

SUMMARY
Background
We conducted a prospective, randomised, double-blind trial to evaluate the efficacy of imatinib rechallenge in patients with metastatic and/or unresectable gastrointestinal stromal tumors (GIST) following objective progression of prior approved tyrosine kinase inhibitor (TKI) therapy.
Method
Between July 2010 and January 2013, 81 patients with prior benefit from first-line imatinib (initial response or stable disease for ≥6 months) and subsequent progression on at least imatinib and sunitinib were randomly assigned in a 1:1 ratio (by computer-generated randomisation list and the central coordinating center using telephone in a double-blind manner; randomised block permutation methods with block size of 2, 4, and 6; stratified by treatment line and performance status) to receive best supportive care with either imatinib 400 mg/day (n=41) or a placebo (n=40). At the time of disease progression, determined by the investigators, crossover to open-label imatinib was allowed. The primary endpoint was progression-free survival (PFS) determined by blinded external radiology review. Secondary endpoints included the disease control rate at 12 weeks, overall survival, and safety. All analyses were based on the full analysis set. This study is registered with ClinicalTrials.gov, number NCT01151852.
Findings
The median PFS was 1·8 months (95% confidence interval [CI], 1·7–3·6) with imatinib as compared with 0·9 months (95% CI, 0·9–1·7) with placebo (hazard ratio for progression or death, 0·46; 95% confidence interval [CI], 0·27–0·76; p=0·005, two-sided). In the placebo arm, 37 patients (93%) crossed over to open-label imatinib after progression. The most common grade 3 or higher adverse events of imatinib resumption was anemia (12 of 41, 29%), fatigue (four of 41, 10%), and hyperbilirubinemia (three of 41, 7%).
Interpretation
Despite prior resistance to imatinib, resumption of imatinib significantly improves PFS in GIST patients after disease progression on at least imatinib and sunitinib, demonstrating that residual bulk disease contains clones with continued sensitivity.
Keywords: imatinib, resistance, gastrointestinal stromal tumors
INTRODUCTION
Gastrointestinal stromal tumors (GIST) are the most common mesenchymal tumors of the digestive tract. More than 90% of GISTs are characterized by mutations in the KIT or platelet-derived growth factor receptor α (PDGFRA) genes, resulting in the constitutive activation of kinase signaling.1–3
Imatinib mesylate, an oral tyrosine kinase inhibitor (TKI) with activity against KIT, PDGFRA, ABL, and DDR, is currently the standard first-line therapy for patients with metastatic and/or unresectable GIST.4 Extended follow-up results of the initial pivotal B2222 study have demonstrated the long-term efficacy of imatinib treatment with a median time-to-progression of 2 years and a median overall survival (OS) of approximately 5 years.5 In the setting of imatinib failure, the phase 3 trial of sunitinib resulted in a median time-to-progression of about 7 months, leading to the approval of sunitinib as the standard second-line therapy for GIST.6 Regorafenib has been recently approved as the third-line therapy based on the results of an international phase 3 trial which documented significant improvement in progression-free survival (PFS) with regorafenib compared with placebo (4·8 vs. 0·9 months) after prior failure of at least imatinib and sunitinib.7
Despite recent improvements in the management of GIST, most patients eventually experience failure of all available standard therapies due to evolution of polyclonal disease that results in progression of TKI-resistant GIST.8 For these patients, reintroduction of imatinib dosing has been commonly practiced and is recommended in many treatment guidelines with the goal of delaying disease progression and palliating symptoms by targeting GIST clones which retain sensitivity to imatinib inhibition.9–11 This strategy has been based on anecdotal findings that the withdrawal of imatinib in patients with an imatinib-refractory GIST induced acute exacerbation or appearance of symptoms, and a “flare” phenomenon on 18F-fluorodeoxyglucose (FDG)-positron emission tomography (PET) imaging (i.e. a rapid upregulation of metabolic activity even within previously dormant tumor lesions).12, 13 However, no prospective randomised clinical trial had been conducted to evaluate rigorously the efficacy of an imatinib rechallenge in this setting.
To address this, we report here a randomised phase 3 trial entitled “Rechallenge of Imatinib in GIST Having no effective Treatment” (RIGHT) which was designed and conducted to determine the efficacy and safety of imatinib, in comparison with a placebo, in patients with metastatic and/or unresectable GIST who had initial benefit from imatinib and subsequently evolved to progression of GIST despite having treatment with at least imatinib and sunitinib.
METHODS
This investigator-initiated study consisted of a randomised, placebo-controlled, double-blind, phase 3 trial accrued at a single institution (Asan Medical Center, Seoul, Korea, designed and conducted jointly with planned external blinded radiology review at the Dana-Farber Cancer Institute and Harvard Medical School, Boston, MA, USA). The research was approved by the institutional review boards of both centers, and conducted in accordance with the Declaration of Helsinki and the Good Clinical Practice guidelines (ClinicalTrials.gov identifier: NCT01151852). All participants provided written informed consent before enrolment. This trial was initiated and designed by two academic investigators (Y-KK, GDD).
Patients
Patients with histologically proven metastatic and/or unresectable GISTs were eligible for enrolment in this study if their tumors had progressed during active treatment with at least prior imatinib and sunitinib sequentially, in accordance with the Response Evaluation Criteria In Solid Tumor (RECIST) version 1·0.14 Documented clinical benefit (i.e. lack of primary resistance) with prior first-line imatinib therapy, defined as complete response (CR), partial response (PR), and stable disease (SD) for at least 6 months, was also required for eligibility. Other inclusion criteria were an age ≥18 years, an Eastern Cooperative Oncology Group (ECOG) performance status of 0–3, at least one measurable lesion, and adequate hematologic, hepatic, and renal function.
Study design and procedures
Eligible patients were randomly assigned in a 1:1 ratio to the imatinib or placebo arms using a random permuted block method. Randomization was stratified by the ECOG performance status (0–1 vs. 2–3) and by the number of prior lines of TKI therapy (i.e. ≤ 2 vs. ≥ 3). Randomization was performed in a double-blind manner, so that the allocated treatment was masked to both the investigators and patients. Patients were randomly assigned by a computer-generated list using a random block permutation method with block size of 2, 4, and 6. Random sequence was kept concealed from investigators. Allocation was done centrally at the coordinating center by telephone. For patients assigned to the imatinib arm, a once daily dose of 400 mg of imatinib was given. In the placebo group, patients each received four capsules of placebo which had the same appearance as the imatinib. Both active imatinib and matching placebo were provided by Novartis Oncology, Basel, Switzerland. Doses of study drugs were modified or interrupted for grade 3–4 hematololgical toxicities (excluding anemia) and grade 2–4 non-hematological toxicities. Other anti-cancer treatments, such as chemotherapy, radiotherapy, other targeted therapy, and surgical resections, were not permitted during the period in which the study treatment was masked. Best supportive care was given to all patients in both trial arms. Masked study treatments continued until patients showed disease progression according to a local investigator assessment, unacceptable toxicities, or if they withdrew consent.
Baseline assessments were performed within two weeks before randomization and in each case included medical history, physical examination, laboratory tests, a computed tomography (CT) scan with contrast enhancement, and a fusion FDG-PET/CT scan. Assessment of toxicities, physical examinations, and laboratory tests were performed at 2 weeks, 4 weeks and every 4 weeks thereafter. For response assessments, CT scans were performed every 4 weeks for the first 4 months of study and every 8 weeks thereafter. Additional imaging was performed if clinically indicated.
Tumor responses were initially determined by the local investigators in accordance with RECIST 1·0.14 Treatment decisions were based on local on-site radiology review. All imaging data were subsequently collected, anonymized and centrally reviewed later in a double-blind manner by two external academic radiology reviewers (NR, JJ). On an average, 4 imaging time-points per patient were available (range, 2–8). Response assessment was determined in a blinded central review using RECIST 1·1 (total 740 target lesions).15 For the assessment of study treatment compliance, the plasma imatinib trough concentration was measured after 2 weeks on study drug for patients in both arms, and the results were masked from both the investigators and patients. Adverse events were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 3·0.
At the time of progression as assessed by the local investigators, the treatment group was unmasked. Patients were then allowed to crossover to receive unblinded imatinib therapy at a 400 mg dose once daily if they had been originally assigned to the placebo arm, or to continue imatinib as open-label treatment if they had been initially assigned to the imatinib arm. In both cases, crossover was at the discretion of the investigator in shared decision-making with the patient.
Statistical analysis
The primary endpoint of the trial was PFS per blinded external radiology central review. Secondary endpoints included the disease control rate (defined as the proportion of CR, PR, and SD for at least 12 weeks), OS, and safety profile. This trial was designed for 80 patients to be randomly assigned to either an imatinib or placebo treatment group. The trial had a power of 85% and Two-sided overall alpha of 0·05 to detect a hazard ratio of 0·50 in PFS at 12 weeks. Two sets of interim analyses were planned when 26 events (progression or death) and 55 events occurred, using the O’Brien-Fleming stopping boundaries with a two-sided alpha of 0·001 for the first interim analysis and 0·017 for the second interim analysis. A final analysis for the comparison of PFS between the imatinib and placebo arms was scheduled to take place after 76 events or at 9 weeks after the accrual of the last patient, whichever occurred first, and was performed using the log-rank test with a two-sided alpha of 0·044 after adjusting for the two interim analyses.
All efficacy parameters were analyzed based on the full analysis set, which included those patients who received at least one dose of the study drug. PFS was defined as the time from randomization to the documentation of disease progression or death, and OS as time from randomization to death from any cause. Patients were censored if they were progression-free and alive at the last follow-up. Kaplan-Meier curves were used to estimate PFS and OS, and compared using the log-rank test. Stratified and crude Cox proportional hazard model was used to estimate hazard ratios and 95% confidence intervals (CI) for comparing the efficacy between treatment groups in the primary and subgroup analyses. The results of stratified analyses were presented here and those of crude analyses were in the web appendix. The proportionality of the Cox regression was checked by plotting log(−log(survival). All statistical analyses were performed by a dedicated biostatistician (B-HN) using SAS version 9·1 (SAS Institute Inc., Cary, NC) and SPSS version 18·0 (SPSS, Chicago, IL).
Role of the funding source
Novartis Oncology supported this study by providing the study drugs (imatinib and matching placebo) and measurement of plasma imatinib trough concentration, as well as financial support to perform the blinded external radiology review, but had no role in the design or conduct of this study, the collection, management, analysis, and interpretation of the data, nor the preparation, review, or approval of the manuscript, or decision to submit the study for publication. The corresponding author (Y-KK) had full access to all the date in the study and had final responsibility for the decision to submit for publication.
RESULTS
Patient characteristics
Between July 20, 2010 and January 17, 2013, a total of 81 patients were randomly assigned to the imatinib (41) or placebo (40) arms (Figure 1). Baseline characteristics were well balanced between the two groups (Table 1). Approximately 40% of patients in both arms received three or more lines of prior TKI therapies, which included (beyond prior imatinib and sunitinib) nilotinib, sorafenib, regorafenib and dovitinib. About 60% of the patients received first-line imatinib at 400 mg/day for at least 2 years. After progression on first-line imatinib at 400 mg/day, most of our patients (76 of 81, 94%) received escalated dose of imatinib (600 mg/day or 800 mg/day); all in the imatinib arm and 88% (35 of 40) in the placebo arm. A KIT exon 11 mutation was the most common primary genotype, reported in 80% of the patients.
Figure 1.
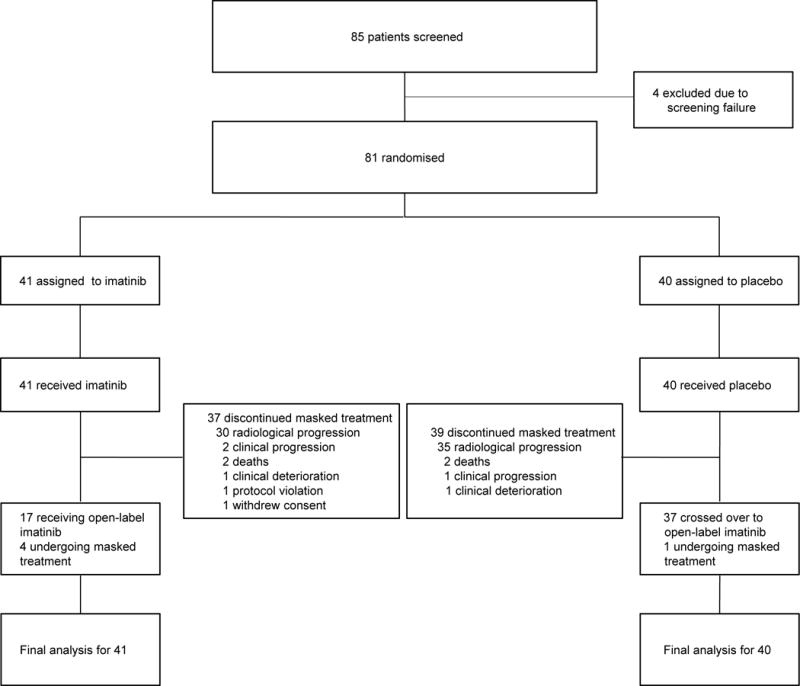
CONSORT diagram
Table 1.
Baseline patient characteristics
| Characteristic | Imatinib arm (%), n=41 |
Placebo arm (%), n=40 |
|---|---|---|
| Median age, years (IQR) | 57 (52–65) | 61 (54–67) |
| Male gender | 29 (71) | 26 (65) |
| ECOG performance status | ||
| 0–1 | 28 (68) | 28 (70) |
| 2–3 | 13 (32) | 12 (30) |
| Primary site | ||
| Stomach | 16 (39) | 13 (33) |
| Small bowel | 20 (49) | 25 (62) |
| Others | 5 (12) | 2 (5) |
| Previous third or more lines of therapy | 16 (39) | 16 (40) |
| Prior nilotinib | 7 (17) | 9 (22) |
| Prior regorafenib or sorafenib | 5 (12) | 10 (25) |
| Prior dovitinib | 7 (17) | 3 (8) |
| Duration of previous first-line imatinibtherapy (400 mg/day) | ||
| ≥6 and <12 months | 3 (7) | 5 (13) |
| ≥12 and <24 months | 14 (34) | 10 (25) |
| ≥24 months | 24 (59) | 25 (62) |
| Duration of previous second-line sunitinib | ||
| ≥6 months | 26 (63) | 18 (45) |
| Primary genotype | N=38 | N=39 |
| KIT exon 11 mutation | 31 (82) | 30 (77) |
| KIT exon 9 mutation | 4 (10) | 5 (13) |
| Others | 3 (8) | 4 (10) |
ECOG, Eastern Cooperative Oncology Group
The assessment of the plasma imatinib trough concentration after 2 weeks of study treatment was available in 40 patients of the imatinib arm and 38 patients of the placebo arm. The median imatinib trough concentration was 1840 ng/mL (range, 670–5820) in the imatinib arm. In the placebo arm, no plasma imatinib was detectable in 36 patients and two patients had a detectable but very low imatinib plasma level (108 ng/mL and 380 ng/mL). These data indicated an acceptable adherence to the study treatment regimens among the enrolled patients.
Efficacy
An independent data monitoring committee reviewed the two preplanned sets of interim analyses and both times recommended continuation of the study. The trial database was locked for the final analysis in March 2013 according to predetermined criteria (9 weeks after the enrollment of the last patient): 72 events (35 in the imatinib arm and 37 in the placebo arm) were determined by local investigator review, and 64 events were confirmed by subsequent blinded central review. Most patients that the tumor progression was determined by investigators but not confirmed by blinded external radiological review had an increase in tumor size that positioned on the borderline of definition of progressive disease according to the RECIST. For the primary analysis, the median follow-up period was 5·2 months (interquartile range [IQR], 3·4–9·4) in living patients. Per central review, 30 patients (73%) in the imatinib arm and 34 patients (85%) in the placebo arm showed disease progression or died.
The median PFS outcomes determined by blinded external central radiologic assessment was 1·8 months (95% CI, 1·73·6) in the imatinib arm, compared with 0·9 months (95% CI, 0·9–1·7) in the placebo arm. The hazard ratio for disease progression or death in the imatinib arm was 0·46 (95% CI, 0·27–0·78; p=0·005), indicating a 54% risk reduction (Figure 2A). The estimated proportion of patients who were progression-free at 4 weeks, 8 weeks, and 12 weeks was 73% (n=30), 42% (17), and 32% (13) in the imatinib arm, respectively, as compared with 43% (17), 15% (6) and 5% (2) in the placebo arm, respectively. The differences at each time point were statistically significant (p=0·005, p=0·008, and p=0·003, respectively). Local investigator assessment showed a median PFS of 1·8 months (95% CI, 1.7–2.7) in the imatinib arm and 1·7 months (95% CI, 0.9–1.8) in the placebo group (hazard ratio=0·56, 95% CI, 0·35–0·93; p=0·02; Supplementary figure S1). Secondary efficacy endpoints of this study were summarized in Table 2. No patient in both arms achieved an objective response in either the blinded central radiologic review or local investigator assessment.
Figure 2. Progression-free survival (A) and overall survival (B).
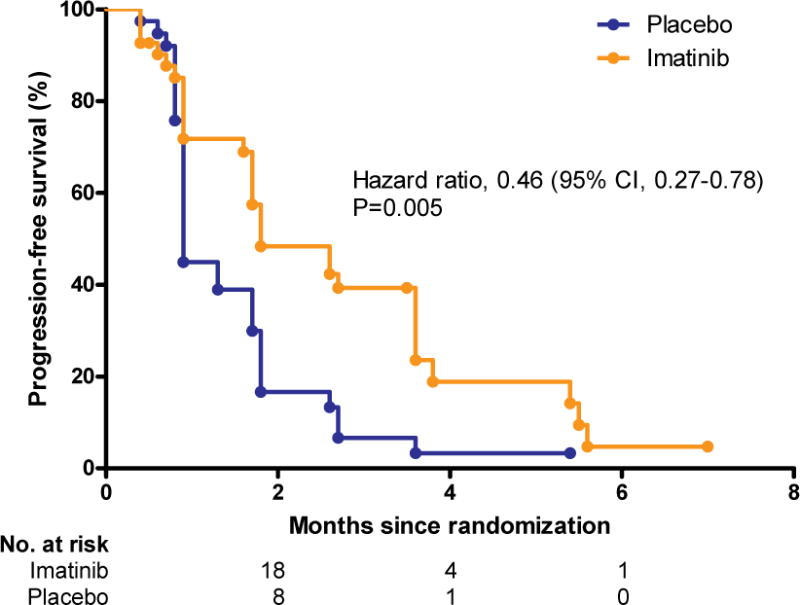
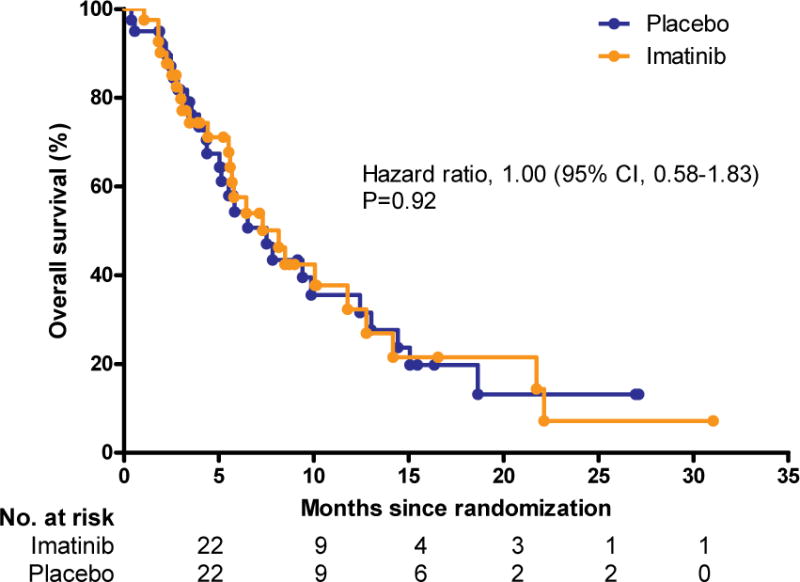
CI, confidence interval
Table 2.
Secondary efficacy endpoints
| Imatinib arm (N=41) | Placebo arm (N=40) | P value | |
|---|---|---|---|
|
| |||
| No. (%) | No. (%) | ||
| Response | 0·03 | ||
| Complete response/Partial response | 0 | 0 | |
| Stable disease | 17 (42) | 6 (15) | |
| Progressive disease | 19 (46) | 29 (73) | |
| Not evaluable | 5 (12) | 5 (13) | |
| Disease control ≥12 weeks* | 13 (32) | 2 (5) | 0·003 |
| Time to progression | Median 1·8 months (95% CI, 1·7–3·6) |
Median 0·9 months (95% CI, 0·9–1·7) |
0·002 HR, 0·48 (95% CI, 0·28–0·82) |
| Overall survival | Median 8·2 months (95% CI, 5·5–12·8) |
Median 7·5 months (95% CI, 4·4–12·4) |
0·92; HR, 1·00 (95% CI, 0·58–1·83) |
Complete response or partial response plus stable disease lasting for at least 12 weeks
HR, hazard ratio; CI, confidence interval
Prespecified subgroup analyses in the blinded central radiologic review revealed that benefits of imatinib in terms of PFS were observed across all subgroups and that there was no significant interaction between the efficacy of imatinib and patient characteristics (Figure 3).
Figure 3. Subgroup analysis of progression-free survival.
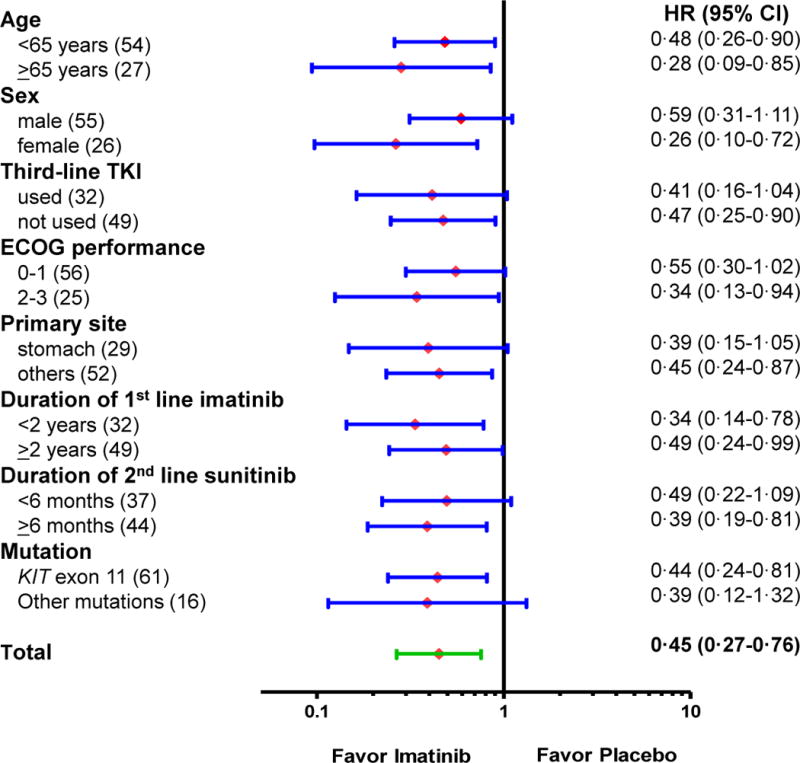
HR, hazard ratio. CI, confidence interval.
Of the 40 patients initially assigned to the placebo arm, 37 of these participants (93%) crossed over to an open-label imatinib. Thirty five of these patients had objective tumor progression confirmed by the blinded central radiologic review. Disease progression in the remaining two patients was only determined by local investigator assessment. The median PFS after crossover to open-label imatinib was 1·7 months (95% CI, 1·52·0) by local investigator assessment (Supplementary Figure S2). A crossover to imatinib was not done in three patients, because of death without progression in two patients and non-progression with placebo in one patient. Further TKI therapy including nilotinib, dovitinib, sorafenib, and regorafenib after failure of crossover to imatinib resumption was given in eight patients (20%) originally assigned to the placebo arm.
Of the 41 patients initially assigned to the imatinib arm, 17 patients (41%) continued open-label imatinib and 9 (22%) received other TKIs (nilotinib, n=5; sorafenib or regorafenib, n=3; and dovitinib, n=1). Further anti-cancer therapies were not given after a failure of imatinib in 11 patients. The remaining four patients in the imatinib arm received ongoing treatment with imatinib without progression. The median OS was estimated at 8·2 months (95% CI, 5·5–12·8) in the imatinib arm and 7·5 months (95% CI, 4·4–12·4) in the placebo arm (Figure 2B). There was no significant difference found between the two groups (hazard ratio for death with imatinib, 1·00; 95% CI, 0·58–1·83; p=0·92).
Safety
Analyses for adverse events were performed for all randomised patients. Adverse events that occurred in 10% or more of the trial participants are listed in Table 3. There was no treatment-related death in either arm. The most common adverse event of any grade was anemia, which occurred in 66% of the imatinib arm and 45% of the placebo arm patients, followed by edema in 44% of the imatinib arm and 13% of the placebo arm cases. Although severe grades of adverse events were rare overall, grade 3 or 4 anemia (29%), fatigue (10%), and hyperbilirubinemia (7%) were more frequent in the imatinib arm compared with the placebo arm. However, none of patients underwent a dose modification or discontinuation of their study treatment due to drug-related adverse events.
Table 3.
Adverse events occurring in ≥10% of patients
| Adverse events | Imatinib arm (n=41) |
Placebo arm (n=40) |
P value |
|---|---|---|---|
| Any toxicity | |||
| All grades | 41 (100%) | 39 (98%) | 0·49 |
| Grade 3 or 4 | 20 (49%) | 7 (18%) | 0·004 |
| Neutropenia | |||
| All grades | 12 (29%) | 5 (13%) | 0·10 |
| Grade 3 or 4 | 1 (2%) | 0 | 1·00 |
| Anemia | |||
| All grades | 27 (66%) | 18 (45%) | 0·08 |
| Grade 3 or 4 | 12 (29%) | 3 (8%) | 0·02 |
| Thrombocytopenia | |||
| All grades | 8 (20%) | 3 (8%) | 0·19 |
| Grade 3 or 4 | 0 | 0 | 1·00 |
| Edema | |||
| All grades | 18 (44%) | 5 (13%) | 0·003 |
| Grade 3 or 4 | 0 | 0 | 1·00 |
| Fatigue | |||
| All grades | 15 (37%) | 5 (13%) | 0·02 |
| Grade 3 or 4 | 4 (10%) | 0 | 0·12 |
| Anorexia | |||
| All grades | 14 (34%) | 8 (20%) | 0·21 |
| Grade 3 or 4 | 1 (2%) | 1 (3%) | 1·00 |
| Nausea | |||
| All grades | 13 (32%) | 1 (3%) | 0·0007 |
| Grade 3 or 4 | 0 | 0 | 1·00 |
| Vomiting | |||
| All grades | 13 (32%) | 2 (5%) | 0·003 |
| Grade 3 or 4 | 0 | 0 | 1·00 |
| Constipation | |||
| All grades | 6 (15%) | 4 (10%) | 0·74 |
| Grade 3 or 4 | 0 | 0 | 1·00 |
| Diarrhea | |||
| All grades | 5 (12%) | 4 (10%) | 1·00 |
| Grade 3 or 4 | 0 | 0 | 1·00 |
| Hyperbilirubinemia | |||
| All grades | 10 (24%) | 6 (15%) | 0·40 |
| Grade 3 or 4 | 3 (7%) | 1 (3%) | 0·62 |
| Azotemia | |||
| All grades | 8 (20%) | 4 (10%) | 0·35 |
| Grade 3 or 4 | 0 | 0 | 1·00 |
DISCUSSION
This trial demonstrates that resumption of imatinib dosing after prior failure of imatinib and other TKIs for metastatic GIST can significantly improve PFS and disease control rates at 12 weeks when compared with placebo. The median PFS with imatinib was twice that for patients receiving a placebo (1·8 months vs 0·9 months), and we found that compared with a placebo, a rechallenge of imatinib led to a 55% risk reduction for progression or death with an estimated improvement of 26·7% in the disease control rate at 12 weeks. In the placebo arm, the median PFS after crossover to imatinib was 1·7 months. This outcome was almost identical to that found for the blinded imatinib treatment in the imatinib arm, which likely confounded the ability of this crossover trial to detect the potential benefit with imatinib in terms of OS.
The discarding of an anti-cancer agent following disease progression has been a traditional paradigm in the management of cancer patients. In the era of targeted therapy that has a different mode of activity compared to conventional cytotoxic chemotherapy, however, continued treatment with targeted agents beyond progression has been suggested to have potential clinical benefit, particularly in patients with HER-2-positive breast cancer and colorectal cancer.16, 17 In our present study, we demonstrate that the re-introduction of imatinib dosing can, in fact, delay tumor progression compared with a placebo in patients with a multiple TKI-refractory GIST. The beneficial effects of a rechallenge therapy with imatinib are likely due to a continuous kinase inhibition by this drug in the bulk of the GIST cells which have retained their sensitivity to imatinib. However, the uncontrolled progression of imatinib-resistant tumor cells almost certainly explains the short-lived benefits of this strategy (approximately 1 month improvement in median PFS). The recent phase 3 GRID trial tested regorafenib versus placebo in a similar therapeutic setting to that of our current trial and reported a median PFS of 4·8 months.7 This is superior to that of imatinib as reported in our present study, whilst the median PFS in the placebo arms of our current trial and in the GRID trial was identical at 0·9 months.
There is little doubt that it is most preferable for patients who experience resistance to standard TKI therapy with objective disease progression despite all standard treatments to participate in clinical trials. The opportunities for receiving new investigational agents, however, are not available in substantial proportions of patients due to their ineligibility for clinical reasons such as a poor performance status or abnormal organ function, and also due to the lack of availability of appropriate trials across the world. For such GIST patients who have no available therapeutic option of proven benefit, the results of our study indicate that kinase inhibition is reasonable to continue through an imatinib rechallenge (if imatinib was effective previously in the patient) in order to suppress the growth of residual bulk of imatinib-sensitive GIST cells. Our results also suggest that the efficacy of new investigational drugs in future randomised trials for TKI-resistant GIST patients might reasonably be compared against imatinib as a comparator, rather than placebo.
Because median PFS in the imatinib arm and that after crossover to imatinib in the placebo arm were almost identical (1·8 months vs. 1·7 months), optimal timing of imatinib resumption may be a practical issue. In this study, response evaluation was performed very frequently (CT scans every 4 weeks for the first 4 months and every 8 weeks thereafter) to estimate the accurate efficacy of this strategy and reduce the potential delay of effective treatment in the placebo arm. In daily practice setting, however, it is difficult to follow up patients as done in this study. Therefore, although it is difficult to state this issue definitely, we believe that the earlier imatinib resumption may be more beneficial in these patients. Furthermore, as our results suggest that the continuation of kinase inhibition is necessary to delay tumor progression, resumption of imatinib and its continuation even after progression might be a practical way of treating patients with TKI-refractory GIST who had no other available therapeutic option. We are now conducting the quality of life analyses in this study population to see whether imatinib resumption help to improve quality of life. The results of those analyses, which will be presented elsewhere in detail, may be informative to support the benefit of imatinib resumption in this setting.
The safety profile of the imatinib rechallenge intervention in our trial was consistent with that in previous clinical trials.4 Although the disease status of our current patients was far advanced, imatinib was found to be well tolerated and we observed no occurrence of adverse events which would require a dose modification or discontinuation of treatment. Considering that drug tolerability is one of major concerns in salvage treatment, imatinib may have an advantage over other approved TKIs as a rechallenge agent.
Although this study was conducted in a single center, our study cohort does likely reflect the general GIST population in Korea, considering that this trial was well known to patients via the non-profit patient support group and patients were very eager to participate in this trial because there were no available approved options in advanced GIST following failure of both imatinib and sunitinib. Furthermore, data worldwide in GIST support the fact that there is no significant difference in terms of prevalence and characteristics of this disease among different ethnic groups and different countries. Therefore, application of our results in different ethnic groups or geographic regions might not be limited.
In this study, repeat biopsy for genotyping at the time of imatinib resumption was not mandatory, and no information on the post-resistance genotype of patients is available. Previous studies have indicated that tumor biopsies do not provide a comprehensive assessment of the mutational status of GIST following the development of TKI resistance, since resistant clones are distinctly different across lesions and even within single lesions from individual GIST patients.8 Therefore, lack of information on the post-resistance genotype might not limit the interpretation of our results.
In summary, our current study reveals that a resumption of imatinib therapy improves PFS in patients with an unresectable or metastatic GIST after failure of prior treatment with multiple TKIs, including at least imatinib and sunitinib. This evidence suggests that the continuous suppression of kinase activity may be necessary even after a treatment failure with all approved TKIs. Although this may appear to contradict certain long-standing dogmas in medical oncology, the biological rationale of targeting the bulk of residual tumor cells which retain sensitivity to imatinib is compelling and now documented by the outcomes demonstrated in this clinical trial.
Supplementary Material
Panel: Research in context.
Systematic review
When we planned the study, a resumption of imatinib dosing has been commonly practiced with the goal of delaying disease progression and palliating symptoms by targeting tumor clones which retain sensitivity to imatinib inhibition for patients with gastrointestinal stromal tumors (GIST) after failure of all approved tyrosine kinase inhibitors (TKIs). In several countries, however, this therapeutic strategy has not been approved for reimbursement due to the lack of evidence for efficacy. We searched PubMed articles and the abstracts of relevant oncology congresses with the terms “gastrointestinal stromal tumor”, “metastatic”, “unresectable”, “imatinib”, “rechallenge” and “refractory”. All studies for a rechallenge of imatinib for TKI-refractory GIST were conducted by retrospective analysis and mostly based on the small sample sizes. However, there was no placebo-controlled, randomised study to evaluate the efficacy of imatinib in the setting of failure of at least standard first-line imatinib and second-line sunitinib.
Interpretation
Our study demonstrates that reintroduction of imatinib dosing after prior failure of imatinib and other TKIs for advanced GIST can significantly improve progression-free survival when compared with placebo. The study shows that a continuous kinase inhibition by a rechallenge of imatinib in the bulk of GIST cells which retain their sensitivity to imatinib is effective therapeutic strategy for GIST patients after failure of all approved treatments. Our results also suggest that the efficacy of new investigational drugs in future randomised trials for TKI-resistant GIST patients might reasonably be compared against imatinib as a comparator, rather than placebo.
Acknowledgments
The authors thank all of the patients who participated in this trial, the Korean GIST Patient Advocacy Group for its support, and the clinical staff of the Asan Medical Center for their commitment to this study.
Funding Supported in part by Novartis Oncology, Basel, Switzerland
Funding/Support: This study was supported in part by Novartis Oncology, with additional support from the Ludwig Center at Dana-Faber/Havard.
Y-KK has received honoraria and a research grant from Novartis and Bayer. GDD_has received consultant and research support from Bayer, Novartis, Pfizer, Sanofi-Aventis, and Glaxo-Smith-Kline; and has been a consultant for Foundation Medicine, Ariad, Kolltan Pharmaceuticals, and Blueprint Medicines.
Footnotes
Conflict of Interest Disclosures
The other authors declare no potential conflict of interests relevant to this article.
Author Contributions: Dr. Kang had full access to all of the data in this study and takes responsibility for the integrity of the data and the accuracy of the analyses.
Study concept and design: Kang, Demetri
Acquisition of data: Kang, Ryu, Ryoo, Kim, Lee, Ramaiya, Jagannathan, Demetri
Analysis and interpretation of data: Kang, Ryu, Yoo, Nam, Demetri
Drafting of the manuscript: Kang, Yoo, Demetri
Critical revision of the manuscript for important intellectual content: Kang, Ryu, Yoo, Ryoo, Kim, Lee, Nam, Ramaiya, Jagannathan, Demetri
Statistical analysis: Nam
Obtained funding:
Study supervision: Kang
Previous Presentation: Presented in part at the 2013 ASCO annual meeting, May 31-June 4, 2013 in Chicago, United States.
References
- 1.Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):577–80. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 2.Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708–10. doi: 10.1126/science.1079666. [DOI] [PubMed] [Google Scholar]
- 3.Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11(12):865–78. doi: 10.1038/nrc3143. [DOI] [PubMed] [Google Scholar]
- 4.Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472–80. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 5.Blanke CD, Demetri GD, von Mehren M, et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26(4):620–5. doi: 10.1200/JCO.2007.13.4403. [DOI] [PubMed] [Google Scholar]
- 6.Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329–38. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]
- 7.Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295–302. doi: 10.1016/S0140-6736(12)61857-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wardelmann E, Merkelbach-Bruse S, Pauls K, et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res. 2006;12(6):1743–9. doi: 10.1158/1078-0432.CCR-05-1211. [DOI] [PubMed] [Google Scholar]
- 9.National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology Soft Tissue Sarcoma (Version 3.2012) Accessed February 23, 2013. [Google Scholar]
- 10.European Sarcoma Network Working Group. Gastrointestinal stromal tumors: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(Suppl 7):vii49–55. doi: 10.1093/annonc/mds252. [DOI] [PubMed] [Google Scholar]
- 11.Kang YK, Kang HJ, Kim KM, et al. Clinical practice guideline for accurate diagnosis and effective treatment of gastrointestinal stromal tumor in Korea. Cancer Res Treat. 2012;44(2):85–96. doi: 10.4143/crt.2012.44.2.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Italiano A, Cioffi A, Coco P, et al. Patterns of care, prognosis, and survival in patients with metastatic gastrointestinal stromal tumors (GIST) refractory to first-line imatinib and second-line sunitinib. Ann Surg Oncol. 2012;19(5):1551–9. doi: 10.1245/s10434-011-2120-6. [DOI] [PubMed] [Google Scholar]
- 13.Van den Abbeele A, Badawi R, Manola J, et al. Effects of cessation of imatinib mesylate (IM) therapy in patients (pts) with IM-refractory gastrointestinal stromal tumors (GIST) as visualized by FDG-PET scanning. J Clin Oncol. 2004;14(suppl):198. [Google Scholar]
- 14.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92(3):205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 15.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45(2):228–47. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 16.von Minckwitz G, du Bois A, Schmidt M, et al. Trastuzumab beyond progression in human epidermal growth factor receptor 2-positive advanced breast cancer: a german breast group 26/breast international group 03–05 study. J Clin Oncol. 2009;27(12):1999–2006. doi: 10.1200/JCO.2008.19.6618. [DOI] [PubMed] [Google Scholar]
- 17.Grothey A, Sugrue MM, Purdie DM, et al. Bevacizumab beyond first progression is associated with prolonged overall survival in metastatic colorectal cancer: results from a large observational cohort study (BRiTE) J Clin Oncol. 2008;26(33):5326–34. doi: 10.1200/JCO.2008.16.3212. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.