

Abstract
Objectives
To demonstrate that nitric oxide (NO) contributes to free radical generation after epicardial shocks and to determine the effect of a nitric oxide synthase (NOS) inhibitor, NG-nitro-L-arginine (L-NNA), on free radical generation. Background: Free radicals are generated by direct current shocks for defibrillation. NO reacts with the superoxide (O2•−) radical to form peroxynitrite (O = NOO−), which is toxic and initiates additional free radical generation. The contribution of NO to free radical generation after defibrillation is not fully defined.
Methods and results
Fourteen open chest dogs were studied. In the initial eight dogs, 40 J damped sinusoidal monophasic epicardial shocks was administered. Using electron paramagnetic resonance, we monitored the coronary sinus concentration of ascorbate free radical (Asc•−), a measure of free radical generation (total oxidative flux). Epicardial shocks were repeated after L-NNA, 5 mg/kg IV. In six additional dogs, immunohistochemical staining was done to identify nitrotyrosine, a marker of reactive nitrogen species-mediated injury, in post-shock myocardial tissue. Three of these dogs received L-NNA pre-shock. After the initial 40 J shock, Asc•− rose 39 ± 2.5% from baseline. After L-NNA infusion, a similar 40 J shock caused Asc•− to increase only 2 ± 3% from baseline (P < 0.05, post-L-NNA shock versus initial shock). Nitrotyrosine staining was more prominent in control animals than dogs receiving L-NNA, suggesting prevention of O = NOO− formation.
Conclusions
NO contributes to free radical generation and nitrosative injury after epicardial shocks; NOS inhibitors decrease radical generation by inhibiting the production of O = NOO−.
Keywords: Nitric oxide, Free radicals, Defibrillation, Ascorbate, Nitrotyrosine
1. Introduction
Although defibrillation is a lifesaving intervention, direct current (DC) shocks themselves ause myocardial toxicity and injury [1]. The mechanism of DC shockinduced myocardial injury is not fully understood. Recent evidence has implicated free radical generation as a contributing factor. Using electron paramagnetic resonance (EPR), our laboratory has shown that free radicals are generated following DC shocks [2]; this does not require a preceding period of ventricular fibrillation [2]. Our previous study demonstrated that the generation of free radicals is energy-dependent; a higher energy shock produces a greater increase in the generation of free radicals than a lower energy shock. Repeated shocks at the same energy generate equivalent rises in free radical concentration [2].
Nitric oxide (NO), a relatively stable free radical, has been proposed to contribute to myocardial reperfusion injury. NO is produced from L-arginine via the enzyme nitric oxide synthase (NOS); this process can be blocked with competitive inhibitors of NOS such as NG-nitro-L-arginine (L-NNA).
NOS is found in endothelial cells where NO has beneficial effects—it promotes vasodilation, preserves endothelial function, and is involved in the regulation of the coronary circulation. However, NO reacts with superoxide to produce peroxynitrite (O = NOO−), a toxic, highly reactive compound capable of producing irreversible cellular injury [3]. Previous research has suggested that superoxide (O2−) is formed after DC shocks to the heart [2].
We hypothesized that by administering an NOS inhibitor, L-NNA, the amount of NO available to react with DC shock-induced superoxide would be less, which in turn would decrease the generation of toxic O = NOO−.
The purpose of this study was to demonstrate that NO contributes to oxidant injury after DC shocks and that this damage can be diminished by administering L-NNA. We used EPR to measure the ascorbate free radical (Asc•−), a real-time marker of total oxidative stress [4], and performed immunohistochemical staining to identify the presence of nitrotyrosine, a byproduct resulting from the presence of O = NOO−.
2. Methods
2.1. Animal preparation
An open chest canine model was used. Fourteen dogs were anesthetized with ketamine 5 mg/kg and xylazine 2 mg/kg given intramuscularly. The animals were intubated and mechanically ventilated. Tidal volume, respiratory rate, and FiO2 were adjusted according to results from frequent blood gas measurements to maintain physiologic pH (7.35–7.45), and pO2 > 100 Torr. Anesthesia was maintained throughout the study with intermittent doses of intravenous pentobarbital 20 mg/kg. The left femoral artery and vein were cannulated as were the left and right internal jugular veins. A Dacron woven 7 French Gensini catheter was inserted under fluoroscopic guidance into the coronary sinus. A left lateral thoracotomy was performed, the pericardium was incised, and a pericardial sling constructed. A 5 French pigtail catheter was inserted through the femoral artery sheath for arterial blood sampling. The animal was given 10,000 units of heparin IV to prevent the coagulation of blood in the spectrometer and catheter.
2.2. Electron paramagnetic resonance
We used a real-time method for the detection of Asc• as previously described in detail [5] to monitor Asc•− concentration in eight dogs. Asc•− is a resonance-stabilized tricarbonyl species that is readily formed from the one-electron oxidation of ascorbate, Asc•−. Asc•− is the terminal small molecule antioxidant. Nearly every oxidative species in a biological system bring about the oxidation of Asc•−. Thus, the concentration of Asc•− is an excellent measure of the total oxidative stress in the animal [4,5]. The following is a brief review of the method.
A Varian E-4 EPR spectrometer with a TM110 cavity and an aqueous flat cell were used to measure the Asc•− signal. The lower end of the flat cell was connected, via Teflon tubing (OD: 0.5 mm), to a manifold with multiple ports. The coronary sinus catheter and the femoral artery catheter were connected to different ports of the manifold. The upper end of the flat cell was connected to the femoral vein with a variable speed infusion pump. The total transit time of blood from coronary sinus to flat cell was approximately 5 s.
The EPR instrument settings were as follow: nominal power, 40 mW; modulation amplitude, 1 G; time constant, 1 s; and scan rate 1 G/24 s. Asc•− concentrationwas proportional to signal amplitude, with 1 mm of signal height corresponding to 0.073 nmol/l Asc•− in the blood with our instrument settings. 3-Carboxy proxyl (Aldrich Chemical Co., Milwaukee, WI) was used as a concentration standard. One gram of bolus ascorbic acid, followed by constant infusion of ascorbic acid, was administered to augment the Asc•− signal, which is normally weakly detectable in arterial blood but not detectable in coronary sinus blood. The infusion rate was adjusted to achieve a steady state in the arterial and coronary sinus Asc•− signals. The arterial Asc•− signal was usually about 14 nmol/l; the coronary sinus Asc•− signal was usually about 8 nmol/l.
2.3. Nitrotyrosine immunohistochemistry
In six additional dogs, myocardium subjected to DC shocks was evaluated by immunohistochemistry. Three dogs received L-NNA pre-shock while the other three dogs received no L-NNA to serve as controls. After the experiment was completed, the heart was removed and perfused with a 4% formaldehyde buffer (500 ml). Left ventricular myocardial biopsies were obtained, cut into 2 mm sections, and post-fixed for 2 h in formaldehyde buffer. Tissue sections were processed through graded alcohols to paraffin blocks. Sections (4 mm) were cut from each block and mounted on Superfrost histology slides (Fisher Scientific, Pittsburgh PA). Sections were deparaffined, rehydrated, and then treated with Antigen Unmasking Solution (Vector Laboratories, Burlingame, CA). After washing, sections were blocked with horse serum for 1 h. Immunohistochemical staining for nitrotyrosine was performed using a Vectastain ABCAP kit (Vector Laboratories, Burlingame, CA). Antinitrotyrosine (Upstate Biotechnology, Inc., Lake Placid, NY) was used at a dilution of 1:1000 and sections were incubated overnight at 4°C. After washing, the slides were incubated with biotinylated anti-mouse antibody for 30 min, then washed and incubated for 30 min with Vectastain ABC-AP reagent. The next day, tissue sections were rinsed with phosphate-buffered saline and processed for the demonstration of immunoreactive protein by treating slides with alkaline phosphatase substrate solution and counterstaining with nuclear fast red. The presence of immunoreactive protein was assessed microscopically by an independent reviewer.
2.4. Protocol
Asc•− was continuously measured from the coronary sinus blood until a stable signal was obtained. This varied between animals but was approximately 8nM. Sampling was then switched to femoral artery blood until a stable signal was achieved, approximately 14 nM. Atropine (0.4 mg, IV) was given immediately prior to the shock to prevent post-shock atrioventricular block, bradycardia, and hypotension [6]. Hand-held electrode paddles were used to cradle the heart, with the pressure of the paddles against the heart kept as constant as possible. A commercial defibrillator (Codemaster; Hewlett-Packard, Andover, MA) delivered a DC 40 J monophasic damped sinusoidal waveform shock. Sampling of the coronary sinus blood continued for 9 min before switching to the femoral artery for approximately 5 min. Following the return of hemodynamics and Asc•− signals to baseline, 5 mg/kg of L-NNA was infused intravenously over 8 min. After the L-NNA infusion was completed, the above sequence was repeated. We have previously shown that repeated shocks of equal energy generate similar Asc•− rises in the absence of an intervention [2]; thus, any decline in Asc•− production after the second shock can be attributed to the intervention, L-NNA, given before that shock, not to repeated shocks. Based on the above observation, we administered both no L-NNA (control) shocks and L-NNA shocks on the same animal in order to reduce the animal numbers used and to minimize inter-animal variation.
ECG and arterial pressures were monitored continuously throughout the study. Ventilator settings were adjusted to maintain arterial pO2, pCO2, and pH within a physiological range.
2.5. Statistical analysis
A two-way repeated measures analysis, with treatment (L-NNA or control) and study phase (pre-shock or post-shock), was used to test for the effect of L-NNA at each study phase and test for mean change over the study phase within each treatment. The P-value for the post hoc pair-wise comparisons of means of L-NNA versus control at each study phase was adjusted using Bonferroni’s method to account for the number of tests performed. Bonferroni adjustment was also applied to the P-values of the tests comparing the mean responses between the study phases within each treatment group. A Bonferroni-adjusted P-value B/0.05 was considered statistically significant.
3. Results
The hemodynamic data showed that there were no significant differences in systolic and diastolic arterial pressures in the no L-NNA condition and L-NNA condition just prior to the shocks and after the shocks (no L-NNA pre-shock: 92 ± 6/67 ±4 mmHg, no L-NNA post-shock: 99 ± 5/72 ± 4 mmHg; L-NNA pre-shock: 91 ± 9/61 ± 6 mmHg, L-NNA post-shock: 88 ± 6/56 ± 3 mmHg).
Changes in coronary sinus Asc•− concentrations are summarized in Fig. 1 and Table 1. Since the baseline Asc•− concentration varied slightly for each shock and each dog, we show the raw Asc•− data in Table 1 and the normalized data as percent change in Fig. 1. At baseline (i.e. before shock), there was no significant difference between the Asc•− levels before the first shock (i.e. L-NNA not yet administered) and the second shock (i.e. L-NNA given). Following a 40 J coronary sinus shock Asc•− levels began to increase at 1min after the no L-NNA shocks but did not rise until 4 min after the L-NNA shocks. The difference between the no L-NNA and L-NNA shocks reached significance at 6 min post-shock (P = 0.). The peak coronary sinus Asc•− concentration rise was 39 ± 3% from baseline after the no L-NNA shock, while only a 2 ± 3% rise from baseline was found in the L-NNA group. While the peak rise in coronary sinus Asc•− reflects myocardial free radical flux at a given time point, the total integrated area under the curve for each group represents the total myocardial oxidative stress during the post-shock period. The area under the no L-NNA curve was 333 (arbitrary units) versus 79 (arbitrary units) in the LNNA curve, with an overall reduction in total myocardial free radical flux of 76%. These changes in peak and overall Asc•− concentration are not simply due to repeated shocks, since we have previously shown that shocks at the same energy without other interventions generate equal rises in Asc•− concentration [2]. Thus, the lower Asc•− rise we observed on the post-L-NNA shock must be due to the intervention, L-NNA. The arterial concentrations of Asc•− were similar just prior to the shocks in the no L-NNA (17.1 ± 0.9 nMol/l) and LNNA (15.0 ± 1.2nMol/l) shocks and showed no significant change after the shocks (16.3 ± 1.1 nMol/l versus 14.8 ± 1.0 nMol/l).
Fig. 1.
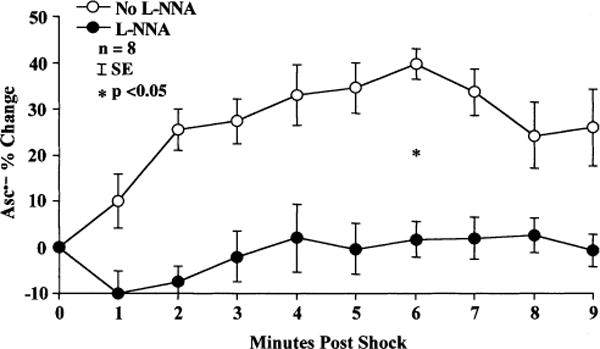
Coronary sinus Asc•− concentration increases following a 40 J DC monophasic damped sinusoidal waveform shock. Dogs that received L-NNA had a significantly lower rise in coronary sinus Asc•− levels than no L-NNA dogs.
Table 1.
Ascorbate free radical data (nM)
| Dog number | Baseline | 1 min | 2 min | 3 min | 4 min | 5 min | 6 min | 7 min | 8 min | 9 min |
|---|---|---|---|---|---|---|---|---|---|---|
| No L-NNA (control shocks) | ||||||||||
| 1 | 6.94 | 8.32 | 7.88 | 7.30 | 7.01 | 7.45 | 9.93 | 9.05 | 9.64 | 9.93 |
| 2 | 7.30 | 8.18 | 8.76 | 9.34 | 9.05 | 10.2 | 10.2 | 10.1 | 8.61 | 9.93 |
| 3 | 5.77 | 7.59 | 7.59 | 7.88 | 8.47 | 7.01 | 7.88 | 7.88 | 7.88 | 8.18 |
| 4 | 7.23 | 9.49 | – | 9.93 | 9.34 | 11.1 | 10.4 | 8.47 | 6.72 | 7.59 |
| 5 | 6.94 | 6.86 | 9.64 | 9.05 | 10.5 | 9.78 | 10.8 | 11.4 | 9.49 | 8.76 |
| 6 | 9.05 | 9.05 | – | – | 12.0 | 11.4 | 11.7 | 11.7 | 11.4 | 11.7 |
| 7 | 6.72 | 6.72 | 8.32 | 8.47 | 9.78 | 9.05 | 9.64 | 8.76 | 9.93 | 9.93 |
| 8 | 7.88 | 6.86 | – | – | – | 12.0 | 9.93 | 9.64 | 7.59 | 6.13 |
| Mean | 7.22 | 7.88 | 8.44 | 8.66 | 9.45 | 9.75 | 10.1 | 9.62 | 8.91 | 9.02 |
| S.D. | 0.95 | 1.05 | 0.80 | 0.97 | 1.57 | 1.81 | 1.10 | 1.36 | 1.50 | 1.72 |
| L-NNA given before shocks | ||||||||||
| 1 | 7.37 | 8.03 | 6.57 | 5.55 | 4.96 | 6.42 | 6.72 | 6.72 | 6.86 | 6.42 |
| 2 | 8.83 | 7.15 | 8.45 | 9.64 | 12.1 | 10.8 | 10.5 | 10.8 | 10.8 | 10.2 |
| 3 | 7.59 | 6.72 | 6.72 | 6.72 | 7.01 | 6.13 | 7.59 | 6.42 | 8.18 | 7.74 |
| 4 | 9.13 | 8.91 | 8.18 | 7.59 | 8.18 | 7.74 | 8.18 | 8.45 | 7.88 | 7.88 |
| 5 | 7.01 | 7.01 | 6.23 | 6.42 | 6.57 | 6.42 | 6.42 | 7.15 | 7.30 | 7.01 |
| 6 | 7.52 | 6.57 | 8.47 | 8.47 | 8.18 | 8.03 | 8.47 | 8.47 | 7.59 | 7.59 |
| 7 | 8.40 | 7.88 | 7.88 | 9.05 | 9.34 | 9.05 | 8.76 | 8.18 | 8.76 | 9.05 |
| 8 | 8.54 | 5.40 | 7.01 | 9.93 | 9.93 | 9.93 | 9.05 | 9.64 | 8.76 | 8.18 |
| Mean | 8.05 | 7.21 | 7.45 | 7.92 | 8.29 | 8.07 | 8.21 | 8.23 | 8.27 | 8.01 |
| S.D. | 0.77 | 1.07 | 0.90 | 1.60 | 2.21 | 1.74 | 1.32 | 1.49 | 1.22 | 1.18 |
Immunohistochemical staining for the presence of nitrotyrosine, a marker of reactive itrogen speciesmediated injury, including O = NOO− was performed in six dogs. Photomicrographs of the immunohistochemistry staining are shown in Fig. 2. Nitrotyrosine staining was much heavier in the control (no L-NNA) (Fig. 2A) dogs than the dogs receiving L-NNA (Fig. 2B), suggesting that L-NNA blunted the formation of O = NOO−.
Fig. 2.
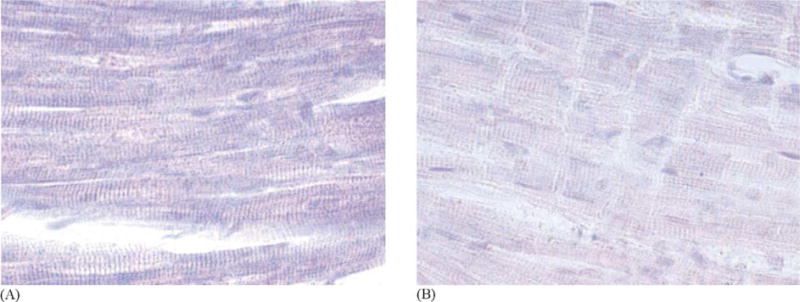
(A) Representative photomicrograph of myocardium subjected to a 40 J shock without receiving L-NNA and incubated with antinitrotyrosine antibody. Heavy nitrotyrosine staining of the cytoplasm (purple stain) is evident. (B) Representative photomicrograph of myocardium subjected to a 40 J shock and incubated with anti-nitrotyrosine antibody. This dog received L-NNA prior to the shock. Only minimal nitrotyrosine staining is present. Compare with (A).
4. Discussion
4.1. Major findings
The major findings of this study are as follows: (1) the NO = O = NOO− pathway contributes to free radical generation from DC shocks; and (2) use of the NOS inhibitor L-NNA decreases the production of free radicals following DC shocks.
4.2. Defibrillation injury
Myocardial injury following DC shocks has been well documented. Doherty et al. [7] showed technetium-99M pyrophosphate uptake consistent with myocardial injury following transthoracic shocks. Warner et al. [1] described pathologic changes of epicardial necrosis that began 2 = 24 h post-shock and evolved over weeks to become scar. Microscopically, dehiscence of the intercalated disks between myocytes occurs accompanied by cellular edema and necrosis. Ultrastructural changes found immediately post-shock include intracellular edema, vacuole development, and mitochondrial degeneration. Over the next 24 h, glycogen depletion occurs with progressive mitochondrial damage, disruption of myofibrils, and dilation of sarcoplasmic reticulum and ttubules [8]. Functional cardiac abnormalities after shocks have also been described including regional contractile abnormalities after epicardial shocks, atrioventricular block, ectopy, supraventricular and ventricular tachycardia, pulseless electrical activity, and refractory ventricular fibrillation [8–11]. Profound but potentially reversible myocardial failure after resuscitation from cardiac arrest has been described by Tang et al. [12] and Gazmuri et al. [13].
4.3. Mechanisms of defibrillation injury
The mechanisms of defibrillation injury are not well established. Vanvleet and Tacker [8] proposed that increased intramyocardial temperature caused by repeated shocks might contribute to post-shock injury.
Others have suggested that a transient increase in cell membrane permeability may occur in the presence of high-intensity electrical fields. Tovar and Tung [14] demonstrated a steep increase in membrane conductance (electroporation). The time course of membrane recovery was highly variable with different defibrillation waveforms. Such electroporation could result in cellular edema, loss of cellular enzymes, and electrolyte fluxes predisposing to arrhythmia. Myocardial injury can be induced by free radicals, a mechanism implicated in ischemia_/reperfusion injury [15]. Our experiments concerned NO and its reaction product O = NOO−. NO is generated from L-arginine by the enzyme NOS in the presence of nicotinamideadenine-dinucleotide phosphate (NADPH) and molecular oxygen by the following reaction:
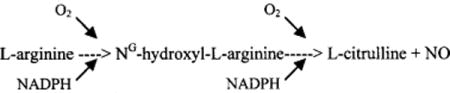
NO is a relatively stable radical but can combine with superoxide to produce O = NOO−. The role of NO and O = NOO− has been studied in ischemia-reperfusion injury. Several authors have found NO to be deleterious during reperfusion [16–20]. Wang and Zweier [21] observed that Ng-nitro-L-arginine methyl ester (LNAME) inhibited NO generation by 70–80% and doubled the recovery of contractile function in isolated perfused rat hearts subjected to 30 min of global ischemia. Zhang et al. [22], in an in vivo closed-chestcanine model of ischemia-reperfusion, used EPR measurements measurements of coronary sinus ascorbate radicals to show that L-NNA decreased free radical generation and nitrotyrosine accumulation, and ameliorated post-reperfusion myocardial “stunning”. NO is a compound of multiple biologic activities and may be a “double-edged sword”. On one hand, NO helps maintain endothelial function, prevents leukocyte adhesion, vasodilates, regulates coronary blood flow, and inhibits platelet aggregation, and can even serve as a chain-breaking antioxidant [23]. This may account for the beneficial effects of NO that many other investigators have emphasized [24–28]. On the other hand, O = NOO− resulting from the reaction of NO and O2•− can be a cellular toxin inducing lipid peroxidation and protein damage, and promoting cellular dysfunction and myocardial stunning. Beckman and Koppenol [3] have proposed that NO may also be a major pathway for the generation of hydroxyl radical (HO+), an oxidizing radical implicated in cellular injury:
It may be that at physiologic levels NO has important beneficial effects but at higher concentrations, especially in a state of increased superoxide production such as ischemia-reperfusion, high levels of O = NOO− are produced and cellular injury occurs.
Does this occur during defibrillation? Free radicals have been shown to occur after DC shocks in an energy dose-dependent manner. A preceding period of ventricular fibrillation is not required [2]. Trouton et al. [29] found that mitochondrial oxygen consumption decreased following DC shocks and suggested that mitochondrial dysfunction and free radical generation might contribute to cellular injury. Trouton et al. [30] further showed that, at voltage gradients great enough to produce myocardial necrosis, electrical discharges did not directly depress mitochondrial function, suggesting other secondary mechanisms for mitochondrial dysfunction.
Using an electron paramagnetic spin resonance method to monitor in real-time the production of free radicals, Caterine et al. [2] previously demonstrated that free radicals were generated when epicardial DC countershocks were delivered to heart and there was a significant linear relation between the shock energy and percent free radical increase. The free radicals generated were similar whether the heart was in ventricular fibrillation or in sinus rhythm, and repeated shocks generated similar Asc•− rise after each shock [2], supporting our conclusion in this study that the blunting of the Asc•− rise after the L-NNA pretreatment may be attributed to L-NNA, not simply to a repeated shock. In addition, Ponderoso et al. [31] demonstrated that NO caused inhibition of mitochondrial enzyme activity. Xie and Wolin [32] showed that blocking NOS limited reoxygenation-induced inhibition of myocyte mitochondrial respiration. Thus, it appears that the NO pathway is involved in cellular dysfunction and injury.
In this study, we have confirmed our earlier work [2] showing that DC shocks generate free radicals, and have extended that work by demonstrating that O = NOO−, a toxic oxidant, is produced during defibrillation. Inhibiting NOS with L-NNA prior to delivery of the shock significantly reduces post-shock NO, O = NOO−, and subsequent free radical generation.
5. Limitations
While this study showed decreased free radical generation following open chest shocks, we do not know if similar results would be obtained following transthoracic shocks. Only a relatively small portion of total transchest current actually traverses the heart when transthoracic shocks are given [33]; this may result in much less myocardial free radical generation.
Only a single L-NNA dose was administered based on experiments published in the literature; it is not known whether different doses may have different effects on free radical and NO/O = NOO− generation or on myocardial function.
Our shocks were administered without a preceding period of ventricular fibrillation. It is not known what effect L-NNA administration would have after prolonged fibrillation and hypoxia, a situation commonly encountered in clinical cardiac arrests.
L-NNA was given before shocks. It is not known if LNNA would provide a similar effect if given post-shock, a property that would be clinically useful.
Nitrotyrosine, a byproduct of O = NOO−, was demonstrated by immunohistochemistry. The method of nitrotyrosine immunohistochemistry we employed shows qualitative differences, but it is not quantitative. No statistical analysis of nitrotyrosine staining was performed.
Finally, this study used only monophasic damped sine wave shocks. It is not known what effect alternative defibrillation waveforms such as biphasic shocks may have on free radical generation.
6. Conclusions
Free radicals and related oxidants (O = NOO−) are generated by DC shocks and are known to be toxic to the myocardium. NO contributes to this free radical production. Pre-shock administration of the NOS inhibitor L-NNA significantly diminishes the coronary sinus concentrations of oxidants and nitrating species post-shock. Whether L-NNA may have a clinical value utility in ameliorating myocardial dysfunction after defibrillation will require additional studies.
Acknowledgments
Supported by NHLBI grants HL-07121 (C.B.C.), HL53284 (R.E.K.), HL62984 (F.J.M.), HL03669 (F.J.M.), CA84462 (G.R.B., S.M.M.), and AHA grant number 0020656Z (Y.Z.).
Abbreviations
- EPR
electron paramagnetic resonance
- DC
direct current
- NO
nitric oxide
- NOS
nitric oxide synthase
- L-NNA
Ng-nitro-L-arginine
- O = NOO−
peroxynitrite
- Asc•−
ascorbate free radical
- AscH−
one-electron oxidation of ascorbate
- HO•
hydroxyl radical
- L-NAME
Ng-nitro-L-arginine methyl ester
- NADPH
nicotinamide-adenine-dinucleotide phosphate
Footnotes
doi of original article 10.1016/S0300-9572(02)00413-6.
References
- 1.Warner ED, Dahl C, Ewy GA. Myocardial injury from transthoracic defibrillator countershock. Arch Pathol. 1975;99:55–9. [PubMed] [Google Scholar]
- 2.Caterine MR, Spencer KT, Pagan-Carlo LA, et al. Direct current shocks to the heart generate free radicals: an electron paramagnetic resonance study. J Am Coll Cardiol. 1996;28:1598–609. doi: 10.1016/s0735-1097(96)00333-6. [DOI] [PubMed] [Google Scholar]
- 3.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and the ugly. Am J Physiol. 1996;271:C1424–37. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 4.Buettner GR, Jurkiewicz BA. Ascorbate free radical as a marker of oxidative stress: an EPR study. Free Radic Biol Med. 1993;14:49–55. doi: 10.1016/0891-5849(93)90508-r. [DOI] [PubMed] [Google Scholar]
- 5.Sharma MK, Buettner GR, Spencer KT, et al. Ascorbyl free radical as a real-time marker of free radical generation in briefly ischemic and reperfused hearts: an electron paramagnetic resonance study. Circ Res. 1994;74:650–8. doi: 10.1161/01.res.74.4.650. [DOI] [PubMed] [Google Scholar]
- 6.Pansegrau DG, Abboud FM. Hemodynamic effects of ventricular defibrillation. J Clin Invest. 1970;49:282–97. doi: 10.1172/JCI106238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doherty PW, McLaughlin PR, Billingham M, et al. Cardiac damage produced by direct current countershock applied to the heart. Am J Cardiol. 1979;43:225–32. doi: 10.1016/s0002-9149(79)80008-9. [DOI] [PubMed] [Google Scholar]
- 8.Vanvleet JF, Tacker WA. Cardiac damage from transchest and ICD defibrillator shocks. In: Tacker WA, editor. Defibrillation of the Heart (Chapter 12) St Louis, MO: Mosby; 1994. pp. 259–98. [Google Scholar]
- 9.Kerber RE, Martin JB, Gascho JA, et al. Effect of direct-current countershocks on regional myocardial contractility and perfusion. Circulation. 1981;63:323–32. doi: 10.1161/01.cir.63.2.323. [DOI] [PubMed] [Google Scholar]
- 10.Weaver WD, Cobb LA, Copass MK, et al. Ventricular defibrillation—a comparative trial using 175-J and 320-J shocks. N Engl J Med. 1982;307:1101–7. doi: 10.1056/NEJM198210283071801. [DOI] [PubMed] [Google Scholar]
- 11.Anderson GL, Reiser J. Electrophysiologic changes induced by external transthoracic countershock. J Electrocardiol. 1983;16(2):191–8. doi: 10.1016/s0022-0736(83)80023-5. [DOI] [PubMed] [Google Scholar]
- 12.Tang W, Weil MH, Sun S, et al. Progressive myocardial dysfunction after cardiac resuscitation. Crit Care Med. 1993;21:1046–50. doi: 10.1097/00003246-199307000-00022. [DOI] [PubMed] [Google Scholar]
- 13.Gazmuri RJ, Weil MH, Bisera J, et al. Myocardial dysfunction after successful resuscitation from cardiac arrest. Crit Care Med. 1996;24:992–1000. doi: 10.1097/00003246-199606000-00020. [DOI] [PubMed] [Google Scholar]
- 14.Tovar O, Tung L. Electroporation and recovery of cardiac cell membrane with rectangular voltage pulses. Am J Physiol. 1992;263:H1128–36. doi: 10.1152/ajpheart.1992.263.4.H1128. [DOI] [PubMed] [Google Scholar]
- 15.Bolli R, Jerrodi MO, Patel BS, et al. Direct evidence that oxygen-derived free radicals contribute to post-ischemic myocardial dysfunction in the intact dog. Proc Natl Acad Sci USA. 1989;86:4695–9. doi: 10.1073/pnas.86.12.4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matheis G, Sherman MP, Buckberg GD, et al. Role of L-argininenitric oxide pathway in myocardial reoxygenation injury. Am J Physiol. 1992;262:H617–20. doi: 10.1152/ajpheart.1992.262.2.H616. [DOI] [PubMed] [Google Scholar]
- 17.Patel VC, Yellon DM, Singh KJ, et al. Inhibition of nitric oxide limits infarct size in the in situ rabbit heart. Biochem Biophys Res Commun. 1993;194:234–8. doi: 10.1006/bbrc.1993.1809. [DOI] [PubMed] [Google Scholar]
- 18.Morita K, Sherman MP, Buckberg GD, et al. Studies of hypoxemic/reoxygenation injury without aortic clamping. V. Role of the L-arginine-nitric oxide pathway: the nitric oxide paradox. J Thorac Cardiovasc Surg. 1995;11:1200–11. doi: 10.1016/s0022-5223(95)70006-4. [DOI] [PubMed] [Google Scholar]
- 19.Naseem SA, Kontos MC, Rao PS, et al. Sustained inhibition of nitric oxide by Ng-nitro-L-arginine improves myocardial function following ischemia/reperfusion in isolated perfused rat heart. J Mol Cell Cardiol. 1995;27:419–26. doi: 10.1016/s0022-2828(08)80038-7. [DOI] [PubMed] [Google Scholar]
- 20.Ma XL, Lopez BL, Liu GL, et al. Peroxynitrite aggravates myocardial reperfusion injury in the isolated perfused rat heart. Cardiovasc Res. 1997;36:195–204. doi: 10.1016/s0008-6363(97)00179-x. [DOI] [PubMed] [Google Scholar]
- 21.Wang P, Zweier J. Measurement of nitric oxide and peroxynitrite generation in the post-ischemic heart. J Biol Chem. 1996;271:29223–30. doi: 10.1074/jbc.271.46.29223. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Bissing JW, Xu L, et al. Nitric oxide synthase inhibitors decrease coronary sinus free radical concentration and ameliorate myocardial stunning in an ischemia–reperfusion model. J Am Coll Cardiol. 2001;38:546–54. doi: 10.1016/s0735-1097(01)01400-0. [DOI] [PubMed] [Google Scholar]
- 23.Kelley EE, Wagner BA, Buettner GR, et al. Nitric oxide inhibits iron-induced lipid peroxidation in HL-60 cells. Arch Biochem Biophys. 1999;1:97–104. doi: 10.1006/abbi.1999.1386. [DOI] [PubMed] [Google Scholar]
- 24.Pabla R, Buda AJ, Flynn DM, et al. Nitric oxide attenuates neutrophil-mediated myocardial contractile dysfunction after ischemia and reperfusion. Circ Res. 1996;78:65–72. doi: 10.1161/01.res.78.1.65. [DOI] [PubMed] [Google Scholar]
- 25.Lefer DJ, Nakamishi K, Johnston WE, et al. Antineutrophil and myocardial protecting actions of a novel nitric oxide donor after acute myocardial ischemia and reperfusion in dogs. Circulation. 1993;88:2337–50. doi: 10.1161/01.cir.88.5.2337. [DOI] [PubMed] [Google Scholar]
- 26.Lefer DJ, Scalia R, Campbell B, et al. Peroxynitrite inhibits leukocyte –endothelial cell interactions and protects against ischemia–reperfusion injury in rats. J Clin Invest. 1997;99:684–91. doi: 10.1172/JCI119212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nossuli TO, Hayward R, Scalia R, et al. Peroxynitrite reduces myocardial infarct size and preserves coronary endothelium after ischemia and reperfusion in cats. Circulation. 1997;96:2317–24. doi: 10.1161/01.cir.96.7.2317. [DOI] [PubMed] [Google Scholar]
- 28.Hasebe N, Shen Y, Vatner S. Inhibition of endothelium-derived relaxing factor enhances myocardial stunning in conscious dogs. Circulation. 1993;88:2862–71. doi: 10.1161/01.cir.88.6.2862. [DOI] [PubMed] [Google Scholar]
- 29.Trouton TG, Allen JD, Young LK, et al. Metabolic changes and mitochondrial dysfunction early following transthoracic countershock in dogs. Pacing Clin Electrophysiol. 1989;11:1827–34. doi: 10.1111/j.1540-8159.1989.tb01869.x. [DOI] [PubMed] [Google Scholar]
- 30.Trouton TG, Barry JJ, Allen JD, et al. Failure of countershocktype pulses in vitro to adversely alter mitochondrial oxidative phosphorylation. Ann Emerg Med. 1992;21:132–6. doi: 10.1016/s0196-0644(05)80146-5. [DOI] [PubMed] [Google Scholar]
- 31.Ponderoso JJ, Carreras MC, Lisdero C, et al. Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch Biochem Biophys. 1996;328:85–92. doi: 10.1006/abbi.1996.0146. [DOI] [PubMed] [Google Scholar]
- 32.Xie YW, Wolin MS. Role of nitric oxide and its interaction with superoxide in the suppression of cardiac muscle mitochondrial respiration: involvement in response to hypoxia/reoxygenation. Circulation. 1996;94:2580–6. doi: 10.1161/01.cir.94.10.2580. [DOI] [PubMed] [Google Scholar]
- 33.Lerman BB, Deale OC. Relation between transcardiac and transthoracic current during defibrillation in humans. Circ Res. 1990;67:1420–6. doi: 10.1161/01.res.67.6.1420. [DOI] [PubMed] [Google Scholar]