase silica gel chromatography (2-10% MeOH in CH2Cl2) to give the title compound as an oil. 1H NMR (400 MHz, chloroform-d) δ 9.11 (s, 1H), 8.92 (d, J = 5.29 Hz, 1H), 8.76 (d, J = 8.03 Hz, 1H), 8.05 (dd, J = 5.77, 7.73 Hz, 1H), 7.95 (d, J = 3.92 Hz, 1H), 7.58 (d, J = 3.72 Hz, 1H), 4.54 (s, 2H), 4.41 (s, 2H). LCMS: tR = 0.24 min, 100%. MS m/z = 230 (M + H)+.
5-(((4-Chloro-benzyl)(pyridin-3-ylmethyl)amino)methyl)thiophene-2-carbonitrile hydrochloride salt (16)
General Method A was used to give the title compound (20 mg, 57 μmol, 28%) as a white solid using 4-chloro-benzaldehyde and 5-(((pyridin-3-ylmethyl)amino)methyl)thiophene-2-carbonitrile. 1H NMR (400 MHz, chloroform-d) δ 8.66 (s, 1H), 8.57 (d, J = 5.08 Hz, 1H), 8.28 (d, J = 8.01 Hz, 1H), 7.73 (dd, J = 5.47, 8.01 Hz, 1H), 7.58 (d, J = 3.91 Hz, 1H), 7.32-7.40 (m, 2H), 7.24-7.33 (m, 2H), 7.05 (d, J = 3.71 Hz, 1H), 3.87 (s, 2H), 3.76 (s, 2H), 3.64 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 153.1, 149.8, 148.6, 139.2, 137.2, 136.2, 133.6, 131.8, 130.3, 128.4, 126.4, 123.6, 114.6, 106.9, 56.2, 54.3, 51.6. LCMS: tR = 0.77 min, 100%. MS m/z = 354 (M + H)+. HRMS calcd. for C19H20N3O3S (M + H)+ 354.0826, found 354.0829.
General Procedure for reductive aminations Method B (used for compounds 17-21). 1-(4-bromo-5-methylthiophen-2-yl)-N-(4-chlorobenzyl)-N-(pyridin-3-ylmethyl)methanamine (17)
The 4-bromo-5-methylthiophene-2-carbaldehyde (9 mg, 0.3 mmol) was weighed into a 2 mL microwave vial. In a 20 mL scintilation vial [(4-chlorophenyl)methyl](3-pyridinylmethyl)amine (70 mg, 0.3 mmol) × 8 was weighed. To this 16 mL of the stock solution (7.5 mL of CH2Cl2, 7.5 mL of ethanol and 3.0 mL of acetic acid) was added. 2.0 mL of this was added to the reaction along with polymer supported sodium cyanoborohydride 2 mM/g (300 mg, 4.77 mmol). The reactions were heated to 120 °C in the microwave for 10 min each. The reaction was filtered through a disposable filter funnel to remove the sodium cyanoborohydride and evaporated overnight under reduced pressure. The resulting solid was dissolved in DMSO and purified by reverse phase prep-HPLC (20%-100% MeCN in water with 0.5% TFA) to give to give the title compound as a white solid (72 mg, 170 μmol, 54%). 1H NMR (400 MHz, DMSO-d6) δ 8.49-8.73 (m, 2H), 8.10 (d, J = 7.83 Hz, 1H), 7.66 (dd, J = 5.29, 7.63 Hz, 1H), 7.37 (s, 5H), 6.90 (s, 1H), 3.67 (d, J = 3.72 Hz, 4H), 3.57 (s, 2H), 2.29 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 158.0, 155.1, 145.4, 144.4, 141.1, 139.5, 136.9, 133.5, 131.8, 130.5, 128.4, 125.3, 107.6, 56.1, 53.7, 51.6, 14.5. LCMS: tR = 0.98 min, 100%. MS m/z = 421 (M + H)+. HRMS calcd. for C19H1979Br35ClN2S (M + H)+ 421.0141, found 421.0139.
1-(Benzo[b]thiophen-2-yl)-N-(4-chlorobenzyl)-N-(pyridin-3-ylmethyl)methanamine (18)
General Method B was used to give the title compound (24 mg, 62 μmol, 12%) as a white solid using benzo[b]thiophene-2-carbaldehyde and [(4-chlorophenyl)methyl](3-pyridinylmethyl)amine. 1H NMR (400 MHz, chloroform-d) δ 8.74 (s, 1H), 8.61 (d, J = 4.30 Hz, 1H), 8.10 (d, J = 7.81 Hz, 1H), 7.77-7.86 (m, 1H), 7.66-7.75 (m, 1H), 7.53 (dd, J = 5.18, 7.71 Hz, 1H), 7.29-7.41 (m, 6H), 7.17 (s, 1H), 3.87 (s, 2H), 3.72 (s, 2H), 3.65 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 153.6, 148.2, 147.2, 147.1, 139.5, 139.1, 138.1, 137.5, 131.8, 130.4, 128.4, 124.4, 124.2, 124.1, 123.3, 122.6, 122.5, 56.1, 54.0, 52.4. LCMS: tR = 0.93 min, 100%. MS m/z = 379 (M + H)+. HRMS calcd. for C22H2035ClN2S (M + H)+ 379.1036, found 379.1038.
1-(3-(1H-Pyrrol-1-yl)thiophen-2-yl)-N-(4-chlorobenzyl)-N-(pyridin-3-ylmethyl)methanamine (19)
General Method B was used to give the title compound (44 mg, 110 μmol, 35%) as a white solid using 3-(1H-pyrrol-1-yl)-2-thiophenecarbaldehyde and [(4-chlorophenyl)methyl](3-pyridinylmethyl)amine. 1H NMR (400 MHz, DMSO-d6) δ 8.60 (br. s., 2H), 8.05 (d, J = 7.83 Hz, 1H), 7.65 (dd, J = 5.48, 7.63 Hz, 1H), 7.53 (d, J = 5.48 Hz, 1H), 7.24-7.41 (m, 4H), 7.06 (d, J = 5.48 Hz, 1H), 6.93 (t, J = 2.06 Hz, 2H), 6.16 (t, J = 1.96 Hz, 2H), 3.71 (s, 2H), 3.60 (s, 2H), 3.54 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 145.2, 144.4, 141.0, 137.1, 136.9, 136.6, 131.8, 131.4, 130.4, 128.3, 125.3, 125.2, 124.9, 121.8, 109.1, 56.5, 53.9, 49.6. LCMS: tR = 0.90 min, 100%. MS m/z = 394 (M + H)+. HRMS calcd. for C22H2135ClN3S (M + H)+ 394.1147, found 394.1145.
N-(4-Chlorobenzyl)-1-(pyridin-3-yl)-N-(thiazol-2-ylmethyl)methanamine (20)
General Method B was used to give the title compound (53 mg, 160 μmol, 51%) as a white solid using 1,3-thiazole-2-carbaldehyde and [(4-chlorophenyl)methyl](3-pyridinylmethyl)amine. 1H NMR (400 MHz, DMSO-d6) δ 8.69 (s, 1H), 8.63 (d, J = 4.89 Hz, 1H), 8.15 (d, J = 7.63 Hz, 1H), 7.62-7.76 (m, 3H), 7.34-7.45 (m, 4H), 3.90 (s, 2H), 3.75 (s, 2H), 3.66 (s, 2H). LCMS: tR = 0.67 min, 95%. MS m/z = 330 (M + H)+. 13C NMR (101 MHz, DMSO-d6) δ 169.1, 145.8, 144.8, 142.5, 140.9, 137.0, 136.1, 131.9, 130.5, 128.4, 125.2, 120.6, 56.5, 54.0, 53.9. HRMS calcd. for C17H1735ClN3S (M + H)+ 330.0832, found 330.0836.
N-((1H-Imidazol-2-yl)methyl)-N-(4-chlorobenzyl)-1-(pyridin-3-yl)methanamine (21)
General Method B was used to give the title compound (47 mg, 150 μmol, 47%) as a white solid using 1H-imidazole-2-carbaldehyde and [(4-chlorophenyl)methyl](3-pyridinylmethyl)amine. 1H NMR (400 MHz, methanol-d4) δ 8.76 (d, 1H), 8.63 (dd, J = 1.17, 5.47 Hz, 1H), 8.39 (d, J = 8.01 Hz, 1H), 7.80 (dd, J = 5.57, 7.91 Hz, 1H), 7.37 (s, 2H), 7.22-7.33 (m, 4H), 4.01 (s, 2H), 3.89 (s, 2H), 3.70 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 158.1, 146.9, 145.7, 144.7, 136.4, 131.9, 130.6, 128.2, 124.6, 119.3, 57.5, 55.1, 48.6. LCMS: tR = 0.43 min, 100%. MS m/z = 313 (M + H)+. HRMS calcd. for C17H1835ClN4 (M + H)+ 313.1220, found 313.1220.
General Procedure for reductive aminations Method C (used for compounds 22-24). 5-(((4-Fluorobenzyl)(pyridin-3-ylmethyl)amino)methyl)thiophene-2-carbonitrile (22)
3-Pyridinecarboxaldehyde (0.10 mL, 1.0 mmol) was added to a solution of 4-fluorobenzylamine (0.12 mL, 1.0 mmol) in dichloroethane (3.4 mL) at RT. After stirring for 5 min, acetic acid (0.06 mL, 1.0 mmol) and sodium triacetoxyborohydride (0.31 mg, 1.4 mmol) were added at RT. After stirring overnight at RT, 5-formylthiophene-2-carbonitrile (0.14 g, 1.0 mmol) and an additional portion of sodium triacetoxyborohydride (0.31 g, 1.4 mmol) were added. After stirring for 1 d at RT, the mixture was quenched with sat. aqueous NaHCO3 solution. The organic phase was dried over sodium sulfate and the solvent evaporated in vacuo. The residue was purified over silica gel (20%-80% EtOAc in petroleum ether) and ISOLUTE® SCX-2 SPE column (NH3 solution, 2 N in MeOH) to give the title compound (0.17 g, 0.51 mmol, 51%) as a pale yellow oil. 1H NMR (500 MHz, chloroform-d) δ 8.63-8.57 (m, 1H), 8.57-8.51 (m, 1H), 7.80-7.75 (m, 1H), 7.47 (d, J = 3.7 Hz, 1H), 7.37-7.30 (m, 3H), 7.07-7.00 (m, 2H), 6.92-6.89 (m, 1H), 3.78 (brs, 2H), 3.63 (brs, 2H), 3.59 (brs, 2H). 13C NMR (DEPTQ-135) (101 MHz, chloroform-d) δ 162.4 (d, J = 248.8 Hz), 152.4, 150.2, 149.2, 137.6, 136.3, 133.8, 133.7 (d, 3.2 Hz), 130.3 (d, J = 8.2 Hz), 125.5, 123.7, 115.6 (d, J = 21.4 Hz), 114.6, 109.0, 57.4, 55.4, 52.6. 19F NMR (377 MHz, chloroform-d) δ-114.5-(-114.7) (m, 1F); 19F{H} NMR (377 MHz, chloroform-d) δ-114.6 (s, 1F). UPLC purity (UV detection 210-350 nm): tR = 0.89 min, 100% (Acquity UPLC BEH C18 column, 50 mm × 2.1 mm i.d., 1.7 μm packing diameter at 40 °C, 97% H2O/3% CH3CN → 3% H2O/97% CH3CN gradient with 0.1% v/v formic acid). MS (ESI, +ve) m/z = 338.1 (M + H)+; HRMS (ES+) calcd. for C19H17N3F1S1 (M + H)+ 338.1122, found 338.1128.
5-(((3,4-Difluorobenzyl)(pyridin-3-ylmethyl)amino)methyl)thiophene-2-carbonitrile (23)
General Method C was used to give the title compound (0.42 g, 1.2 mmol, 59%) as pale yellow oil using 3,4-difluorobenzaldehyde (0.23 mL, 2.0 mmol) and 3-(aminomethyl)pyridine (0.21 mL, 2.0 mmol). 1H NMR (400 MHz, chloroform-d) δ 8.59-8.53 (m, 1H), 8.51 (dd, J = 4.8, 1.7 Hz, 1H), 7.74-7.68 (m, 1H), 7.45 (d, J = 3.8 Hz, 1H), 7.28 (ddd, J = 7.9, 4.8, 0.5 Hz, 1H), 7.24-7.15 (m, 1H), 7.15-7.04 (m, 2H), 6.93-6.88 (m, 1H), 3.77 (brs, 2H), 3.61 (brs, 2H), 3.56 (brs, 2H). 13C NMR (DEPTQ-135) (101 MHz, chloroform-d) δ 151.8, 150.5 (dd, J = 248.9, 12.9 Hz), 150.1, 149.8 (dd, J = 248.0, 12.8 Hz), 149.2, 137.6, 136.3, 135.2 (dd, J = 5.2, 3.8 Hz), 133.4, 125.6, 124.5 (dd, J = 6.4, 3.6 Hz), 123.7, 117.3 (dd, J = 17.1, 7.5 Hz), 114.3, 109.0, 57.0, 55.3, 52.4, one carbon missing. 19F NMR (377 MHz, chloroform-d) δ-139.3-(-139.1) (m, 1F), δ-137.2 (ddd, J = 21.8, 11.0, 6.8 Hz, 1F); 19F{H} NMR (377 MHz, chloroform-d) δ-139.2 (d, J = 20.3 Hz, 1F), δ-137.2 (d, J = 20.3 Hz, 1F). UPLC purity (UV detection, 210-350 nm): tR = 0.92 min, 100% (Acquity UPLC BEH C18 column, 50 mm × 2.1 mm i.d., 1.7 μm packing diameter at 40 °C, 97% H2O/3% CH3CN → 3% H2O/97% CH3CN gradient with 0.1% v/v formic acid). MS (ESI, +ve) m/z = 356.1 (M + H)+; HRMS (ES+) calcd. for C19H16N3F2S1 (M + H)+ 356.1028, found 356.1032.
5-(((4-Methoxybenzyl)(pyridin-3-ylmethyl)amino)methyl)thiophene-2-carbonitrile (24)
General Method C was used to give the title compound (0.17 g, 0.5 mmol, 50%) as pale yellow oil using 3-pyridinecarboxaldehyde (0.10 mL, 1.0 mmol) and 4-methoxybenzylamine (0.13 mL, 1.0 mmol). 1H NMR (400 MHz, chloroform-d) δ 8.62-8.56 (m, 1H), 8.52 (dd, J = 4.8, 1.5 Hz, 1H), 7.78-7.72 (m, 1H), 7.46 (d, J = 3.8 Hz, 1H), 7.32-7.25 (m, 3H), 6.92-6.86 (m, 3H), 3.80 (s, 3H), 3.76 (brs, 2H), 3.62 (brs, 2H), 3.57 (brs, 2H). 13C NMR (j-mod) (101 MHz, chloroform-d) δ 159.2, 152.9, 150.2, 149.1, 137.7, 136.4, 134.1, 130.1, 129.8, 125.3, 123.7, 114.7, 114.0, 108.9, 57.5, 55.4, 55.2, 52.3. UPLC purity (UV detection, 210 to 350 nm): tR = 0.87 min, 100% (Acquity UPLC BEH C18 column, 50 mm × 2.1 mm i.d., 1.7 μm packing diameter at 40 °C, 97% H2O/3% CH3CN → 3% H2O/97% CH3CN gradient with 0.1% v/v formic acid). MS (ESI, +ve) m/z = 350.1 (M + H)+; HRMS (ES+) calcd. for C20H20O1N1S1 (M + H)+ 350.1322, found 350.1327.
5-(((2,5-Difluorobenzyl)(pyridin-3-ylmethyl)amino)methyl)thiophene-2-carbonitrile (25)
General Method A was used to give the title compound (23 mg, 60 μmol, 31%) as a white solid using 2,5-difluorobenzaldehyde and 5-(((pyridin-3-ylmethyl)amino)methyl)thiophene-2-carbonitrile. 1H NMR (400 MHz, methanol-d4) δ 8.92 (s, 1H), 8.78 (d, J = 5.48 Hz, 1H), 8.72 (d, J = 8.03 Hz, 1H), 8.06 (dd,J = 6.17, 7.73 Hz, 1H), 7.62 (d, J = 3.72 Hz, 1H), 7.24-7.37 (m, 1H), 7.21 (d, J = 3.13 Hz, 1H), 6.91-7.15 (m, 2H), 4.17 (br. s., 2H), 4.11 (br. s., 2H), 3.93 (br. s., 2H). 13C NMR (101 MHz, DMSO-d6) δ 157.0, 152.3, 139.2, 137.9, 126.9, 126.3, 117.4, 117.2, 116.9, 116.8, 116.1, 116.0, 115.9, 115.8, 114.6, 107.2, 54.1, 52.3, 50.4. LCMS: tR = 0.77 min, 100%. MS m/z = 356 (M + H)+. HRMS calcd. for C19H16F2N3S (M + H)+ 356.1033, found 356.1037.
5-(((2-Fluorobenzyl)(pyridin-3-ylmethyl)amino)methyl)thiophene-2-carbonitrile (26)
General Method A was used to give the title compound (35 mg, 94 μmol, 50%) as a white solid using 2-fluorobenzaldehyde and 5-(((pyridin-3-ylmethyl)amino)methyl)thiophene-2-carbonitrile. 1H NMR (400 MHz, methanol-d4) δ 8.99 (s, 1H), 8.68-8.88 (m,2H), 8.06 (dd, J = 6.07, 7.63 Hz, 1H), 7.65 (d, J = 3.72 Hz, 1H), 7.54 (t, J = 7.24 Hz, 1H), 7.28-7.41 (m, 2H), 7.17 (t, J = 7.44 Hz, 1H), 7.08 (t, J = 9.30 Hz, 1H), 4.38 (br. s., 2H), 4.31 (br. s., 2H), 4.12 (br. s., 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.3, 142.0, 141.6, 139.2, 138.4, 131.4, 129.8, 129.7, 126.8, 126.5, 124.5, 115.5, 115.3, 114.6, 107.1, 104.8, 54.0, 52.2, 50.6. LCMS: tR = 0.53 min, 100%. MS m/z = 338 (M + H)+. HRMS calcd. for C19H17FN3S (M + H)+ 338.1129, found 338.1127.
REV-ERBα NCOR FRET Assay
REV-ERBα was made at GSK. Proteins were chemically biotinylated using standard methods. Biotinylated NCOR1 was purchased from CPC scientific. Streptavidin-labeled APC (CR130-150) and Eu-W1024 labeled Streptavidin (AD0063) were purchased from Perkin Elmer. Compounds were diluted in 100% neat DMSO at 10mM. The compounds were dispensed into an intermediate plate (polypropylene Greiner PP V-bottom: 781280) to make serial dilutions in 100% neat DMSO. Approximately 100 nL of the serial dilution was added to the assay plate. The stock buffer used was a 0.5 M solution of MOPS was made by adding 104 grams of MOPS to 800 mL H2O in a graduated cylinder, using a calibrated pH meter, add increasing amounts NaOH to give a final pH of 7.5. This solution was filtered using a Costar 0.2 μm filtering apparatus. The assay buffer used was prepared by adding 100 mL of 10X MOPS stock solution to graduated cylinder up to 800 mL. NaF (2.09 g), CHAPS (0.03 g), and BSA (0.1 g) were added to the flask. Water was added to give final volume of 1 L. The assay buffer was filtered with a Costar 0.2 μm filtering apparatus. DTT was added to the assay buffer to a final concentration of 10 mM. To a polypropylene costar conical centrifuge tube was added assay buffer, an appropriate amount of biotinylated-NCOR1 from the 100 μM stock solution to give a final concentration of 20 nM. To the above biotinylated NCOR1 solution, an appropriate amount of Europium-labeled streptavidin was added to give a final concentration of 10 nM. The solution was incubated 15 min at RT. In a separate polypropylene tube was added an appropriate amount of biotinylated REV-ERBα protein from the stock solution to give a final concentration of 20 nM. To the biotinylated-REV-ERBα solution was added an appropriate amount of APC-labeled streptavidin to give a final concentration of 10 nM. The resulting solution was incubated 15 minutes at RT. A 20-fold excess biotin from the 10 mM stock solution was added and the resulting solution was incubated 10 minutes at RT. Gently mix the above solutions together to give a final solution containing 20 nM REV-ERBα 10 nM APC and 20 nM NCOR1_10 nM SA_EU. The resulting mixture was incubated for 5 min, and a Thermo Combi Multidrop was used to add 10 uL peptide/REV-ERBα solution to assay plates containing 100nL of test compound. The plate was incubated for 1hr at RT and read on ViewLux in Lance mode for EU/APC. Raw data was analyzed using ABASE (IBDS) software. The data was normalized initially using the following equation: Normalization = 100* ((Basal HTRF - value)/(Basal HTRF - Minimal HTRF). The normalized data was then was fit to a 4 - parameter logistic equation.
Peptide Scan
Using the same reagents as for the NCOR FRET assay, various biotinylated peptides were added to Greiner assay plate to give a final concentration of 20 nM. An appropriate amount of Eu-W1024 was added to the plate to give a final concentration of 20 nM. The plate was incubated for 20 minutes at room temperature. At the same time, in a polypropylene tube add an appropriate amount of biotinylated-REV-ERBα protein from the stock solution to give a final concentration of 20 nM. To the biotinylated-REV-ERBα solution, an appropriate amount of APC-labeled streptavidin was added to give a final concentration of 10 nM. The tube was inverted gently to mix and incubated 20 minutes at room temperature. Following the 20 minute incubations, 20-fold excess biotin from the 10 mM stock solution was added, and the tube was inverted gently to mix. After a 15 minute incubation at room temperature, a Thermo Combi Multidrop was used to add 5 uL of the REV-ERBα solution to the plates containing 100 nL of the various peptides. The plates were read on a ViewLux in Lance mode for EU/APC. The raw data were analyzed using an excel template which averaged the four wells of each peptide and generate a standard deviation.
BMAL Reporter Gene Assay
Stable U2OS cell line expressing mouse Bmal-promoter luciferase was generated by transfecting U2OS cells with pGl4-mouse-Bmal1-luciferase plasmid. The single clones were selected by dilution cloning in culture media (M1) containing 100 ug/mL Hygromycin and the positive clones were determined by measuring luciferase continuously for five days. Freshly passaged cells were seeded at confluence in 384-well format. Robust circadian rhythms were induced by media change, and synchronization was evaluated by measuring the clock-driven luciferase reporter activity over 48 h. Synchronized cells were treated with compound and real time bioluminescent oscillations were recorded for 72 h with measurements every 20 m using the temperature controlled luminometer Orion II. After 72 h of treatment, cells were washed and allowed to recover in full growth media. The recovery oscillations were monitored and used as an internal toxicity counter-screen for off-target effects on cell transcription.
Cell Assays
THP-1 cells were obtained from ECACC. THP-1 cells were sub-cultured between 3×105 cells/mL and 8×105cells/mL in RPMI1640 (PAA) supplemented with 10% FBS (Invitrogen U.K.). Total RNA was prepared using the RNAeasy kit (Qiagen, U.K.) and treated with DNASe (Qiagen, U.K.). Reverse transcription was performed using a high capacity RNA to cDNA kit (Applied Biosystems, USA). The cDNA was analyzed by qPCR using Power Sybr Green master mix (Applied Biosystems, USA) and pre-validated Quantitect primers (Qiagen, U.K.). TATA box binding protein was the house keeping gene. The cell viability assay was performed using the Celltiter-glo kit (Promega, USA). The kit was used according to manufacturer’s instructions. Briefly, cells were incubated for six hours with the chemical compounds. Celltiter-glo was then added for 15 minutes before the luminescence was recorded.
DMPK Experiment
Plasma levels of a cassette of the 5 test compounds were evaluated in C57BL6 mice purchased from SLAC Laboratory Animal Co. (male mice, 22-25 g, n = 3 per time point, total of 9 animals per dosing regimen) administered by either i.v. tail vein injection (1 mg/Kg) or PO oral gavage administration (1 mg/Kg). Compounds were dissolved in a 10% DMSO, 10% Solutol-HS-15, and 80% water solution. 0, 0.083, 0.25, 0.5, 1, 2, 4, 8, and 24 h after dosing, blood was taken. Plasma was generated using standard centrifugation techniques and was frozen at -78 °C. Plasma was mixed with acetonitrile (1:15 (v/v)) and vortexed for 2 min and centrifuged at 14000 rpm for 5 min. An 2 mL aliquot of the supernatant was analyzed for drug levels by liquid chromatography/tandem mass spectrometry.
Supplementary Material
ACKNOWLEDGMENT
We would like to acknowledge G. Bruce Wisely and Tom G. Consler for production of the REV-ERBα protein.
ABBREVIATIONS
- REV-ERBα
NR1D1
- LXRα
Liver X Receptor alpha (NR1H3)
- NCOR
Nuclear Receptor Co-Repressor 1
- SMRT
Nuclear Receptor Co-Repressor 2
- PGC1-β
Peroxisome proliferator-activated receptor gamma coactivator 1-beta
- IL-6
Interleukin 6
- THP-1
Human Acute Monocytic Leukemia Cell Line
- LPS
Lipopolysaccharides
- ABCA1
ATP-binding cassette sub-family A, member 1
- GAL4
Regulatory protein GAL4
- BMAL
Brain and muscle aryl hydrocarbon receptor nuclear translocator (ARNT)-like
- U2OS
human osteosarcoma U2OS cell line
- SAR
Structure activity relationship
- AcOH
acetic acid
Footnotes
ASSOCIATED CONTENT
Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).Bass J, Takahashi JS. Science. 2010;330:1349. doi: 10.1126/science.1195027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Takahashi JS, Hong HK, Ko CH, McDearmon EL. Nat Rev Genet. 2008;9:764. doi: 10.1038/nrg2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Welsh DK, Takahashi JS, Kay SA. Annu Rev Physiol. 2010;72:551. doi: 10.1146/annurev-physiol-021909-135919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Liu C, Li S, Liu T, Borjigin J, Lin JD. Nature. 2007;447:477. doi: 10.1038/nature05767. [DOI] [PubMed] [Google Scholar]
- (5).Raghuram S, Stayrook KR, Huang P, Rogers PM, Nosie AK, McClure DB, Burris LL, Khorasanizadeh S, Burris TP, Rastinejad F. Nat Struct Mol Biol. 2007;14:1207. doi: 10.1038/nsmb1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Bugge A, Feng D, Everett LJ, Briggs ER, Mullican SE, Wang F, Jager J, Lazar MA. Genes Dev. 2012;26:657. doi: 10.1101/gad.186858.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Chen H, Chu G, Zhao L, Yamauchi N, Shigeyoshi Y, Hashimoto S, Hattori MA. Biochem Biophys Res Commun. 2012;420:374. doi: 10.1016/j.bbrc.2012.02.164. [DOI] [PubMed] [Google Scholar]
- (8).Cho H, Zhao X, Hatori M, Yu RT, Barish GD, Lam MT, Chong LW, DiTacchio L, Atkins AR, Glass CK, Liddle C, Auwerx J, Downes M, Panda S, Evans RM. Nature. 2012;485:123. doi: 10.1038/nature11048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Gibbs JE, Blaikley J, Beesley S, Matthews L, Simpson KD, Boyce SH, Farrow SN, Else KJ, Singh D, Ray DW, Loudon AS. Proc Natl Acad Sci U S A. 2012;109:582. doi: 10.1073/pnas.1106750109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Grant D, Yin L, Collins JL, Parks DJ, Orband-Miller LA, Wisely GB, Joshi S, Lazar MA, Willson TM, Zuercher WJ. ACS Chem Biol. 2010;5:925. doi: 10.1021/cb100141y. [DOI] [PubMed] [Google Scholar]
- (11).Solt LA, Wang Y, Banerjee S, Hughes T, Kojetin DJ, Lundasen T, Shin Y, Liu J, Cameron MD, Noel R, Yoo SH, Takahashi JS, Butler AA, Kamenecka TM, Burris TP. Nature. 2012;485:62. doi: 10.1038/nature11030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Glass CK, Saijo K. Nat. Rev. Immunol. 2010;10:365. doi: 10.1038/nri2748. [DOI] [PubMed] [Google Scholar]
- (13).Kalaany NY, Mangelsdorf DJ. Annu. Rev. Physiol. 2006;68:159. doi: 10.1146/annurev.physiol.68.033104.152158. [DOI] [PubMed] [Google Scholar]
- (14).Shibata N, Glass CK. Circ. J. 2010;74:2045. doi: 10.1253/circj.cj-10-0860. [DOI] [PubMed] [Google Scholar]
- (15).Hann MM, Keseru GM. Nat Rev Drug Discov. 2012;11:355. doi: 10.1038/nrd3701. [DOI] [PubMed] [Google Scholar]
- (16).Meng QJ, McMaster A, Beesley S, Lu WQ, Gibbs J, Parks D, Collins J, Farrow S, Donn R, Ray D, Loudon A. J Cell Sci. 2008;121:3629. doi: 10.1242/jcs.035048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.