

Abstract
Lowering low‐density lipoprotein cholesterol (LDL‐C) is a cornerstone for the prevention of atherosclerotic heart disease, improving clinical outcomes and reducing vascular mortality in patients with hypercholesterolemia. The clinical benefits of LDL‐C reduction appear to extend even to patients starting with LDL‐C as low as 60–80 mg/dL prior to initiating therapy. Statins are the first‐line agents for treating hypercholesterolemia and are effective in reducing LDL‐C, but many patients are unable to achieve their optimal lipid targets despite intensive statin therapy. Therefore, there has been a strong impetus for the development of novel pharmacologic agents designed to lower LDL‐C further in patients already on statin therapy. Genetic mutations resulting in altered cholesterol homeostasis provide valuable information regarding novel approaches for treating hypercholesterolemia. To that end, mutations in proprotein convertase subtilisin/kexin type 9 (PCSK9) were linked to altered levels of LDL‐C, illustrating this protein's role in lipid metabolism. PCSK9 promotes degradation of the LDL receptor, preventing its transport back to the cell surface and thereby increasing circulating LDL‐C. Conversely, inhibition of PCSK9 can profoundly decrease circulating LDL‐C, and thus is an attractive new target for LDL‐C–lowering therapy. AMG 145 is a fully human monoclonal immunoglobulin G2 antibody that binds specifically to human PCSK9 and inhibits its interaction with the low‐density lipoprotein receptor. In this manuscript, we describe the rationale and design of LDL‐C Assessment with PCSK9 Monoclonal Antibody Inhibition Combined With Statin Therapy–Thrombolysis In Myocardial Infarction 57 (LAPLACE‐TIMI 57; NCT01380730), a 12‐week, randomized, double‐blind, dose‐ranging, placebo‐controlled study designed to assess the safety and efficacy of AMG 145 when added to statin therapy in patients with hypercholesterolemia. Clin. Cardiol. 2012. doi: 10.1002/clc.22014
This trial was supported by Amgen. Payal Kohli, Nihar R. Desai, Robert P. Giugliano, Timothy Abrahamsen, Shannon McDonald and Marc S. Sabatine are members of the TIMI Study Group, which received research grant support from Amgen for the conduct of this trial. Payal Kohli has received honorarium for consultation from Daiichi‐Sankyo. Robert P. Giugliano has received honoraria for lectures and consultation from Amgen, Merck, Regeneron, and Sanofi‐Aventis, and research‐grant support from Merck for work related to lipid‐lowering therapies. Marc S. Sabatine has received research‐grant support from AstraZeneca, Bristol‐Myers Squibb/Sanofi‐Aventis Joint Venture, Merck, and Pfizer and honoraria for lectures and consultation from Amgen, Bristol‐Myers Squibb/Sanofi‐Aventis Joint Venture, GlaxoSmithKline, Merck, and Pfizer. Jae B. Kim, Ransi Somaratne, Fannie Huang, Beat Knusel, Scott M. Wasserman, and Robert Scott are employees and stockholders of Amgen, Inc.
Payal Kohli, MD, and Nihar R. Desai, MD, MPH, contributed equally to this work.
The authors have no other funding, financial relationships, or conflicts of interest to disclose.
Introduction
Morbidity and mortality from cardiovascular disease impose a significant burden on healthcare resources and remain the number one cause of death worldwide.1 There is a robust inverse relationship between low‐density lipoprotein cholesterol (LDL‐C) and the occurrence of major vascular events.2., 3., 4., 5. To that end, reducing circulating levels of LDL‐C has consistently been shown to reduce the risk of major vascular events, including vascular death, in patients with hypercholesterolemia,3., 6., 7. both in primary as well as secondary prevention. More recently, this finding also appears to apply to patients who already have low baseline LDL‐C values (eg, <60–80 mg/dL) whether in a primary8 or secondary prevention setting.9 Importantly, pharmacologically achieving LDL‐C concentrations <40 mg/dL appears to be safe and well‐tolerated.8., 10., 11., 12., 13., 14. Although statins, the first‐line agents for treating hypercholesterolemia, are effective in reducing LDL‐C, many patients are unable to achieve their optimal lipid targets despite intensive statin therapy.7., 15. With the advent of guidelines endorsing lower LDL‐C targets (LDL‐C <70 mg/dL or <1.8 mmol/L) in very‐high‐risk patients, there is an increasing awareness of the limits of LDL‐C lowering that can be achieved with statin monotherapy and consequently the need for adjunctive therapy. Therefore, there has been a strong impetus for the development of new pharmacologic agents to lower LDL‐C further in patients already being treated with statins.
Inhibiting Proprotein Convertase Subtilisin/Kexin Type 9
One novel approach to decreasing circulating LDL‐C is inhibition of LDL‐receptor (LDL‐R) regulation and recycling. Gain‐of‐function mutations in the proprotein convertase subtilisin/kexin type 9 (PCSK9) gene cause autosomal dominant hypercholesterolemia, with elevated LDL‐C and associated premature coronary artery disease.16 Conversely, loss‐of‐function mutations in PCSK9 are associated with a lifelong decrease in LDL‐C (28%–40% lower) and lower risk of coronary heart disease (47%–88% lower).17., 18., 19., 20., 21. Individuals with 2 loss‐of‐function alleles, resulting in no detectable PCSK9 levels, typically have very low plasma LDL‐C levels (<20 mg/dL) without any apparent adverse clinical consequences from absence of PCSK9.20., 21.
In animal models, administration of PCSK9 inhibitors markedly lowers LDL‐C.14., 15. Similarly, in gene‐inactivation studies, PCSK9−/− mice demonstrated a 4‐fold decrease in the accumulation of cholesteryl esters than did wild‐type mice, whereas transgenic mice overexpressing PCSK9 developed more severe aortic lesions and had an accumulation of cholesteryl esters in the aorta.22
The mechanism by which PCSK9 influences LDL‐C levels has not been fully elucidated. However, PCSK9 routes the LDL‐R to lysosomal degradation, instead it being recycled to the hepatocyte cell surface, thereby regulating the cell‐surface expression of LDL‐R and removal of LDL‐C from the circulation. Further, low intrahepatocyte levels of cholesterol lead to sterol regulatory element‐binding protein activation with consequent increased expression of LDL‐R and PCSK9.23,24 This phenomenon raises the possibility that PCSK9 up‐regulation may play a role in the diminishing returns of LDL‐C lowering seen with increasing doses of statins. Results from a randomized controlled trial of a high‐potency statin vs. placebo have demonstrated up‐regulation of PCSK9 following treatment with statins.25 To that end, inhibiting PCSK9 in murine models results in increased sensitivity to statins, suggesting possible synergy between these 2 pathways in regulating LDL‐C and revealing a novel therapeutic target in patients on statins.14
AMG145: A Monoclonal Antibody Inhibitor of Proprotein Convertase Subtilisin/Kexin Type 9
AMG 145 (Amgen, Thousand Oaks, CA) is a fully human monoclonal antibody (immunoglobulin G2) that binds specifically to human PCSK9. This binding prevents the PCSK9–LDL‐R interaction, leading to a near‐complete absence of detectable unbound PCSK9 immediately after administration and concurrent reduction in circulating LDL‐C by 50% to 70% following a single dose of ≥70 mg in healthy volunteers. The duration of this effect is dose dependent and the LDL‐C nadir is observed within 2 weeks of dosing.26 Phase Ib data27 in subjects on stable statin therapy demonstrated a dose‐dependent decrease in LDL‐C and unbound PCSK9 with increasing subcutaneous doses of AMG 145. Low‐density lipoprotein cholesterol was lowered by up to 81% at maximal doses, over and above the LDL‐lowering achieved with statin alone.27 Similar findings were observed from data of another PCSK9 antibody.28., 29. Regarding the safety and tolerability, both phase Ia26 and phase Ib27 data reported no serious adverse events in the AMG 145 group compared with placebo, no discontinuations from the studies related to adverse events, and only 1 case of transaminase elevation >3× upper limit of normal. There were no trends indicative of a clinically important effect of AMG 145 on hepatic‐function tests. There were also no cases of creatine kinase elevations (>5×) that were considered related to study drug. Notably, the subcutaneous injections were well‐tolerated with no severe injection‐site reactions.
In this report, we describe LDL‐C Assessment with PCSK9 Monoclonal Antibody Inhibition Combined With Statin Therapy–Thrombolysis In Myocardial Infarction 57 (LAPLACE‐TIMI 57), a phase II trial designed to assess safety, tolerability, and efficacy of different doses and dosing frequencies of AMG 145 in patients with hypercholesterolemia on statin therapy to define the optimal regimen(s) for future studies.
Study Design and Objectives
LAPLACE–TIMI 57 (NCT01380730) is a 12‐week, randomized, double‐blind, dose‐ranging, placebo‐controlled study designed to assess the safety, tolerability, and efficacy of AMG 145 when added to ongoing statin therapy in subjects with hypercholesterolemia (Figure 1). The primary objective of the study is to assess the effect of 12 weeks of therapy with AMG 145, administered every 2 weeks (Q2W) or every 4 weeks (Q4W), on percent change from baseline in LDL‐C as compared with placebo. In addition, secondary objectives include evaluation of the safety and tolerability of the study treatment, characterization of the pharmacokinetics of AMG 145, and the effects of AMG 145 on other lipid parameters (e.g. non–high‐density lipoprotein cholesterol [HDL‐C], apolipoprotein B [ApoB], total cholesterol/HDL‐C, and ApoB/ApoA1).
Figure 1.
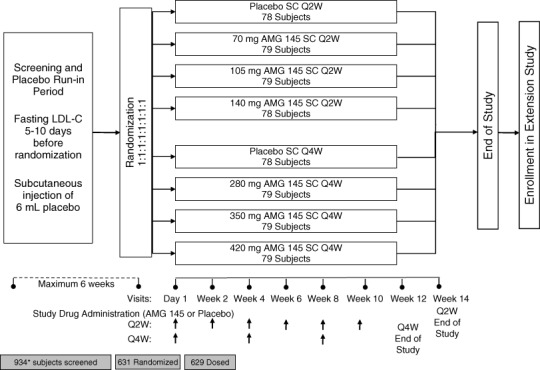
Schematic of trial design. Vertical arrows indicate study‐drug injections. Subjects successfully completing the screening and placebo run‐in were randomized to one of 8 parallel treatment arms. All subjects attended study visits every 2 weeks. Those subjects randomized to one of the 4 Q2W treatment arms received study drug every 2 weeks. Those subjects randomized to one of the 4 Q4W treatment arms received study drug every 4 weeks. The end‐of‐study visit occurs 4 weeks after the last administration of study drug, at week 14 for those subjects in the Q2W arm, and at week 12 for those subjects in the Q4W arm. Abbreviations: LDL‐C, low‐density lipoprotein cholesterol; Q2W, every 2 weeks; Q4W, every 4 weeks; SC, subcutaneous. *One subject was screened twice.
Study Population
Subjects age 18 to 80 years (inclusive) with known hypercholesterolemia (LDL‐C ≥85 mg/dL) on statin therapy (with or without concomitant ezetimibe) were eligible for screening. If a subject was on a nonstatin lipid‐modifying agent (other than ezetimibe) prior to enrollment, the investigator could withdraw this therapy and allow for a 4‐week “wash‐out” period prior to screening the subject again for enrollment. Subjects with a recent acute coronary syndrome, severe heart failure, or a recent serious arrhythmia were excluded, as were subjects with severe chronic kidney disease or other major medical comorbidities. The full inclusion and exclusion criteria are listed in Table 1.
Table 1.
Eligibility Criteria
| Inclusion criteria |
|---|
| Age 18–80 years, inclusive |
| Stable dose of statin ± ezetimibe for 4 weeks |
| Fasting LDL‐C ≥85 mg/dL (≥2.2 mmol/L) |
| Fasting TG ≤400 mg/dL (<4.5 mmol/L) |
| Key exclusion criteria |
| Cardiovascular |
| NYHA class III–IV heart failure or last known LVEF <30% |
| Serious cardiac arrhythmia within 3 months (VT, AF with rapid ventricular rate, SVT) poorly controlled with medication |
| MI/UA, PCI, CABG, stroke, DVT/PE within 3 months |
| Planned surgery or PCI |
| SBP >160 and/or DBP >100 |
| Type 1 DM; newly diagnosed or poorly controlled type 2 DM (HbA1C ≥8.5%) |
| Current therapeutic anticoagulation (nb, antiplatelet agents, eg, aspirin, clopidogrel are permitted) |
| Medications |
| Lipid‐regulating medications (niacin >200 mg/d, red yeast rice, ω‐3 >1000 mg/d, fibrates) within 6 weeks; systemic cyclosporine, systemic steroids, or isotretinoin within 3 months |
| Laboratory |
| TSH < LLN or >1.5× ULN |
| eGFR <30 ml/min/1.73m2 |
| AST or ALT >2× ULN |
| CK >3× ULN |
| Known concurrent illness within 3 months |
| Infection |
| GI, endocrine, metabolic, or hematological dysfunction |
| Malignancy (except non‐melanoma skin cancers, cervical cancer in situ, ductal carcinoma in situ, or stage I prostate cancer) within 5 years |
| Other |
| Premenopausal female subjects who are pregnant or breastfeeding or have inadequate birth control |
| Sensitivity to AMG 145 or anti‐PCSK9 therapy |
| Inability to provide informed consent or attend follow‐up visits |
| Unreliability as a participant or alcohol/drug dependence in the past year |
| Currently enrolled in another investigational device or drug study, <30 days since ending another investigational device or drug study |
Abbreviations: AF, atrial fibrillation; ALT, alanine aminotransferase; AST, aspartate aminotransferase; CABG, coronary artery bypass graft; CK, creatine kinase; DBP, diastolic blood pressure; DM, diabetes mellitus; DVT, deep vein thrombosis; eGFR, estimated glomerular filtration rate; GI, gastrointestinal; HbA1C, glycated hemoglobin; LDL‐C, low‐density lipoprotein cholesterol; LLN, lower limit of normal; LVEF, left ventricular ejection fraction; MI, myocardial infarction; NYHA, New York Heart Association; PCI, percutaneous coronary intervention; PE, pulmonary embolism; SBP, systolic blood pressure; SVT, supraventricular tachycardia; TG, triglycerides; TSH, thyroid‐stimulating hormone; UA, unstable angina; ULN, upper limit of normal; VT, ventricular tachycardia.
Treatment Protocol
After signing informed consent, eligible subjects entered a screening phase that lasted for up to 6 weeks. During this period, fasting LDL‐C was measured, safety laboratory values were assessed, and the subject received a subcutaneous (SC) placebo injection of 6 mL of normal saline (administered as 3 injections of 2 mL each). Subjects who met all eligibility criteria and tolerated the placebo injection were eligible for randomization.
Subjects were randomized in an equal fashion across all 8 treatment arms (Figure 1). Randomization was stratified by screening LDL‐C (<130 mg/dL vs ≥130 mg/dL) and concomitant use of ezetimibe. Enrollment of subjects with fasting LDL‐C of 85 to 100 mg/dL was to be limited to ≤20% of the overall cohort. The treatment arms were designed to study 2 dosing frequencies: every 2 weeks (Q2W) and every 4 weeks (Q4W). To maintain blinding within each dosing frequency, the Q2W and Q4W each had its own matching placebo arm. Within the Q2W group, there were 3 active treatment arms, with doses of 70 mg, 105 mg, and 140 mg (each consisting of a 2‐mL injection administered SC Q2W); within the Q4W group, the doses were 280 mg, 350 mg, and 420 mg (each consisting of 6 mL administered SC Q4W) (Figure 1). All subjects returned to the investigative center every 2 weeks for study visits (regardless of the dosing frequency of study drug), during which they were examined and assessed for adverse events (AEs), concurrent treatment, and dietary changes, and provided blood samples for measurements of plasma lipids and collection of safety laboratories. Throughout the duration of the study, assessment of unblinded lipids, lipoprotein profiles, or high‐sensitivity C‐reactive protein was prohibited. Laboratory assessments were conducted centrally at the study core laboratories (Medpace Reference Laboratories [MRL], Cincinnati, OH, for lipids; and Covance Central Laboratory Services, Inc., Indianapolis, IN, for safety laboratories).
Subjects were instructed to maintain their diet and exercise regimen for the duration of the study. No change in the statin regimen was permitted from 4 weeks prior to randomization to the final study visit. Likewise, ezetimibe was permitted at the discretion of the investigator up to 4 weeks prior to randomization, but it could not be added, changed, or discontinued between 4 weeks prior to randomization and the end of the study. Nonstatin lipid‐modifying therapies, including red yeast rice, niacin >200 mg daily, fibrates, or ω‐3 fatty acids >1000 mg daily, were not permitted at any time during the study. Vitamin A derivatives, cyclosporine, systemic steroids, or hormone therapy were not allowed due to their effect on lipids. The final study visit procedures, including a review of AEs, were conducted 4 weeks after the last injection for each dosing frequency (at 14 weeks for subjects on every 2 week injections and at 12 weeks for subjects on monthly injections). Subjects who discontinued the study drug prematurely were to be followed until the end of the study, including measurement of LDL‐C at all study visits. At the conclusion of the study, each subject who successfully completed LAPLACE‐TIMI 57 was given the option of enrolling in an ongoing open‐label extension (Open Label Study of Long‐Term Evaluation Against LDL‐C Trial [OSLER], NCT01439880) receiving open‐label AMG 145 and standard of care or standard of care alone, randomized in a 2:1 fashion, respectively.
Study Endpoints
The primary endpoint of LAPLACE‐TIMI 57 is the percent change in LDL‐C, measured using ultracentrifugation, from baseline (defined as the mean of the 2 most recent fasting concentrations measured through the central laboratory prior to and on the day of randomization) to week 12. Secondary efficacy endpoints include absolute change in LDL‐C from baseline to week 12, percent change in non–HDL‐C and ApoB from baseline to week 12, and percent change in the ratio of total cholesterol/HDL‐C and Apo B/ApoA1 from baseline to week 12. Additional exploratory endpoints include the absolute and percent change from baseline at each scheduled visit in various lipid and nonlipid parameters, as well as the incidence of clinical events including death, cardiac or cerebrovascular ischemic events, coronary and noncoronary revascularizations, and hospitalizations for heart failure. Key safety endpoints include the subject incidence of AEs and the incidence of anti‐AMG 145 antibodies. Pharmacokinetics endpoints include the serum concentration of PCSK9 and AMG 145 at selected visits, as well as the maximum concentration of AMG 145 after administration (Cmax), the duration of maximum concentration after administration (Tmax), and the area under the curve (AUCt) obtained between weeks 8 and 12.
Statistical Design and Analysis
We estimated that 600 subjects, approximately 75 subjects in each of the 8 treatment groups, would provide approximately 99% power to detect at least a 20% reduction in LDL‐C with AMG 145 (either Q2W or Q4W) compared with placebo.
For each dosing frequency, the primary efficacy endpoint will be analyzed using an analysis of covariance (ANCOVA) model to assess the efficacy of each AMG 145 dose group to placebo. The ANCOVA model will include terms for the treatment group and stratification factors. The highest dose will be compared with placebo using the 0.05 significance level. If the highest AMG 145 dose is found to reach statistical significance, the next‐highest dose will be assessed using a closed testing procedure. Testing of the doses will continue in descending strength until the 0.05 statistical significance is not met or the lowest dose within the dosing frequency is tested, whichever occurs first. Missing values will be imputed using the last observation carried forward approach.
Analyses will be performed in all randomized subjects who have received ≥1 dose of study drug by randomized treatment group (a modified intention‐to‐treat analysis). Additional analyses for the primary endpoint will be performed using on‐treatment analysis sets. Analyses of secondary endpoints will be similar to the primary analysis for the primary endpoint with no adjustment for multiple comparisons, at a significance level of 0.05. For the exploratory endpoints, lipid parameters will be examined by scheduled visit and the subject incidence of adjudicated events will be summarized.
For the analysis of safety data, the subject incidence of treatment‐emergent AEs will be tabulated by system organ class and preferred term by randomized treatment group. Adverse events will be coded using version 14.0 or higher of the Medical Dictionary for Regulatory Activities. The incidence and percentages of subjects who develop anti‐AMG 145 antibodies (binding and neutralizing) will be reported.
Independent Data Monitoring Committee
An external independent data monitoring committee was established to formally review the accumulating data from this and other ongoing phase II studies with AMG 145 to ensure there is no avoidable increased risk for harm to subjects (see Appendix). Analyses for the data monitoring committee will be provided by an independent biostatistical group that is external to TIMI and Amgen.
Independent Clinical Endpoint Committee
Although only a limited number of cardiovascular events are expected in this study, an independent clinical endpoint committee blinded to treatment allocation will adjudicate all deaths, suspected cardiac or cerebrovascular ischemic events, coronary and noncoronary revascularizations, hospitalizations for heart failure, and suspected adverse muscle events using standard endpoint definitions30 (see Appendix).
Substudies
Pharmacokinetics:
The objectives of this substudy are to further characterize the pharmacokinetic parameters of AMG 145 and PCSK9 levels after multiple SC dose administrations. In addition to the pharmacokinetic samples collected every 2 weeks in all subjects in the trial, additional pharmacokinetic samples will be obtained at weeks 9 and 11 in ≥120 subjects (approximately 15 subjects per treatment arm).
Biomarkers and Pharmacogenetics:
All subjects enrolled in LAPLACE‐TIMI 57 will participate in the biomarker substudy, for which 14.5 mL of blood will be collected at baseline and weeks 2, 4, and 12 for the analysis of biomarkers related, but not limited, to PCSK9 signaling, LDL‐R turnover, cholesterol metabolism, inflammation, and plaque stability. There is also an optional pharmacogenetic substudy that will allow exploration of the association of relevant genetic variants with lipid levels, the response to PCSK9, inflammatory biomarkers, and other parameters.
Study Organization
LAPLACE‐TIMI 57 is a collaboration between the TIMI Study Group (an Academic Research Organization based at Brigham and Women's Hospital and Harvard Medical School) and Amgen, the sponsor. A joint leadership team consisting of members of the TIMI Study Group and Amgen oversees all aspects of the trial (see Appendix). The study is being conducted in 102 sites across 5 countries: Canada, Czech Republic, Denmark, Hungary, and the United States.
Results
Subject recruitment began on July 6, 2011, and was completed on December 22, 2011. A total of 934 subjects were screened, and from this group 631 subjects were randomized into the trial. Baseline characteristics of the 629 subjects who received ≥1 dose of study drug are shown in Table 2. Results are based on the interim data snapshot taken February 2, 2012.
Table 2.
Baseline Characteristics (N = 629)
| Demographic | |
|---|---|
| Age, y, median (IQR) | 62 (55, 67) |
| Female sex, n (%) | 319 (51) |
| Race, n (%) | |
| White | 559 (89) |
| Asian | 13 (2) |
| Black | 50 (8) |
| Other | 7 (1) |
| Cardiac risk factors | |
| Clinical atherosclerotic disease, n (%) | 221 (35) |
| CAD | 187 (30) |
| PVD or cerebrovascular disease | 66 (11) |
| DM, n (%) | 102 (16) |
| Hypertension, n (%) | 435 (69) |
| Current smoking, n (%) | 100 (16) |
| Metabolic syndrome, n (%)a | 271 (43) |
| BMI, kg/m2, median (IQR) | 29.0 (26.1, 32.8) |
| Prerandomization screening lipids | |
| Total cholesterol, mg/dL, median (IQR) | 199 (178, 222) |
| LDL‐C, mg/dL, median (IQR) | 119 (106, 138) |
| HDL‐C, mg/dL, median (IQR) | 50 (42, 63) |
| TG, mg/dL, median (IQR) | 122 (92, 161) |
| Baseline lipid‐lowering therapy, n (%) | |
| Simvastatin | 277 (44) |
| Atorvastatin | 158 (25) |
| Rosuvastatin | 129 (21) |
| Other statin | 61 (10) |
| Ezetimibe | 50 (8) |
Abbreviations: BMI, body mass index; CAD, coronary artery disease; DBP, diastolic blood pressure; DM, diabetes mellitus; HDL‐C, high‐density lipoprotein cholesterol; IQR, interquartile range; LDL‐C, low‐density lipoprotein cholesterol; N, number of subjects randomized and dosed; PVD, peripheral vascular disease; SBP, systolic blood pressure; TG, triglycerides. a Metabolic syndrome is defined by ≥3 of the following: elevated waist circumference (≥102 cm for non‐Asian men, ≥88 cm for non‐Asian women, ≥90 cm for Asian men, or ≥80 cm for Asian women); TG ≥150 mg/dL; HDL‐C <40 mg/dL in men or <50 mg/dL in women; SBP ≥130 mm Hg, DBP ≥85 mm Hg, or history of hypertension; fasting glucose ≥110 mg/dL or type 2 DM. Results are based on interim data snapshot taken February 2, 2012.
Discussion
PCSK9 is a newly recognized mediator of LDL‐C levels through its role in LDL‐C receptor degradation.31 Genetic studies have identified loss‐of‐function mutations in PCSK9 that are associated with increased density of LDL‐R, higher rate of clearance of LDL‐C from plasma, very low levels of LDL‐C, and low rates of cardiovascular events.14., 15. Thus, inhibition of PCSK9 has become an attractive target to reduce LDL‐C.26., 27., 32. AMG 145 is a fully human monoclonal antibody that inhibits PCSK9–LDL‐R binding and profoundly lowers LDL‐C when given either as monotherapy or in combination with statin therapy. The LAPLACE‐TIMI 57 trial is studying the LDL‐C–lowering efficacy and the safety of AMG 145 in subjects with hypercholesterolemia being treated with statins and has important implications for the dose selection and design of a definitive phase III cardiovascular outcomes trial in subjects with cardiovascular disease at high risk for events.
Appendix
TIMI Study Group, Brigham and Women's Hospital, Boston, MA: Marc S. Sabatine (study chairman), Robert P. Giugliano (principal investigator), Payal Kohli (co‐investigator), Nihar Desai (co‐investigator), Suzanne E. Morin (director of operations), Timothy Abrahamsen (senior project director), Shannon McDonald (project director), Samantha Berg (research assistant), Alexandria Cavalieri (research assistant), Andrea Dedonato (research assistant), Mia DiFabbio (research assistant).
Amgen, Thousand Oaks, CA: Rob Scott (vice president, global development), Scott M. Wasserman (executive medical director, global development), Moetaz Albizem (director, global study management), Ransi Somaratne (clinical research medical director), Jae Kim (clinical research medical director), Beat Knusel (development clinical director), Fannie Huang (biostatistics manager), Sarah Hubbard (clinical research study manager), Paul‐Michael Johnson (clinical research study manager), Hayley Sloan (global study management, senior associate).
Clinical Events Committee, Boston, MA: Stephen D. Wiviott (chairman).
Independent Data Monitoring Committee: Charles H. Hennekens, (chairman), W. Virgil Brown, Barry Davis.
References
- 1. Lloyd‐Jones D, Adams RJ, Brown TM, et al. Executive summary: heart disease and stroke statistics—2010 update: a report from the American Heart Association. Circulation. 2010;121:948–954. [DOI] [PubMed] [Google Scholar]
- 2. Reiner Z, Catapano AL, De Backer G, et al. ESC/EAS Guidelines for the management of dyslipidaemias: the task force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Eur Heart J. 2011;32:1769–1818. [DOI] [PubMed] [Google Scholar]
- 3. Baigent C, Blackwell L, Emberson J, et al; Cholesterol Treatment Triallists' Collaborators. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta‐analysis of data from 170 000 participants in 26 randomised trials. Lancet. 2010;376: 1670–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baigent C, Keech A, Kearney PM, et al; Cholesterol Treatment Triallists' Collaborators. Efficacy and safety of cholesterol‐lowering treatment: prospective meta‐analysis of data from 90 056 participants in 14 randomised trials of statins [published corrections appear in Lancet. 2005;366:1358 and 2008;371:2084]. Lancet. 2005;366:1267–1278. [DOI] [PubMed] [Google Scholar]
- 5. Kearney PM, Blackwell L, Collins R, et al; Cholesterol Treatment Triallists' Collaborators. Efficacy of cholesterol‐lowering therapy in 18 686 people with diabetes in 14 randomised trials of statins: a meta‐analysis. Lancet. 2008;371:117–125. [DOI] [PubMed] [Google Scholar]
- 6. Ballantyne CM. Low‐density lipoproteins and risk for coronary artery disease. Am J Cardiol. 1998;82:3Q–12Q. [DOI] [PubMed] [Google Scholar]
- 7. O'Keefe JH Jr, Cordain L, Harris WH, et al. Optimal low‐density lipoprotein is 50 to 70 mg/dL: lower is better and physiologically normal. J Am Coll Cardiol. 2004;43:2142–2146. [DOI] [PubMed] [Google Scholar]
- 8. Hsia J, MacFadyen JG, Monyak J, et al. Cardiovascular event reduction and adverse events among subjects attaining low‐density lipoprotein cholesterol <50 mg/dL with rosuvastatin. The JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). J Am Coll Cardiol. 2011;57:1666–1675. [DOI] [PubMed] [Google Scholar]
- 9. Giraldez RR, Giugliano RP, Mohanavelu S, et al. Baseline low‐density lipoprotein cholesterol is an important predictor of the benefit of intensive lipid‐lowering therapy: a PROVE IT‐TIMI 22 (Pravastatin or Atorvastatin Evaluation and Infection Therapy‐Thrombolysis In Myocardial Infarction 22) analysis. J Am Coll Cardiol. 2008;52:914–920. [DOI] [PubMed] [Google Scholar]
- 10. Wiviott SD, Cannon CP, Morrow DA, et al. Can low‐density lipoprotein be too low? The safety and efficacy of achieving very low low‐density lipoprotein with intensive statin therapy: a PROVE IT‐TIMI 22 substudy [published correction appears in J Am Coll Cardiol. 2006;47:472]. J Am Coll Cardiol. 2005;46: 1411–1416. [DOI] [PubMed] [Google Scholar]
- 11. Wiviott SD, Mohanavelu S, Raichlen JS, et al. Safety and efficacy of achieving very low low‐density lipoprotein cholesterol levels with rosuvastatin 40 mg daily (from the ASTEROID Study). Am J Cardiol. 2009;104:29–35. [DOI] [PubMed] [Google Scholar]
- 12. Leeper NJ, Ardehali R, deGoma EM, et al. Statin use in patients with extremely low low‐density lipoprotein levels is associated with improved survival. Circulation. 2007;116:613–618. [DOI] [PubMed] [Google Scholar]
- 13. Bakker‐Arkema RG, Nawrocki JW, Black DM. Safety profile of atorvastatin‐treated patients with low LDL‐cholesterol levels. Atherosclerosis. 2000;149:123–129. [DOI] [PubMed] [Google Scholar]
- 14. Athyros VG, Tziomalos K, Karagiannis A, et al. Aggressive statin treatment, very low serum cholesterol levels and haemorrhagic stroke: is there an association? Curr Opin Cardiol. 2010;25: 406–410. [DOI] [PubMed] [Google Scholar]
- 15. Waters DD, Brotons C, Chiang CW, et al. Lipid treatment assessment project 2: a multinational survey to evaluate the proportion of patients achieving low‐density lipoprotein cholesterol goals. Circulation. 2009;120:28–34. [DOI] [PubMed] [Google Scholar]
- 16. Abifadel M, Varret M, Rabes JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–156. [DOI] [PubMed] [Google Scholar]
- 17. Cohen JC, Boerwinkle E, Mosley TH Jr, et al. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–1272. [DOI] [PubMed] [Google Scholar]
- 18. Cohen J, Pertsemlidis A, Kotowski IK, et al. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37:161–165. [DOI] [PubMed] [Google Scholar]
- 19. Zhao Z, Tuakli‐Wosornu Y, Lagace TA, et al. Molecular characterization of loss‐of‐function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet. 2006;79:514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hooper AJ, Marais AD, Tanyanyiwa DM, et al. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis. 2007;193:445–448. [DOI] [PubMed] [Google Scholar]
- 21. Benn M, Nordestgaard BG, Grande P, et al. PCSK9 R46L, low‐density lipoprotein cholesterol levels, and risk of ischemic heart disease: 3 independent studies and meta‐analyses. J Am Coll Cardiol. 2010;55:2833–2842. [DOI] [PubMed] [Google Scholar]
- 22. Denis M, Marcinkiewicz J, Zaid A, et al. Gene inactivation of proprotein convertase subtilisin/kexin type 9 reduces atherosclerosis in mice. Circulation. 2012;125:894–901. [DOI] [PubMed] [Google Scholar]
- 23. Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29:431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dubuc G, Chamberland A, Wassef H, et al. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis‐regulated convertase‐1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2004;24:1454–1459. [DOI] [PubMed] [Google Scholar]
- 25. Awan Z, Seidah NG, MacFadyen JG, et al. Rosuvastatin, proprotein convertase subtilisin/kexin type 9 concentrations, and LDL cholesterol response: the JUPITER trial. Clin Chem. 2012;58:183–189. [DOI] [PubMed] [Google Scholar]
- 26. Dias C SA, Smith B, Emery M, et al. A Phase 1, Randomized, double‐blind, placebo‐controlled, ascending single dose study to evaluate the safety, tolerability and pharmacodynamics of AMG145. Circulation (Suppl). 2011;124:A10701. [Google Scholar]
- 27. Dias C SA, Cooke B, Uy S, et al. Effects of AMG145, a fully human monoclonal antibody against PCSK9, on low‐density lipoprotein cholesterol in subjects taking statins: A phase 1, randomized, double‐blind, placebo‐controlled, ascending Study. J Am Coll Cardiol (Suppl). 2012;59:E1379. [DOI] [PubMed] [Google Scholar]
- 28. Stein EA, Mellis S, Yancopoulos GD, et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J Med. 2012;366: 1108–1118. [DOI] [PubMed] [Google Scholar]
- 29. McKenney JM, Koren MJ, Kereiakes DJ, et al. Safety and efficacy of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease, SAR236553/REGN727, in patients with primary hypercholesterolemia receiving ongoing stable atorvastatin therapy. J Am Coll Cardiol. 2012;10.1016/j.jacc.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 30. Scirica BM, Bhatt DL, Braunwald E, et al. The design and rationale of the saxagliptin assessment of vascular outcomes recorded in patients with diabetes mellitus‐thrombolysis in myocardial infarction (SAVOR‐TIMI) 53 study. Am Heart J. 2011;162:818. e6–825.e6. [DOI] [PubMed] [Google Scholar]
- 31. Horton JD, Cohen JC, Hobbs HH. Molecular biology of PCSK9: its role in LDL metabolism. Trends Biochem Sci. 2007;32:71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Swergold G, Renard R, Nadler D, Wu R, Mellis S. Safety, lipid, and lipoprotein effects of REGN727/SAR236553, a fully‐human proprotein convertase subtilisin kexin 9 (PCSK9) monoclonal antibody administered intravenously to healthy volunteers. Circulation (Suppl). 2010;122:A23251. [Google Scholar]