

Abstract
Transforming growth factor β (TGFβ) can act either as a tumor promoter or a tumor suppressor in a context-dependent manner. High levels of TGFβ are found in prostate cancer tissues and correlate with poor patient prognosis. We recently identified a novel TGFβ-regulated signaling cascade in which TGFβ type I receptor (TβRI) is activated by the E3 ligase TNF-receptor-associated factor 6 (TRAF6) via the Lys63-linked polyubiquitination of TβRI. TRAF6 also contributes to activation of TNF-α-converting enzyme and presenilin-1, resulting in the proteolytic cleavage of TβRI and releasing the intracellular domain of TβRI, which is translocated to the nucleus to promote tumor invasiveness. In this report, we provide evidence that Lys178 of TβRI is polyubiquitinated by TRAF6. Moreover, our data suggest that TRAF6-mediated Lys63-linked ubiquitination of the TβRI intracellular domain is a prerequisite for TGFβ regulation of mRNA for cyclin D1 (CCND1), expression, as well as for the regulation of other genes controlling the cell cycle, differentiation, and invasiveness of prostate cancer cells.
Keywords: cell cycle, cyclin D1, EMT, invasion, prostate cancer, Snail1, TRAF6, transforming growth factor β
Introduction
Aggressive prostate cancer is a lethal disease due to the invasion and metastasis of disseminated cancer cells.1 Transforming growth factor β (TGFβ) plays pivotal roles in controlling cell proliferation, apoptosis, and the final fate of metazoan cells.2,3 In malignancies, TGFβ is initially a tumor suppressor, since it inhibits cell proliferation and induces apoptosis, but at later stages of tumor progression, it is often produced at high levels by tumor and stromal cells and promotes tumor progression.2,3
TGFβ brings together 2 pairs of TGFβ type II and TGFβ type I receptors (TβRI) to form a heterotetrameric complex.4 TβRI is activated via phosphorylation of its glycine/serine-rich domain by TGFβ type II receptor, thereby inducing the activation of several downstream signaling pathways.5-7 Activated TβRI propagates signals through the receptor-associated Smads (R-SMAD; SMAD2, and SMAD3) by phosphorylation of their extreme C termini.8,9 Activated Smads form complexes with the co-Smad Smad4, translocating to the nucleus to regulate the transcription of certain genes.10-13 TGFβ also transduces signals through non-Smad signaling pathways such as those involving extracellular signal-regulated kinase, Jun N-terminal kinases, p38 mitogen-activated protein kinases (MAPKs), and other pathways.3,14 Recent investigations revealed that TβRI has a consensus binding motif for the E3 ubiquitin ligase TRAF6. Activation of TRAF6 is induced by ligand-induced hetero-oligomerization of the TβR complex. Thereafter, catalytically active TRAF6 in turn catalyzes Lys63-linked polyubiquitination of TGFβ activated kinase 1 (TAK1), which activates the p38 and JNK MAPK pathways, leading to apoptosis and the epithelial-mesenchymal transition (EMT).11,15,16
Various post-translational modifications, such as phosphorylation, acetylation, and ubiquitination, determine the function and role of proteins and can thereby control specific cellular processes such as apoptosis, cell-cycle progression, and gene regulation.17 Ubiquitination is one such post-translational modification in which ubiquitin is covalently bound to an acceptor lysine of the target protein.18 Covalent attachment of Lys48-linked polyubiquitin to target proteins promotes their proteasomal degradation, whereas Lys63-linked polyubiquitination modulates the activity or trafficking of the substrate protein.19-22
We recently reported a mechanism whereby TGFβ promotes the invasiveness of cancer cells.23-25 TGFβ induces the activation of TRAF6, which ubiquitinates TβRI in a Lys63-dependent manner and promotes proteolytic cleavage of the receptor by TNF α-converting enzyme (TACE) and presenilin-1, resulting in the release of the intracellular domain (ICD) of TβRI.23-25 The TβRI ICD is then translocated to the nucleus, where it associates with the transcriptional co-activator p300 and promotes tumor-cell invasion by inducing the expression of genes encoding TβRI, Snail1, and matrix metallopeptidase 2(MMP2).23,24,26 In the present study, we identified the acceptor lysine in TβRI that is ubiquitinated by TRAF6, and determined the biological significance of this ubiquitination.
Results
Lys178 is the acceptor lysine for Lys63-linked polyubiquitination of TβRI
Previously, we reported that TβRI is polyubiquitinated in a Lys63-dependent manner by TRAF6.23 We observed that Lys178 is the only lysine near the transmembrane region of TβRI in close proximity to the TRAF6 consensus binding site, suggesting that it could be the acceptor lysine for polyubiquitination.15 To investigate whether Lys178 is an acceptor lysine, we created a point mutant in the wild-type, constitutively active TβRI (c.a.TβRI) in which Lys178 was mutated to arginine.
We performed in vivo ubiquitination assays in PC-3U cells transiently transfected with C-terminally hemagglutinin (HA)-tagged wild-type TβRI (HA-TβRI) or with the HA-tagged K178R mutant TβRI (HA-K178R); these cells were or were not exposed to TGFβ. HA-TβRI and HA-K178R proteins were immunoprecipitated with anti-HA antibodies and subjected to immunoblotting with antiserum against Lys63-linked polyubiquitin. Stimulation of cells with TGFβ resulted in Lys63-linked polyubiquitination in cells transfected with HA-TβRI, but not in cells transfected with HA-K178R (Fig. 1A).
Figure 1.
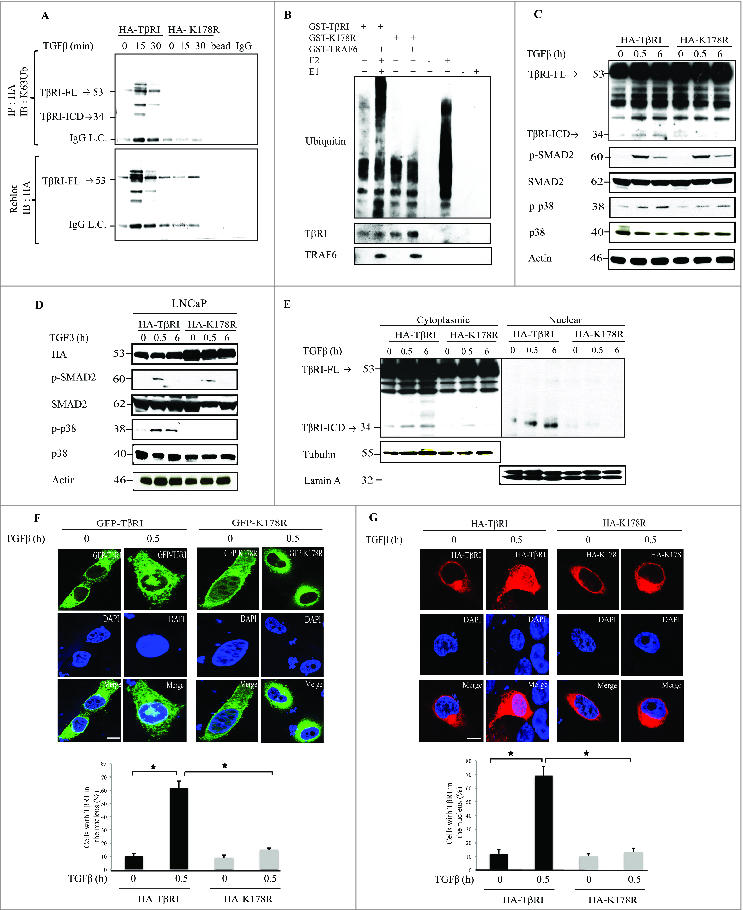
For figure legend, see page 557. Figure 1 (see previous page). TGFβ induces TRAF6-mediated ubiquitination of wild-type TβRI but not of the K178R mutant of TβRI. (A) In vivo ubiquitination assay performed in PC-3U cells transiently transfected with wild-type HA-TβRI or mutant HA-K178R and treated with TGFβ as indicated. Complexes immunoprecipitated (IP) with anti-HA antibodies were immunoblotted (IB) with a Lys63-linked polyubiquitin-specific antibody (TβRI- FL: Full length TGFβ Receptor I, TβRI-ICD: TGFβ Receptor I intracellular domain). The IP filter was stripped and reblotted with antiserum against HA. IgG light chain (IgG L.C). (B) In vitro ubiquitination assays were performed with recombinant GST-TβRI or GST-K178R in the presence or absence of GST-TRAF6; samples were subjected to SDS-PAGE, followed by IB with anti-polyubiquitin (Ub) antibodies. Only GST-TβRI, and not GST-K178R, was ubiquitinated in the presence of TRAF6. Reaction mixtures without E2 or without E1 were used as negative controls. (C) A portion of the total cell lysate was subjected to IB against HA, p-p38, and p-SMAD2 with specific antisera. The filters were then reblotted with antisera against p38 and SMAD2. Actin was used as a control for equal protein loading onto the gel. (TβRI- FL: Full length TGFβ Receptor I, TβRI-ICD: TGFβ Receptor I intracellular domain) (D) LNCaP cells transiently transfected with HA-TβRI or HA-K178R were exposed to TGFβ as indicated. Total cell lysates were subjected to IB with antisera against p-p38 and p-SMAD2. The filter was then reblotted with antisera against p38 and SMAD2; IB with HA antiserum was used as a control for transfection. (E) PC-3U cell lysates were transfected with HA-TβRI or HA-K178R and exposed to TGFβ as indicated; cytoplasmic and nuclear fractions were subjected to SDS-PAGE, then IB with HA antibody. Membranes were re-blotted with β-tubulin and lamin A as controls for the cytoplasmic and nuclear fractions, respectively. (TβRI- FL: Full length TGFβ Receptor I, TβRI-ICD: TGFβ Receptor I intracellular domain) (F) PC-3U cells were transfected with GFP-TβRI or GFP-K178R, exposed to TGFβ as indicated, and subjected to immunofluorescence. Green, wild-type TβRI or K178R tagged with GFP. Blue, nuclei (2-(4-amidinophenyl)-1H-indole-6-carboxamidine; (DAPI)). Scale bars, 20 µm. Below, percentage of cells with nuclear TβRI. (*P < 0.05, representative of 3 independent experiments) (G) PC-3U cells were transfected with HA-TβRI or HA-K178R, treated as indicated, and subjected to immunofluorescence and confocal imaging. Red, anti-HA antibodies; blue, nuclei (DAPI). Scale bars, 20 µm. Below, percentage of cells with nuclear TβRI. (*P < 0.05, representative of 3 independent experiments).
To further validate that Lys178 is the acceptor lysine for polyubiquitination by TRAF6, we performed in vitro ubiquitination assays. Recombinant glutathione S-transferase (GST) -TβRI or GST-tagged mutant TβRI (GST-K178R) was incubated in the presence or absence of GST-TRAF6 for 1 h at 37°C, and then subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by immunoblotting against polyubiquitin. GST-TβRI was ubiquitinated by TRAF6, but GST-K178R was not (Fig. 1B). Reaction mixtures without E2 or without E1 were used as negative controls (Fig. 1B). No TGFβ-induced ICD formation was observed for HA-K178R, whereas an ICD was formed from HA-TβRI (Fig. 1C). The kinase activity of HA-TβRI K178R was intact in PC-3U cells, as it was found to phosphorylate Smad2, whereas in cells transfected with HA-K178R, we detected reduced phosphorylation of p38 (Fig. 1C).
Next, we repeated the experiments in LNCaP cells, which harbor a non-functional TβRI.27 In LNCaP cells transfected with wild-type TβRI, TGFβ stimulated phosphorylation of both Smad2 and p38, but in cells transfected with the K178R mutant of TβRI, TGFβ stimulated phosphorylation only of Smad2 and not of p38 (Fig. 1D).
Mutation of the acceptor lysine in TβRI inhibits nuclear translocation
To investigate the importance of Lys63-linked polyubiquitination of TβRI for its subcellular localization, we performed a nuclear fractionation assay of cells transfected with the wild-type TβRI or K178R mutant of TβRI, Stimulation of cells with TGFβ resulted in nuclear translocation of TβRI-ICD in cells transfected with HA-TβRI, but not in cells transfected with HA-K178R (Fig. 1E). Next we validated this finding by using immunofluorescence and confocal microscopy. We fused green fluorescent protein (GFP) with the C-termini of wild-type (TβRI-GFP) and mutant (GFP-K178R) TβRI. PC-3U cells were transfected with GFP-TβRI or GFP-K178R and immunofluorescence staining was performed to visualize the receptors with confocal microscopy. Only GFP-TβRI translocated to the nucleus upon TGFβ stimulation; nuclear translocation of GFP-K178R was inhibited (Fig. 1F). Similar results were obtained in PC-3U cells transfected with molecules tagged with HA (Fig. 1G).
TGFβ induces proximity between TβRI and Lys63-polyubiquitin chains
We used the proximity ligation assay (PLA) to investigate the proximity of TβRI to Lys63-polyubiquitin chains. PC-3U cells ectopically expressing HA-TβRI or HA-K178R were treated or not treated with TGFβ, fixed, blocked, and probed with anti-HA antibody (rabbit) and anti-Lys63-polyubiquitin antibody (mouse). TGFβ stimulation led to a significant increase in signal in PC-3U cells transfected with HA-TβRI but not with HA-K178R; very little signal was detected in the latter (Fig. 2A). Taken together, these observations indicate that TGFβ enhanced the proximity between wild-type TβRI and Lys63-polyubiquitin chains, an effect that was not detected for the K178R mutant of TβRI, supporting the hypothesis that Lys178 is the acceptor lysine for Lys63-linked polyubiquitination of TβRI.
Figure 2.
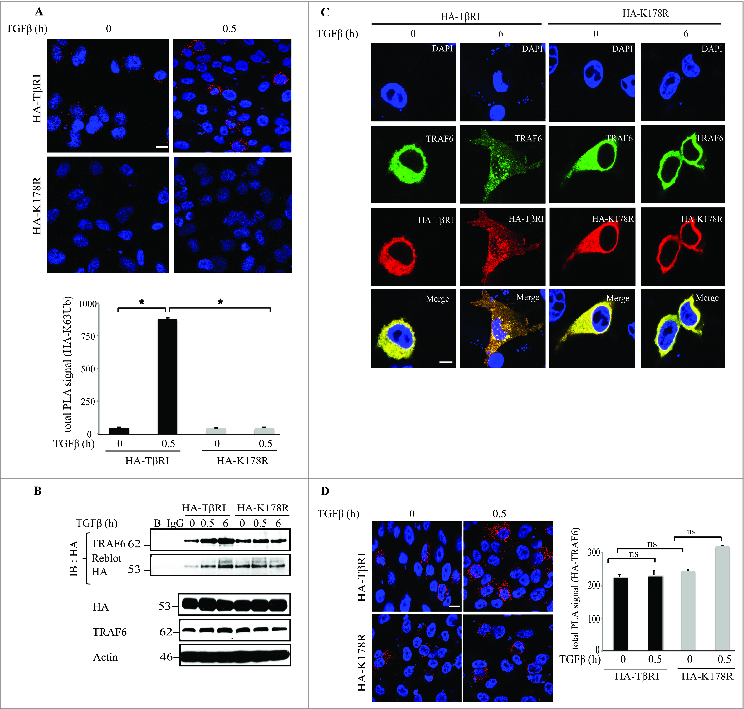
TRAF6 associates with both wild-type and mutant TβRI. (A) PC-3U cells transiently transfected with wild-type HA-TβRI or mutant HA-K178R was treated as indicated. Left, ubiquitinated HA-TβRI was visualized by staining cells with proximity probes directed against Lys63-polyubiquitin and HA (red), followed by ligation and rolling circle amplification of the oligonucleotides. Cell nuclei were stained with 2-(4-amidinophenyl)-1H-indole-6-carboxamidine (DAPI; blue). Right, PLA signal was quantified with the Duolink ImageTool. Data are from 3–5 independent experiments; *P < 0.05. Scale bars, 20 µm. (B) PC-3U cells transfected with HA-TβRI or HA-K178R were treated as indicated, and cell lysates were immunoprecipitated (IP) with anti-HA antibodies and subjected to immunoblotting with antibodies against TRAF6. Light chain-specific secondary antiserum was used to avoid cross-reaction with the IgG heavy chain. The filter was reprobed with HA antiserum as a control. (C) PC-3U cells were transfected with HA-TβRI or HA-K178R. Red, HA; green, TRAF6; blue, nuclei (DAPI). (D) Top, PC-3U cells were transiently transfected with HA-TβRI or HA-K178R and exposed to TGFβ as indicated. The association of TβRI with TRAF6 was visualized by staining cells with proximity probes against TRAF6 and HA (red), followed by ligation and rolling circle amplification. Blue, nuclei. Scale bars, 20 µm. Bottom, quantification of PLA signal from 3–5 independent experiments; *P < 0.05.
We performed immunoprecipitation experiments to investigate whether the point mutation in the K178R mutant TβRI affected its interaction with TRAF6. Importantly, both wild-type and mutant TβRI interacted with TRAF6 (Fig. 2B). Immunofluorescence experiments also confirmed the association of HA-TβRI and HA-K178R with the E3 ligase TRAF6 (Fig. 2C). In addition, PLAs of PC-3U cells transiently transfected with HA-TβRI or HA-K178R showed that both HA-TβRI and HA-K178R associated with TRAF6 in a TGFβ-dependent manner (Fig. 2D).
From these observations, we conclude that K178 in TβRI is the acceptor lysine for Lys63-linked polyubiquitination catalyzed by TRAF6 in response to TGFβ stimulation. Furthermore, this event is crucial for TGFβ- and TRAF6-induced cleavage of TβRI, as well as for nuclear translocation of the TβRI ICD.
TβRI ubiquitination induces gene expression that regulates the cell cycle and cell invasion
We previously reported that TGFβ-induced nuclear localization of the TβRI ICD leads to increased expression of the pro-invasive genes SNAI1 (also known as Snail1), matrix metallopeptidase 2 (MMP2), and TβRI.23,24 To further explore the set of genes regulated by TGFβ-induced and TRAF6-mediated polyubiquitination of TβRI, we performed quantitative reverse transcription polymerase chain reaction (qRT-PCR) to analyze the mRNA expression of Vimentin, Twist1, N-cadherin, p73, cyclin D1 (CCND1), and Plasminogen activator inhibitor-1 (PAI1) (Fig. 3). We found that TGFβ promotes the expression of Vimentin, Twist1, N-cadherin, and cyclin D1 (CCND1) more efficiently in PC-3U cells transfected with HA-TβRI than in cells transfected with HA-K178R (Fig. 3). In contrast, TGFβ induction of the expression of the tumor suppressor gene p73 was higher in HA-K178R-transfected cells than in HA-TβRI-transfected cells (Fig. 3). As expected, both wild-type and mutant TβRI induced the classical TGFβ target gene PAI1 (Fig. 3), demonstrating that the K178R mutation did not interfere with the kinase activity of TβRI and Smad signaling, consistent with data in Figures 1C and D.
Figure 3.
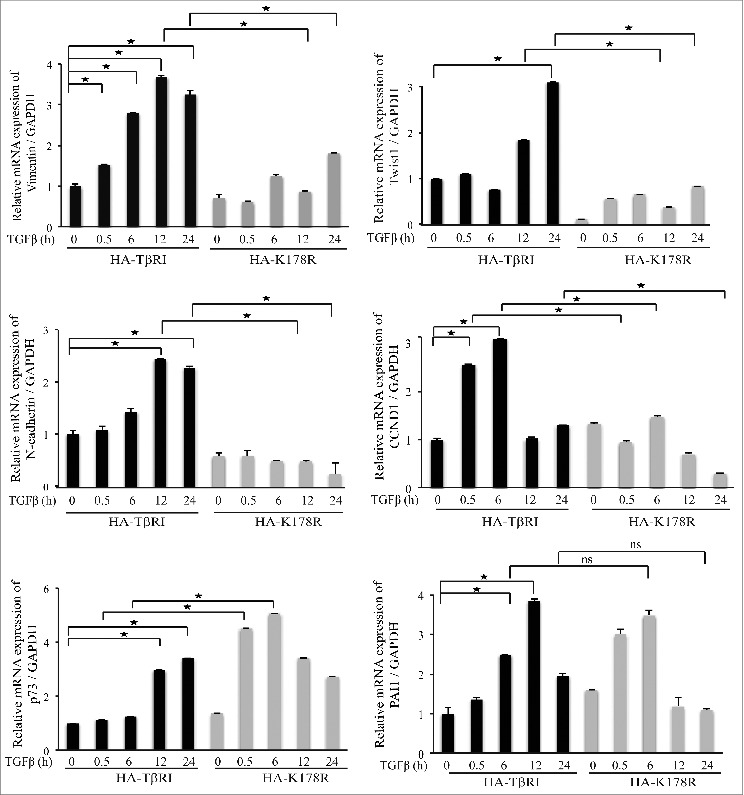
TβRI ubiquitination promotes the expression of Vimentin, Twist1, N-cadherin, and CCND1. qRT-PCR of Vimentin, Twist1, N-cadherin, CCND1, p73, and PAI1 was performed on mRNA extracted from PC-3U cells transiently transfected with HA-TβRI or HA-K178R and treated as indicated (mean ± standard deviation; n = 3; *P < 0.05).
To further understand the role of TRAF6 in TGFβ signaling in prostate cancer cells, PC-3U cells were transfected with control or TRAF6-specific siRNA, and RNA microarray analysis was performed to identify TGFβ-dependent genes regulated by TRAF6 (Fig. 4). These genes include the invasive marker Snail1 and cell cycle regulators like cyclin B1 (CCNB1), Cyclin-A1 (CCNA1), and CCND1. We previously identified Snail1, TGFβRI, and Jagged1 (Jag1) as targets of the TGFβ- and TRAF6-induced Lys63-linked polyubiquitination of TβRI.23,24
Figure 4.
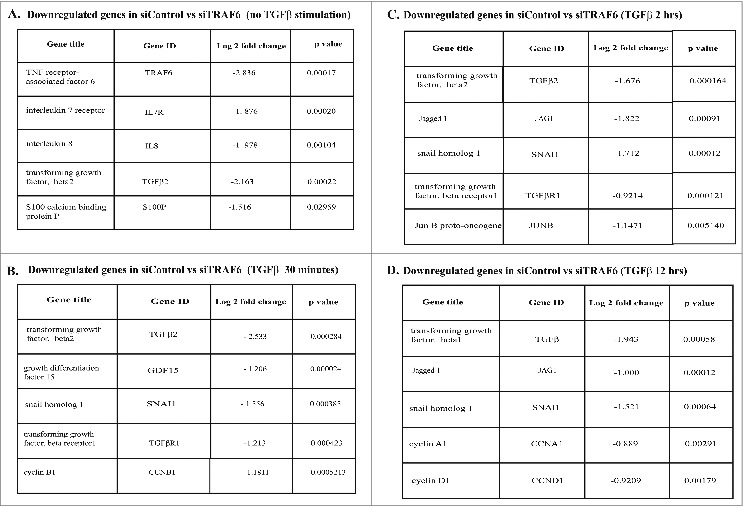
TRAF6-dependent TGFβ-induced gene expression in prostate cancer cells. PC-3U cells transfected with control or TRAF6-specific siRNA, stimulated with TGFβ for the corresponding time points (0, 30 minutes, 2 hours, 12 hours) and cells were harvested. RNA was extracted from the cells and RNA microarray analysis was performed.
We report here that TGFβ exposure increased the mRNA expression of the G0/G1 cell-cycle inducer CCND1 (encoding cyclin D1) in a manner dependent on TRAF6-induced Lys63-linked polyubiquitination of TβRI at K178 (Fig. 3). In this context, to further understand the role of cyclin D1, we investigated the cell-cycle profile of cells transfected with HA-TβRI or HA-K178R. Wild type- and mutant-transfected cell populations were chosen and gated via fluorescence-activated cell sorting; cells positive for pyronin y (a RNA stain) and Hoechst (a DNA stain) were further assessed (Fig. 5A, B). Interestingly, we found that TGFβ exposure for 48 h significantly enhanced the transition of cells from early G0 to G1 in HA-TβRI transfected cells, compared to HA-K178R transfected cells (Fig. 5B, Fig. S1A).
Figure 5.
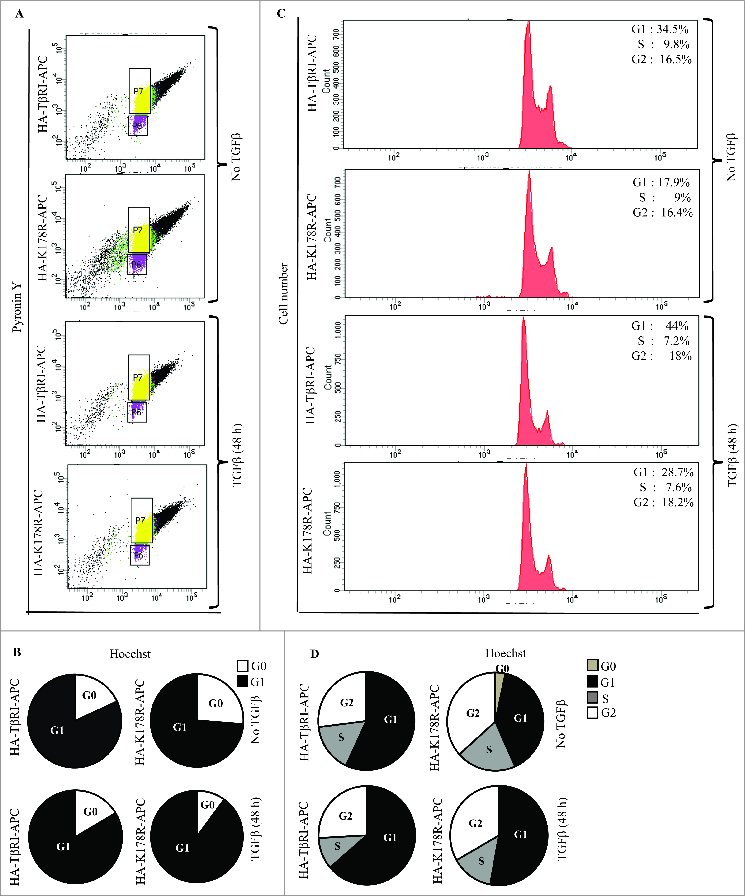
TGFβ exposure affects cell-cycle entry and regulation. (A) PC-3U cells transiently transfected with HA-TβRI (wildtype) or HA-K178R (mutant TβRI) was exposed to TGFβ as indicated. HA-TβRI or HA-K178R cell populations were chosen by staining with HA-allophycocyanin (APC). From this population, cells positive for pyronin y and Hoechst were identified (box plots, n = 10000 cells). (B) PC-3U cells transiently transfected with HA-TβRI or HA-K178R was exposed to TGFβ as indicated. HA-TβRI and HA-K178R cell populations were chosen by staining with HA-allophycocyanin and Hoechst. (C) RNA content of G0/G1 cell-cycle phases of HA-TβRI or HA-K178R cells stained with pyronin y and Hoechst (n = 5 experiments; data shown in A). (D) DNA content of cells in various phases of the cell cycle; cells were transfected with HA-TβRI or HA-K178R and stained with Hoechst (n = 5 experiments; data shown in B).
Next, cells positive for HA-TβRI-allophycocyanin or HA-K178R-allophycocyanin and Hoechst were selected and cell cycle profiles were analyzed. TGFβ treatment for 48 h promoted the cell cycle progression by increased number of cell in G1 in HA-TβRI-transfected cells, compared to HA-K178R-transfected cells. But, then there was also a reduction of cell cycle progression from G1 to S phase in HA-TβRI-transfected cells, while this response was less efficient in HA-K178R-transfected cells (Fig. 5C, D Fig. S1B). This difference could be linked to that HA-K178R-transfected cells without TGFβ treatment, were found to be moderately apoptotic (Fig. S1B), suggesting that the TβRI-TRAF6 pathway also provides pro-survival signals. Statistical analysis of 5 independent experiments revealed a significance difference in mRNA and DNA content during G1 and S between HA-TβRI-transfected cells (48 h TGFβ exposure) and HA-K178R-transfected cells (48 h TGFβ exposure) (Fig. S1B). Taken together, these results suggest that cyclin D1, a critical moderator of the transition of cells from G0 to early G1, is poorly regulated in cells transfected with HA-K178R.
TGFβ-induced nuclear localization of the TβRI-ICD is linked to the invasion behavior of cancer cells.23,24 We therefore investigated the invasive capability of PC-3U cells expressing HA-TβRI or HA-K178R. Only cells expressing HA-TβRI exhibited invasive properties (Fig. 6A) and similar results were obtained from invasion assays with LNCaP cells (Fig. 6B). From these data, we conclude that TGFβ-induced invasiveness of cancer cells is linked to TRAF6-dependent Lys63-linked polyubiquitination of Lys178 of TβRI.
Figure 6.
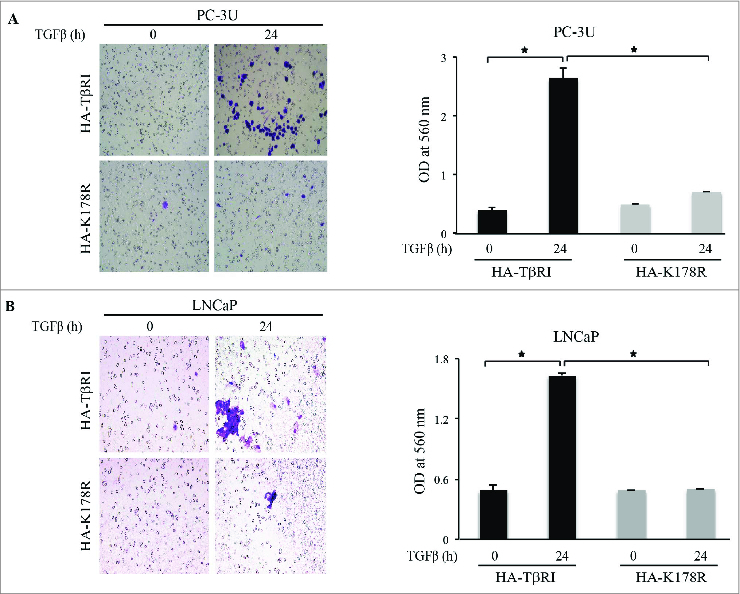
TβRI ubiquitination promotes the invasion of prostate cancer cells. (A) Invasion assay for PC-3U cells transiently transfected with HA-TβRI or HA-K178R and exposed to TGFβ as indicated. Left, cells were visualized by staining with crystal violet. Right, optical density (OD) of invasive PC-3U cells (mean ± standard deviation, n = 5, *P < 0.05). (B) Invasion assay for LNCaP cells transiently transfected with HA-TβRI or HA-K178R and exposed to TGFβ as indicated. Left, invasive cells were stained with crystal violet. Right, OD of invasive LNCaP cells (mean ± standard deviation, n = 5, *P < 0.05).
Discussion
The crucial role of TGFβ signaling during EMT and cancer progression is well established.2,3 TGFβ transduces signals through Smad and non-Smad signaling pathways. The cellular effects regulated through canonical Smad signaling have been thoroughly elucidated, while the biological significance of non-Smad signaling pathways induced by TGFβ still is gradually being unraveled.7,28 In order to enhance or specify TGFβ signaling, TRAF family members (TRAF6 and, recently, TRAF4), have been shown to play important roles in activating the TAK1-p38 MAPK pathway and in positively regulating both Smad-dependent and Smad-independent TGFβ signaling.15,16,29,30
Our previous research identified a non-Smad signaling cascade in which TRAF6 plays a central role.15 TGFβ utilizes TRAF6 to cause Lys63-linked polyubiquitination of the TβRI-ICD, which is associated with proteolytic cleavage by ADAM17/TACE and subsequent intramembranous proteolysis of TβRI by presenilin-1, a catalytic core component of the γ-secretase complex.23,24 The nuclear TβRI ICD binds the transcriptional co-activator p300 and drives the expression of proinvasive genes like Snail1 and MMP2 to promote oncogenic phenotypes such as invasion.23
In the current study, we identified Lys178 as the acceptor lysine for TRAF6-induced TβRI ubiquitination (Fig. 1). We found that mutagenesis of this lysine to arginine inhibited TGFβ-dependent ubiquitination both in vivo and in vitro. Importantly, TRAF6 associated with both TβRI and the K178R mutant of TβRI, but Lys63-polyubiquitination occurred only in cells overexpressing wild-type TβRI. Moreover, immunofluorescence assays indicated that the mutant did not enter the nucleus of HA-K178R-transfected or GFP-K178R-transfected cells, suggesting that Lys63-polyubiquitination is a prerequisite for the cleavage and nuclear trafficking of TβRI, consistent with our previous findings.23,24 Moreover, our data show that TβRI ubiquitination enhances the expression of certain target genes, like Vimentin, twist family bHLH transcription factor 1 (Twist1), and N-cadherin, implicated in EMT and invasion.
Transitions between the phases of the cell cycle are tightly regulated by the expression of cyclins, which enable cell-cycle progression.31 Most importantly, early G1 phase progression is governed by cyclin D1; in contrast, in late G1, cyclin E play a pivotal role in allowing the cell to pass the restriction point and enter S phase.32 Interestingly, we found in this study that mRNA expression of the gene encoding cyclin D1, also was enhanced upon TGFβ treatment in cells transfected with HA-TβRI compared to those transfected with HA-K178R, suggesting that TGFβ promotes both cyclin D1 expression and the invasion phenotype of prostate cancer cells, via this herein elucidated signaling pathway. When cells are arrested during the cell cycle, oncogenic signals such as active Ras or mammalian target of rapamycin (mTOR) can instruct cells to become senescent. Senescent cells are characterized by hypertrophy, high levels of cyclin D and loss of their replicative potential.33-35 Interestingly, Leontieva et al., have recently reported that p21 depletion from senescent cells leads to decreased levels of cyclin D1 and cyclin E, while the cell-cycle progressed from S phase into mitosis but resulted in cell death which occurred during mitotic arrest or after mitotic slippage.36,37 We have previously shown that TRAF6 in a non-canonical, Smad-independent pathway, regulates both the expression of c-Jun, a transcription factor implicated in stress responses, and p21, a classical target gene for TGFβ and p53, in cells treated with TGFβ.25 As cyclin D1 has been reported to contain binding sites for AP-1 transcription factor (heterodimers of the Fos and Jun families),38,39 c-Jun may regulate both the levels of both cyclin D1 and the pro-invasive protein Snail.40,41 Here, we found that TRAF6, via its Lys63-linked polyubiquitination of K178 in TβRI, regulates both EMT-associated genes as well as cyclin D1 expression which might reflect that the cancer cells also become senescent due to conflicting signaling inputs. This could further explain the aggressiveness of these cancer cells (Fig. 5, Fig. S1). Interference with this pathway could influence the tendency for senescence or result in changes of the metabolic status of cells which also is changed during TGFβ-induced EMT.42 Further studies are required to elucidate whether the pathway unraveled in this study, also controls the metabolic status of cancer cells.
In conclusion, although Smad signaling pathways are important, non-Smad signaling pathways also play critical roles in regulating cellular homeostasis in epithelial cells and in cancer initiation, progression, and invasion.23,24 In particular, the E3 ligase TRAF6 plays a key role in TGFβ-induced tumor progression due to its catalytic activity, resulting in liberation of the TβRI-ICD. Here we have demonstrated that TRAF6 promotes Lys63-linked polyubiquitination at K178 of TβRI, leading to its nuclear translocation and subsequent regulation of genes implicated in tumor invasion and progression. The identification of TRAF6 as a key component of the TGFβ-induced invasive program may be an important step in the development of future biomarkers and drugs against aggressive prostate cancer.
Materials and Methods
Cell culture
In this study, we used 2 human prostate cancer cell lines: PC-3U (derived from PC3 cells)43 and LNCaP (from ATCC). These cell lines were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 10% fetal bovine serum, 1% glutamine, and 1% penicillin/streptomycin (all from Sigma). PC-3U and LNCaP cells were starved in medium containing 1% fetal bovine serum for 18 h after transient transfection using Fugene6 (Promega). Ten nanograms per milliliter of TGFβ1 (R&D Systems) were used to stimulate cells.
Antibodies and reagents
Antibodies against the following epitopes were used for immunoblotting, immunoprecipitation, or immunofluorescence: HA (Y11) and ubiquitin (P4D1), from Santa Cruz Biotechnology; p-Smad2, generated at the Ludwig Institute for Cancer Research, Uppsala, Sweden; and Smad2, p-Smad2, p38, p-p38, and HA, from Cell Signaling. Lys63 polyubiquitin mouse monoclonal antibody was obtained from Enzo Life Sciences. An antibody against TRAF6 was obtained from Zymed Laboratories. Horseradish peroxidase-conjugated secondary anti-rabbit and anti-mouse antibodies were purchased from GE Healthcare and Dako, respectively. Light chain-specific anti-rabbit and anti-mouse antibodies were obtained from Jackson Laboratory. Secondary donkey anti-rabbit Alexa Fluor 555 antibodies and goat anti-mouse Alexa Fluor 488 antibodies (Invitrogen) were used for immunofluorescence. Tris-buffered saline and phosphate-buffered saline (PBS) were purchased from Medicago. Protein-G Sepharose was obtained from GE Healthcare, LumiLight Western Blotting Substrate and Pefabloc were purchased from Roche Life Science, and PageRuler Prestained Protein Ladder was obtained from Fermentas.
Immunoblotting and immunoprecipitation
Prior to lysis, cells were washed with ice-cold PBS and lysed in ice-cold RIPA buffer (150 mM NaCl, 50 mM Tris [pH 8.0], 1% Triton X-100, 10% (v/v) glycerol, 1 mM aprotinin, 1 mM Pefabloc, 1 mM sodium orthovanadate). The BCA Protein Assay Kit (Nordic Biolabs) was used to measure protein concentration. Equal amounts of proteins were used for immunoblotting or immunoprecipitation, and SDS-PAGE was conducted using 10%, 12%, or 4–12% bis-Tris polyacrylamide gels using MOPS buffer (Invitrogen). Proteins were transferred using an iBlot machine (Invitrogen).
Plasmids
C-terminally HA-tagged c.a.TβRI (HA-TβRI) was a kind gift from P. ten Dijke and is referred to as “wildtype” in comparison to the K187R mutant of TβRI. The expression vector for HA-K178R was generated by PCR and site-directed mutagenesis; the mutation was confirmed via sequencing. GFP-TβRI was constructed in-house, as previously described.23 Using this plasmid, we created a C-terminally GFP-tagged K178R mutant (GFP-K178R) via site-directed mutagenesis. Expression vectors for GST fusion proteins encoding the complete cytoplasmic parts (amino acid residues 148–503) of c.a.TβRI (T204D) and GST-K178R were also used.
Immunofluorescence
PC-3U cells were washed with PBS and fixed with 4% formaldehyde. Cells were permeabilized with 2% Triton X-100 and blocked in 5% bovine serum albumin. After incubation with primary and secondary antibodies, Fluoromount with 2-(4-amidinophenyl)-1H-indole-6-carboxamidine was used to stain cell nuclei. Confocal images of mounted cells were taken using a Zeiss 710 Meta (Carl Zeiss Microimaging, Inc.) with a digital camera (RET-EXi-F-M-12-C). Images were analyzed with Zen software.
qRT-PCR
An RNeasy Mini Kit (Qiagen) was used to isolate total RNA, and the Thermoscript RT-PCR System (Invitrogen) was used to prepare double-stranded cDNA. qRT-PCR was performed using Power SYBR Green PCR Masterrmix (Applied Biosystems) on an Applied Biosystems 7900HT Fast Real-time PCR system. The following primers were used for qRT-PCR (Table 1).
Table 1.
List of primers used for qRT-PCR experiments
| Gene name | Forward primer | Reverse primer |
|---|---|---|
| PAI 1 | 5′-CTCTCTCTGCCCTCACCAAC | 5′-GTGGAGAGGCTCTTGGTCTG |
| vimentin | 5′-CTCTGGCACGTCTTGACCTT | 5′-GTGAGGTCAGGCTTGGAAAC |
| Twist1 | 5′-TTCCTCTACCAGGTCCTCCA | 5′-TCCATTTTCTCCTTCTCTGGAA |
| N-cadherin | 5′-CCTGCTTCTCTGGGTTCTG | 5′-CACGTATTCTTCCAGCACA |
| p73 | 5′-GGGGACGGAATTCACCAC | 5′-CAGATGCGCCCTCAAAG |
| CCND1 | 5′-CGTGGCCTCTAAGATGAAGG | 5′-CTGGCATTTTGGAGAGGAAG |
In vivo ubiquitination assays
After stimulation with TGFβ, PC-3U cells were washed once in ice-cold PBS, then scraped into 1 mL ice-cold PBS and centrifuged at 2,000 rpm for 5 min. Non-covalent protein interactions were dissociated via treatment in 1% sodium dodecyl sulfate for 10 min at 95 °C. Samples were diluted in ice-cold PBS (1:10) containing 0.5% NP40, 1 mM aprotinin, and 1 mM Pefabloc. Samples were centrifuged at 13,000 rpm for 10 min, and then the supernatants were subjected to immunoprecipitation followed by immunoblotting.
In vitro ubiquitination assay
Reaction mixtures containing 2 µM E1, 2.5 µM E2 Ubc13-Uev1, purified GST-TβRI, or GST-K178R (~0.1 µg) were incubated at 37°C for 1 h in the presence or absence of recombinant GST-TRAF6 (∼0.1 µg) in 20 mM Tris [pH 7.4], 50 mM NaCl (Sigma), 10 mM MgCl2 (Sigma), 10 mM dithiothreitol (Sigma), 10 mM ATP (Sigma), 2.5 µM ubiquitin, and 100 µM MG132 (Sigma). Reactions were stopped by adding non-reducing sample buffer, and the samples were subjected to SDS-PAGE and immunoblotted with antisera against TβRI-(V22) (Santa Cruz Biotechnology), TRAF6 (Invitrogen), and K63-polyubiquitin (Enzo Life Sciences). Reaction mixtures with or without E1 and with or without E2 were used as controls.
Nuclear fractionation assay
Cells grown in 10-cm dishes were transfected with HA-TβRI or HA-K178R, then starved and stimulated with TGFβ for the indicated times. Subcellular fractionation was performed as previously described. Protein concentration was measured using BCA Protein Assay Kit, and equal amounts of protein were used for immunoblotting. Antibodies against lamin A (Cell Signaling) and β-tubulin (Cell Signaling) were used as loading controls for the nuclear and cytoplasmic fractions, respectively.
In situ PLA
Transiently transfected PC-3U cells were treated with TGFβ for 30 min or left untreated, fixed, blocked, and then probed with the primary antibodies indicated. These antibodies were raised in different species and directed toward the intracellular C-terminally HA-tagged domains of TβRI, Lys63-polyubiquitin, or TRAF6. Cells were blocked and incubated with 2 sets of primary antibodies; in addition, the samples were incubated with 2 PLA probes, which were later ligated and amplified. Thereafter, the slides were mounted with Duolink Mounting Medium (Sigma) and evaluated via confocal microscopy (Carl Zeiss). The amplified PLA signals were analyzed using Blob-Finder image analysis software (version 2.5), which was developed by the Center for Image Analysis, Uppsala University, Sweden.
Microarray analysis
Cells were seeded and transfected with control siRNA or TRAF6 siRNA and left untreated or stimulated with TGFβ for 30 min, 2 h, or 12 h. Total RNA was isolated from PC-3U cells transfected with HA-TβRI or HA-K178R using the RNeasy Mini Kit. We used the service at Uppsala Array Platform (Uppsala University) for microarray analysis. RNA quality was confirmed by bioanalyser prior to Affymetrix gene expression analysis. We also used bioinformatics support from Uppsala Array Platform.
Flow cytometry
PC-3U cells transfected with HA-TβRI or HA-K178R were starved and stimulated with TGFβ for 48 h. Cells were harvested, fixed, permeabilized, stained with anti-HA primary antibody (Santa Cruz Biotechnology), and incubated with secondary anti-rabbit allophycocyanin antibody (Jackson Laboratories). Later, cells were stained with pyronin Y (Sigma-Aldrich) to label RNA and Hoechst (Sigma-Aldrich) to label DNA. Fluorescence-activated cell sorting was performed at the Umea Immunology Center on a FACS LSR II (BD Biosciences), and data were analyzed using BD Biosciences software.
Invasion assays
Invasion assays were performed using the CytoSelect™ Cell Invasion Assay (Cell Biolabs, Inc.). The collagen membrane layers of the cell-culture inserts were rehydrated in 300 μL serum-free RPMI-1640, and 2 × 106 cells were seeded on the upper area of the membrane in serum-free RPMI-1640 with or without TGFβ. The lower chamber was filled with 500 μL RPMI supplemented with 10% fetal bovine serum. Non-invasive cells were removed from the upper chamber and cells that had transversed the membrane were stained with cell-staining solution (Cell Biolabs, Inc.). A Leica DMR light microscope was used to image stained invasive cells. Colorimetric quantification was performed by measuring optical density at 560 nm.
Statistical analysis
Mann-Whitney 2-tailed tests were used to calculate p values. Analyses were performed using SPSS Statistics (IBM SPSS Statistics 20 software). Values are expressed as mean ± standard deviation of 3 or more independent experiments performed in triplicate, unless otherwise indicated. P values <0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
We thank all current and previous members of our research group for constructive discussions and support. We appreciate valuable initial experiments carried out by Susanne Grimsby and Noopur Thakur, as well as comments on our manuscript given by Anders Wallenius.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants to ML from the Swedish Medical Research Council (K2013–66X-15284-04-4), the Swedish Cancer Society (13 0688), the Knut and Alice Wallenberg Foundation (2012.0090), ALF-VLL-224051 and Umeå University.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Cook LM, Shay G, Aruajo A, Lynch CC. Integrating new discoveries into the “vicious cycle” paradigm of prostate to bone metastases. Cancer Metast Rev 2014; 33:511-25; PMID:24414228; http://dx.doi.org/10.1007/s10555-014-9494-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol 2012; 13:616-30; PMID:22992590; http://dx.doi.org/10.1038/nrm3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heldin CH, Landstrom M, Moustakas A. Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol 2009; 21:166-76; PMID:19237272; http://dx.doi.org/10.1016/j.ceb.2009.01.021 [DOI] [PubMed] [Google Scholar]
- 4.Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Mechanism of activation of the TGF-beta receptor. Nature 1994; 370:341-7; PMID:8047140; http://dx.doi.org/10.1038/370341a0 [DOI] [PubMed] [Google Scholar]
- 5.Groppe J, Hinck CS, Samavarchi-Tehrani P, Zubieta C, Schuermann JP, Taylor AB, Schwarz PM, Wrana JL, Hinck AP. Cooperative assembly of TGF-beta superfamily signaling complexes is mediated by two disparate mechanisms and distinct modes of receptor binding. Mol Cell 2008; 29:157-68; PMID:18243111; http://dx.doi.org/10.1016/j.molcel.2007.11.039 [DOI] [PubMed] [Google Scholar]
- 6.Massagué J. TGFbeta in Cancer. Cell 2008; 134:215-30; PMID:18662538; http://dx.doi.org/10.1016/j.cell.2008.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mu Y, Gudey SK, Landstrom M. Non-Smad signaling pathways. Cell Tissue Res 2012; 347:11-20; PMID:21701805; http://dx.doi.org/10.1007/s00441-011-1201-y [DOI] [PubMed] [Google Scholar]
- 8.Souchelnytskyi S, Tamaki K, Engstrom U, Wernstedt C, ten Dijke P, Heldin CH. Phosphorylation of Ser465 and Ser467 in the C terminus of Smad2 mediates interaction with Smad4 and is required for transforming growth factor-beta signaling. J Biol Chem 1997; 272:28107-15; PMID:9346966; http://dx.doi.org/10.1074/jbc.272.44.28107 [DOI] [PubMed] [Google Scholar]
- 9.Macias-Silva M, Abdollah S, Hoodless PA, Pirone R, Attisano L, Wrana JL. MADR2 is a substrate of the TGFbeta receptor and its phosphorylation is required for nuclear accumulation and signaling. Cell 1996; 87:1215-24; PMID:8980228; http://dx.doi.org/10.1016/S0092-8674(00)81817-6 [DOI] [PubMed] [Google Scholar]
- 10.Massagué J. TGF-beta signal transduction. 1998; 67 [DOI] [PubMed] [Google Scholar]
- 11.Thakur N, Sorrentino A, Heldin CH, Landstrom M. TGF-beta uses the E3-ligase TRAF6 to turn on the kinase TAK1 to kill prostate cancer cells. Fut Oncol 2009; 5:1-3; PMID:19243289; http://dx.doi.org/10.2217/14796694.5.1.1 [DOI] [PubMed] [Google Scholar]
- 12.Massague J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J 2000; 19:1745-54; PMID:10775259; http://dx.doi.org/10.1093/emboj/19.8.1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moustakas A, Heldin CH. Non-Smad TGF-beta signals. J Cell Sci 2005; 118:3573-84; PMID:16105881; http://dx.doi.org/10.1242/jcs.02554 [DOI] [PubMed] [Google Scholar]
- 14.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003; 425:577-84; PMID:14534577; http://dx.doi.org/10.1038/nature02006 [DOI] [PubMed] [Google Scholar]
- 15.Sorrentino A, Thakur N, Grimsby S, Marcusson A, von Bulow V, Schuster N, Zhang S, Heldin CH, Landstrom M. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat Cell Biol 2008; 10:1199-207; PMID:18758450; http://dx.doi.org/10.1038/ncb1780 [DOI] [PubMed] [Google Scholar]
- 16.Yamashita M, Fatyol K, Jin C, Wang X, Liu Z, Zhang YE. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol Cell 2008; 31:918-24; PMID:18922473; http://dx.doi.org/10.1016/j.molcel.2008.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kirkin V, Dikic I. Ubiquitin networks in cancer. Curr Opin Genet Dev 2011; 21:21-8; PMID:21071203; http://dx.doi.org/10.1016/j.gde.2010.10.004 [DOI] [PubMed] [Google Scholar]
- 18.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem 1998; 67:425-79; PMID:9759494; http://dx.doi.org/10.1146/annurev.biochem.67.1.425 [DOI] [PubMed] [Google Scholar]
- 19.Bennett EJ, Harper JW. DNA damage: ubiquitin marks the spot. Nat Struct Mol Biol 2008; 15:20-2; PMID:18176551; http://dx.doi.org/10.1038/nsmb0108-20 [DOI] [PubMed] [Google Scholar]
- 20.Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature 2009; 458:438-44; PMID:19325623; http://dx.doi.org/10.1038/nature07960 [DOI] [PubMed] [Google Scholar]
- 21.Kravtsova-Ivantsiv Y, Ciechanover A. Non-canonical ubiquitin-based signals for proteasomal degradation. J Cell Sci 2012; 125:539-48; PMID:22389393; http://dx.doi.org/10.1242/jcs.093567 [DOI] [PubMed] [Google Scholar]
- 22.Pickart CM, Fushman D. Polyubiquitin chains: polymeric protein signals. Curr Opin Chem Biol 2004; 8:610-6; PMID:15556404; http://dx.doi.org/10.1016/j.cbpa.2004.09.009 [DOI] [PubMed] [Google Scholar]
- 23.Mu Y, Sundar R, Thakur N, Ekman M, Gudey SK, Yakymovych M, Hermansson A, Dimitriou H, Bengoechea-Alonso MT, Ericsson J, et al. TRAF6 ubiquitinates TGFbeta type I receptor to promote its cleavage and nuclear translocation in cancer. Nat Commun 2011; 2:330; PMID:21629263; http://dx.doi.org/10.1038/ncomms1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gudey SK, Sundar R, Mu Y, Wallenius A, Zang G, Bergh A, Heldin CH, Landstrom M. TRAF6 stimulates the tumor-promoting effects of TGFbeta type I receptor through polyubiquitination and activation of presenilin 1. Sci Signaling 2014; 7:ra2; PMID:24399296; http://dx.doi.org/10.1126/scisignal.2004207 [DOI] [PubMed] [Google Scholar]
- 25.Thakur N, Gudey SK, Marcusson A, Fu JY, Bergh A, Heldin CH, Landstrom M. TGFbeta-induced invasion of prostate cancer cells is promoted by c-Jun-dependent transcriptional activation of Snail1. Cell Cycle 2014; 13; PMID:24945502; http://dx.doi.org/10.4161/cc.29339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gudey SK, Wallenius A, Landstrom M. Regulated intramembrane proteolysis of the TGFbeta type I receptor conveys oncogenic signals. Fut Oncol 2014; 10:1853-61; PMID:24597658; http://dx.doi.org/10.2217/fon.14.45 [DOI] [PubMed] [Google Scholar]
- 27.Piek E, Franzen P, Heldin CH, ten Dijke P. Characterization of a 60-kDa cell surface-associated transforming growth factor-beta binding protein that can interfere with transforming growth factor-beta receptor binding. J Cell Physiol 1997; 173:447-59; PMID:9369958; http://dx.doi.org/10.1002/(SICI)1097-4652(199712)173:3%3c447::AID-JCP17%3e3.0.CO;2-8 [DOI] [PubMed] [Google Scholar]
- 28.Landstrom M. The TAK1-TRAF6 signalling pathway. Int J Biochem Cell Biol 2010; 42:585-9; PMID:20060931; http://dx.doi.org/10.1016/j.biocel.2009.12.023 [DOI] [PubMed] [Google Scholar]
- 29.De Boeck M, ten Dijke P. Key role for ubiquitin protein modification in TGFbeta signal transduction. Upsala J Med Sci 2012; 117:153-65; PMID:22335355; http://dx.doi.org/10.3109/03009734.2012.654858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang L, Zhou F, Garcia de Vinuesa A, de Kruijf EM, Mesker WE, Hui L, Drabsch Y, Li Y, Bauer A, Rousseau A, et al. TRAF4 promotes TGF-beta receptor signaling and drives breast cancer metastasis. Mol Cell 2013; 51:559-72; PMID:23973329; http://dx.doi.org/10.1016/j.molcel.2013.07.014 [DOI] [PubMed] [Google Scholar]
- 31.Tessema M, Lehmann U, Kreipe H. Cell cycle and no end. Virchows Archiv: Int J Pathol 2004; 444:313-23; PMID:14968363; http://dx.doi.org/10.1007/s00428-003-0971-3 [DOI] [PubMed] [Google Scholar]
- 32.Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Minireview: cyclin D1: normal and abnormal functions. Endocrinology 2004; 145:5439-47; PMID:15331580; http://dx.doi.org/10.1210/en.2004-0959 [DOI] [PubMed] [Google Scholar]
- 33.Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging 2012; 4:159-65; PMID:22394614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, Lowe SW. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev 1998; 12:3008-19; PMID:9765203; http://dx.doi.org/10.1101/gad.12.19.3008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007; 8:729-40; PMID:17667954; http://dx.doi.org/10.1038/nrm2233 [DOI] [PubMed] [Google Scholar]
- 36.Leontieva OV, Lenzo F, Demidenko ZN, Blagosklonny MV. Hyper-mitogenic drive coexists with mitotic incompetence in senescent cells. Cell Cycle 2012; 11:4642-9; PMID:23187803; http://dx.doi.org/10.4161/cc.22937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leontieva OV, Demidenko ZN, Blagosklonny MV. MEK drives cyclin D1 hyperelevation during geroconversion. Cell Death Differ 2013; 20:1241-9; PMID:23852369; http://dx.doi.org/10.1038/cdd.2013.86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shen Q, Uray IP, Li Y, Krisko TI, Strecker TE, Kim HT, Brown PH. The AP-1 transcription factor regulates breast cancer cell growth via cyclins and E2F factors. Oncogene 2008; 27:366-77; PMID:17637753; http://dx.doi.org/10.1038/sj.onc.1210643 [DOI] [PubMed] [Google Scholar]
- 39.Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, Arnold A, Pestell RG. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem 1995; 270:23589-97; PMID:7559524; http://dx.doi.org/10.1074/jbc.270.40.23589 [DOI] [PubMed] [Google Scholar]
- 40.Wisdom R, Johnson RS, Moore C. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J 1999; 18:188-97; PMID:9878062[http://dx.doi.org/10.1093/emboj/18.1.188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ryseck RP, Hirai SI, Yaniv M, Bravo R. Transcriptional activation of c-jun during the G0/G1 transition in mouse fibroblasts. Nature 1988; 334:535-7; PMID:3136397; http://dx.doi.org/10.1038/334535a0 [DOI] [PubMed] [Google Scholar]
- 42.Lamouille S, Derynck R. Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol 2007; 178:437-51; PMID:17646396; http://dx.doi.org/10.1083/jcb.200611146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Franzen P, Ichijo H, Miyazono K. Different signals mediate transforming growth factor-beta 1-induced growth inhibition and extracellular matrix production in prostatic carcinoma cells. Exp Cell Res 1993; 207:1-7; PMID:7686495; http://dx.doi.org/10.1006/excr.1993.1156 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.