

Abstract
Polycythemia vera (PV) is an acquired blood disorder, with variable increase of clonal myeloid cells (erythrocytes, granulocytes and platelets) and mostly normal polyclonal T lymphocytes. Most patients have a somatic V617F gain-of-function mutation in JAK2 associated with acquired uniparental disomy (UPD) on chromosome 9p. Yet, the JAK2 V617F mutation is not a PV-initiating event and the family clustering of PV suggests a contribution of inherited genetic events. Using whole-genome SNP arrays, we assayed 34 T-cells and 66 granulocytes (including 32 pairs from the same patients), and identified multiple SNPs around JAK2 that are associated with PV susceptibility (rs11999802, P=1.8×10−8, OR=4.4). We also developed a quantitative measure of the fraction of somatic single nucleotide variants (SNVs) based on allele-specific PCR, and a quantitative measure of somatic UPD based on “fractional copy-neutral loss-of-heterozygosity (LOH)” on SNP arrays. Somatic genomic changes in granulocytes revealed strong genetic heterogeneity, including 9p UPD and chromosomal gain. The magnitude of somatic 9p UPD was strongly associated with V617F dosage (r2=0.74, P=4.8×10−12), suggesting that UPD preferentially increases the V617F subclone. In granulocytes with heterozygous rs11999802 genotypes, UPD increased the relative fraction of germline risk alleles (P=0.03). Thus, germline risk variants at JAK2 predispose to somatic point mutations within JAK2, whose allelic dosage can be further increased by a serial subclonal expansion of allele-specific UPD or copy number alteration, contributing to PV pathogenesis. We argue that PV represents a unique disease model to study the interplay between germline risk variants and convergent mechanisms of somatic mutations.
Introduction
Polycythemia vera [PV, (OMIM #263300)] is an acquired clonal blood disorder characterized by a variably increased number of clonal myeloid cells (erythrocytes, granulocytes, and platelets) and mostly normal polyclonal T lymphocytes. Over 95% of PV patients carry a JAK2 V617F mutation in 2 copies acquired by uniparental disomy (UPD) on chromosome 9p, and this gain-of-function mutation is involved in their PV development (Kralovics et al., 2002). Yet JAK2 V617F mutation is not a PV-initiating event (Kralovics et al., 2006; Nussenzveig et al., 2007), and the family clustering of PV suggests a contribution of inherited genetic events to its pathogenesis (Kralovics et al., 2003). Several studies have utilized whole-genome, high-density SNP arrays to investigate the genetic basis of PV, consistently demonstrating that a specific haplotype surrounding the JAK2 locus is a major risk factor for the development of Philadelphia chromosome negative myeloproliferative neoplasms, including PV (Jones et al., 2009; Kilpivaara et al., 2009; Olcaydu et al., 2009). Chromosomes carrying this specific haplotype tend to have the V617F mutation in JAK2, and this somatic gain-of-function mutation subsequently leads to the development of a myeloproliferative phenotype.
There are two competing hypotheses to explain the association between germline and somatic V617F risk factors in PV, referred to as the hypermutability hypothesis and the fertile background hypothesis (Campbell, 2009). The hypermutability hypothesis specifies that different germline risk alleles are associated with differences in somatic mutation rates at the same homologous chromosome (in cis), in that a specific haplotype may be more “fragile” and thus susceptible to somatic mutagenesis such as DNA replication or proofreading error (Figure 1A). The fertile background hypothesis specifies that somatic mutation rates are the same for chromosomes with germline risk variants or non-risk variants, but those cells in which the mutation occurs in cis to the risk allele gain more selective advantage, and are therefore more likely to undergo clonal expansion (Figure 1B). These hypotheses were difficult to test in previous studies due to the need to look at two distant mutations (Campbell, 2009). However, in our study, we attempted to address these two possibilities by additionally examining large-scale UPD events (shown as copy-neutral loss-of-heterozygosity in SNP arrays), which target both the germline risk variant and somatic V617F mutation and should not have a preferential bias towards a specific chromosome.
Figure 1.
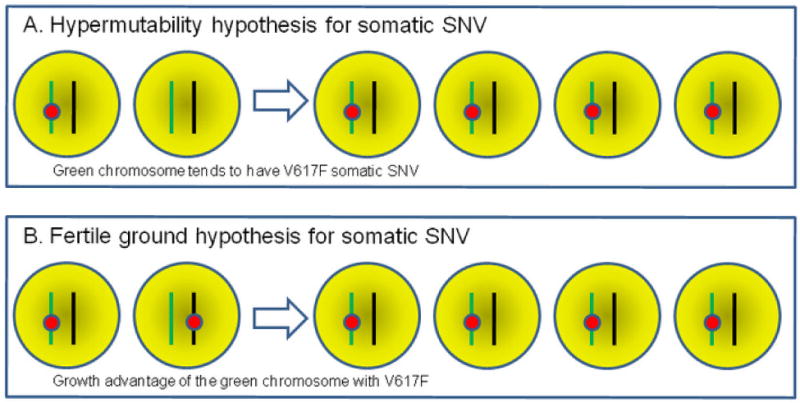
Illustration of the “hypermutability hypothesis” and the “fertile ground hypothesis.” Two homologous chromosomes with different colors are depicted, with the green and black chromosomes carrying germline risk and non-risk alleles, respectively. The red circle represents somatic V617F mutation. SNV, single nucleotide variant.
Additionally, previous genetic studies did not investigate in depth the role of somatic loss-of-heterozygosity (LOH) caused by UPD during mitotic recombination (Kralovics et al., 2002). In fact, given the known high frequency of 9p UPD in granulocytes of PV patients, the specific role of UPD with respect to germline risk variants and somatic single nucleotide variants (SNVs) remains largely unexplored. Therefore, our study aimed to explore the complex relationships between germline SNPs, somatic SNVs, and somatic UPD. Comparing to previous studies, the unique aspect of our study is the utilization of quantitative measure (as opposed to a positive/negative binary measure) of the fraction of somatic SNVs, as well as the quantitative measure of the magnitude of somatic UPD based on SNP genotyping signal intensities.
Materials and Methods
All studied PV subjects fulfilled the current WHO criteria for PV diagnosis (Tefferi et al., 2009). Granulocytes and T lymphocytes were isolated and their DNA were prepared as previously described (Swierczek et al., 2008). The population-based controls were recruited by the Children’s Hospital of Philadelphia (CHOP) clinicians and nursing and medical assistant staff within the CHOP Health Care Network, which includes multiple primary care clinics and outpatient practices. These control subjects were also genotyped on the same technical platform by the same technical staff at the same genotyping center as the PV cases, reducing concerns of potential batch effects. All patients and control subjects gave informed written consent.
We performed an integrative genomic analysis to investigate the intrinsic relationships between germline risk variants, somatic SNVs, somatic UPD, and somatic chromosome duplications at the same locus. Our sample set included 34 T-cells and 66 granulocytes from PV patients (including 32 pairs from the same patients), all of whom were of European ancestry. All DNA samples were genotyped using the Illumina Human610 whole-genome SNP genotyping arrays, which contain over 590,000 SNP markers and 20,000 copy number (CN) markers.
We wish to emphasize several distinct methodological differences between our approach and previous studies. The previous studies dichotomized patients as JAK2 V617F-positive or V617F-negative. However, the very nature of multistep evolution of clonal hematopoiesis to the JAK2 V617F-positive subclone (Kralovics et al., 2006; Nussenzveig et al., 2007) suggests that such dichotomization may not have sufficient precision to describe the true state of somatic SNVs. Some previous studies already demonstrated the value of using quantitative measures of mutation (Guglielmelli et al., 2010). In our study, we utilized a specifically designed allele-specific quantitative polymerase chain reaction (PCR) technology (Nussenzveig et al., 2007) to quantify the fraction of V617F mutations in granulocytes. The major difference between our JAK2 assay and standard allele-specific PCR assays is that the primers in our assay carry the extra mismatch primer together with locked nucleic acid that increases binding affinity for complementary sequences (Stephenson and Zamecnik, 1978), having superior discrimination of wild-type and mutant JAK2 alleles, with ΔCt greater than 14 cycles between matched and mismatched primers. We found that quantitation of JAK2 V617F allele frequency was reproducible (SD=1.5%) and sensitive, i.e., JAK2 V617F allele burdens of 0.1% were detectable in 40ng of DNA (Nussenzveig et al., 2007), which would otherwise be classified as “V617F-negative” by conventional approaches. Second, although large-scale LOH have been described in many studies, typically a dichotomization of “with LOH” or “without LOH” was utilized based on SNP genotype calls. Here we utilized high-density Illumina SNP arrays with well-standardized SNP signal intensity measures, permitting accurate inference of chromosome copy number changes that relies on total intensity and allelic intensity ratio (the ratio of signal intensity for two alternative alleles for a given SNP) (Wang et al., 2007). When there is no copy number change, the allelic intensity ratio can also be used in a quantitative fashion to derive a “fractional LOH” measure that accurately captures the fraction of cells (a subclone) with UPD (Figure 2). The method to derive the “fractional LOH” measure is similar to previously described (Rodriguez-Santiago et al., 2010; Yamamoto et al., 2007), and is available in the PennCNV software package. Briefly, it is a sliding window approach that examines a slice of genomic region for compatibility of BAF (B Allele Frequency) data with fractional LOH of 10%, 20%, 30%, and extending to 100%, and then selects the most likely value of fractional LOH supporting the observed BAF data. BAF is an allelic intensity ratio measure for each SNP, which summarizes the ratio of the B allele intensity versus the total signal intensity. Altogether, the utilization of quantitative measures of both SNVs and UPD allowed us to perform more powerful correlation analysis, despite the modest sample sizes.
Figure 2.
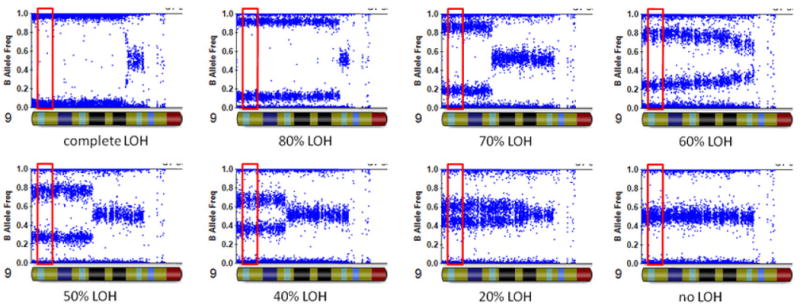
Illustration of the “fractional LOH measure” on eight granulocytes with different levels of copy-neutral LOH in chromosome 9p. The fractional LOH measure was calculated using B Allele Frequency measures for SNPs within 2Mb of the JAK2 gene (red box).
Results
We first studied the role of germline risk variants in PV pathogenesis by performing a genome-wide association study (GWAS) on 34 PV patients (genotypes from polyclonal T-cells) and 3,278 control subjects (genotypes from polyclonal whole blood) of European ancestry. Since the T-cells from PV individuals and whole blood from control subjects are both polyclonal and reflect germline genotypes, they can be compared to each other in genome-wide association studies. Due to the small sample size, results of such analysis are uninterpretable from a genome-wide perspective, so we regarded this analysis as a positive control experiment confirming that JAK2 can be confidently identified as a susceptibility locus. Indeed, we found that over a dozen SNPs surrounding the JAK2 locus demonstrated highly significant association signals by Fisher’s exact test [Table 1, Supplementary Figure 1 (see the attached supplementary figures)]. The most significant SNP is rs11999802 (MAF=24% in controls, P=1.8×10−8), whereas the SNP rs10974993 [the best proxy for rs10974994 reported by Kilpivaara et al. (2009)] also showed significance but was not as strong (MAF=28% in controls, P=5.1×10−7). We stress here that these SNPs most likely do not represent the true risk allele, and that full sequencing of the entire region in a large set of patients may yield more insights about the true causal alleles. These significant SNPs extend far beyond the conventional linkage disequilibrium (LD) block, strongly supporting a hypothetical scenario that a long-range haplotype may exist in patient populations and that the causal mutation may not be within the JAK2 gene itself (Dickson et al., 2010). The most significant marker at the JAK2 locus has a very high odds ratio of 4.4, well above typical effect sizes observed in most other GWAS (Manolio et al., 2008). However, given the observed long-range haplotype, if rare alleles are responsible for the association signal as predicted by “synthetic association,” the true odds ratio is probably even higher than 4.4 (Wang et al., 2010). Besides JAK2, several other loci contain SNPs with genome-wide significance, but manual examination suggested that all of these were isolated signals and were therefore likely to be due to random noise (typical of small sample sizes). In summary, GWAS confirmed that JAK2 predisposes to PV susceptibility, testifying to the quality of phenotype data that we have collected.
Table 1.
The Markers with Highest Association (P<1×10−5) at the JAK2 Locus in Comparison Between PV Patients and Control Subjects.
| Chr | SNP | Position | MAF (case) | MAF (control) | P | Odds ratio |
|---|---|---|---|---|---|---|
| 9 | rs11999802 | 5179773 | 0.5833 | 0.2409 | 1.84E-08 | 4.41 |
| 9 | rs3780373 | 5088223 | 0.6207 | 0.2835 | 1.79E-07 | 4.135 |
| 9 | rs12347727 | 4990811 | 0.5806 | 0.2662 | 2.90E-07 | 3.816 |
| 9 | rs10974993 | 5172159 | 0.6034 | 0.2838 | 5.09E-07 | 3.84 |
| 9 | rs7030260 | 4998070 | 0.5833 | 0.2709 | 5.15E-07 | 3.768 |
| 9 | rs10118930 | 5138278 | 0.6034 | 0.2842 | 5.23E-07 | 3.833 |
| 9 | rs7047795 | 5171467 | 0.6034 | 0.2843 | 5.26E-07 | 3.831 |
| 9 | rs7851556 | 5012807 | 0.5862 | 0.2709 | 7.01E-07 | 3.813 |
| 9 | rs10815149 | 5053701 | 0.5862 | 0.2725 | 7.61E-07 | 3.782 |
| 9 | rs10491651 | 5190060 | 0.569 | 0.2732 | 2.46E-06 | 3.512 |
| 9 | rs16922786 | 5190714 | 0.569 | 0.2732 | 2.47E-06 | 3.512 |
| 9 | rs3780381 | 5104523 | 0.5667 | 0.2783 | 3.62E-06 | 3.391 |
| 9 | rs7862042 | 5258139 | 0.5667 | 0.2786 | 3.67E-06 | 3.387 |
| 9 | rs6476948 | 5194404 | 0.569 | 0.2736 | 4.21E-06 | 3.505 |
We next turn our attention to somatic single nucleotide variants (SNVs), because the JAK2 V617F mutation is a well-known somatic mutation in PV. We used a quantitative method, allele-specific quantitative PCR, to assay the exact fraction of V617F mutations at the JAK2 locus in clonal granulocytes. Previous studies have demonstrated that germline sequence variants around JAK2 are associated with the somatic V617F mutation. Our analyses were restricted to the 32 patients with paired T-cells and granulocytes to eliminate concerns about using inaccurate SNP genotypes from granulocytes, given the frequent 9p UPD events. Possibly due to the small sample size, we did not observe significant association between V617F allelic load and germline SNP alleles (P=0.75 for rs11999802, linear regression, Supplementary Figure 2).
We next utilized the SNP genotyping signal intensity data and studied the role of somatic UPD in PV. Previous studies, utilizing low-density SNP arrays and using genotype calls (rather than signal intensity), have estimated that one third of PV patients carry 9p UPD (Kralovics et al., 2002). Our study, however, relies on high-density SNP genotyping data, and can therefore provide much finer resolution and much higher ability to assay subtle changes of UPD, which we refer to as “fractional LOH” that detects a subclone from the clonal PV granulocytes. This measure can be calculated from examination of the distribution of mode of B Allele Frequency measures, within a user-specified sliding window that covers JAK2. Increasing amounts of UPD result in larger “splitting” of the heterozygous genotype clusters that deviate from the center (0.5), so the mode of the distribution can be used to accurately infer “fractional LOH” in a fairly quantitative manner. In our data, conventional 9p UPD covering JAK2 was observed in 36% of the patients, whereas “fractional LOH” was observed in 14% of the patients; additionally, we also observed 9p chromosomal gain in one patient (Supplementary Figure 3). We emphasize here that when “fractional LOH” is present, many SNPs within the region may still show heterozygous genotypes, as only a small fraction of cells carry homozygous SNPs. Strong associations were detected between “fractional LOH” on JAK2 and somatic V617F load (Figure 3, r=0.74, P=4.8×10−12). We additionally observed that some PV patients carry multiple 9p UPD with different “fractional LOH” measures, suggesting a stepwise subclonal expansion of several UPD events with distinct breakpoints (Supplementary Figure 4). These results suggest that UPD may play a major role in increasing the allelic dosage of V617F, and that previous association studies ignoring UPD events may not be utilizing the data in an optimal fashion.
Figure 3.
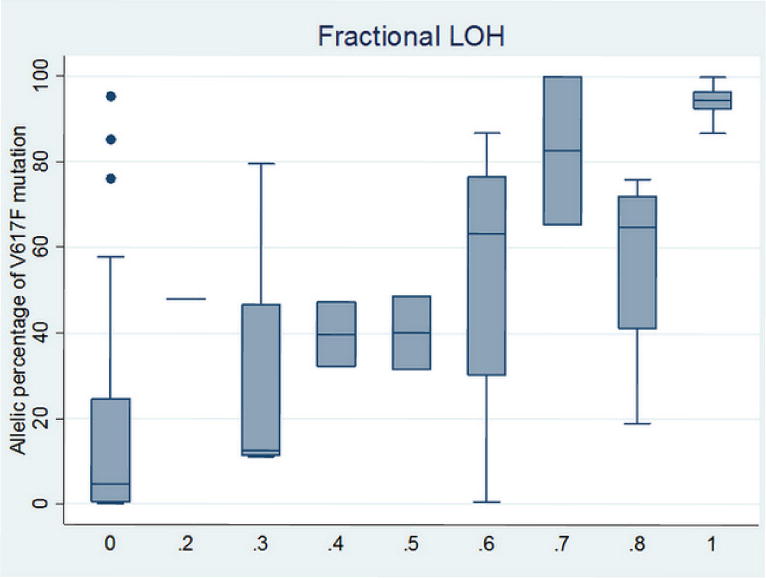
Box plot of the percentage of somatic V617F mutation in JAK2 over each of the fractional LOH measures in granulocytes. Strong correlation between these two measures was detected (r=0.74, P=4.8×10−12).
To examine whether there are any preferential biases in UPD events in targeting one homologous chromosome versus the other, we examined the BAF measure for all granulocytes carrying heterozygous genotypes for rs11999802. We identified a total of 11 granulocytes with heterozygous rs11999802 genotypes, yet 9 of them have a BAF value higher than 0.5 (P=0.03), that is, germline risk alleles had a higher dosage than non-risk alleles in most PV patients. Therefore, in keeping with previous observations on somatic SNVs, our analysis demonstrated that there are also preferential biases for somatic UPD events with respect to germline risk alleles.
Discussion
In the current study, we assayed a collection of PV patients, including some with paired samples of T-cells bearing germline DNA and clonal granulocytes bearing somatic mutations, for somatic mutations at the JAK2 9p locus and its association with 9p germline configuration. Despite the relatively small sample size, our study design generated testable hypotheses and our results offer novel insights relevant to the genesis of the PV phenotype. First, we confirmed that germline sequence variants correlate with both PV susceptibility and somatic JAK2 mutations. Next, we also showed that the magnitude of somatically acquired UPD strongly predicts the allelic dosage of JAK2 mutations in circulating clonal granulocytes. In this context, our minimum detection level was 10% by SNP array, yet we are aware that based on analyses on erythropoietin-independent colonies, virtually all PV patients have some JAK2 V617F homozygous progenitors (Nussenzveig et al., 2007; Scott et al., 2006) that are below the detection limit by our analyses. Finally, we demonstrated that somatic copy number gain can occur in JAK2 to increase the dosage of gain-of-function mutations. The JAK2 locus therefore provides a paradigm on how distinct mechanisms of somatic mutation can work together in a stepwise fashion to increase the allelic dosage of a known gain-of-function mutation resulting in PV, with the potential of leading to oncogenic transformation of a premalignant clonal disorder to an aggressive cancer, such as acute leukemic transformation.
Based on these observations, we can infer a hypothetical model depicting the likely stepwise events that occur in PV pathogenesis (Supplementary Figure 5). We however caution that such a model is somewhat arbitrary and may not be fully supported by current available data. An unknown germline risk factor predisposes to PV susceptibility, and PV-initiating events occur in some blood cells which subsequently acquire clonal expansion. Within these cell populations, germline sequence variants in JAK2 predispose to an increased likelihood of somatic JAK2 V617F mutation in the same homologous chromosome. Cells carrying the V617F mutation have a higher proliferation rate and may undergo clonal expansion, but a fraction of these cells may acquire a UPD at 9p, which further amplifies the allelic dosage of the JAK2 V617F mutation without altering genomic copy numbers. The mechanism of acquiring UPD was due to mitotic recombination, which was under a different mechanism that creates somatic copy number variations (CNVs, DNA replication error). We observed only one patient with somatic CNVs, suggesting that this is a relatively rare JAK2 gain-of-function mechanism, compared to UPD events. Furthermore, in some PV patients, some subset of PV cells may further develop somatic mutations in genes important for DNA replication and repair, and subsequently develop malignancies, such as leukemia, which are characterized by extensive copy number alternations in cells. This intuitive model has two important messages: First, the JAK2 locus is not the only germline risk factor and is unlikely to be the first “trigger” in the pathogenesis of PV, given the extensive subclonality of PV cells (Nussenzveig et al., 2007; Swierczek et al., 2008). This hypothesis is supported by the observation that patients who transform from PV to acute leukemia may not carry clones with the V617F mutation even if V617 can be still present in granulocytes in these patients (Campbell et al., 2006; Jelinek et al., 2005; Theocharides et al., 2007). Second, PV cells are adept in utilizing multiple mechanisms of somatic mutation, including DNA replication/proof reading and mitotic recombination, and, in rare cases, chromosomal duplication due to defects in DNA repair, to increase the allelic dosage of gain-of-function mutations.
Another interesting observation from our study is the strong association between germline risk variants and PV susceptibility. Previous studies, using different genotyping platforms, have demonstrated that a haplotype surrounding JAK2 is associated with PV susceptibility. In our study, without performing any haplotype analysis (which may be arbitrary and highly dependent on marker selection), we can achieve genome-wide significance of individual SNPs using a relatively modest set of samples. As we have previously argued, haplotype-based analysis can help further dissect association signals at loci detected in GWAS, and can help detect underestimation of effect sizes by GWAS hits using SNPs (Wang et al., 2010). Given our small sample size, we elected not to elaborate on comparative haplotype analysis, and focused on the description of somatic mutations described herein.
The hypermutability hypothesis and fertile background hypothesis have been proposed to explain the association between germline and somatic risk factors in PV (Campbell, 2009). These hypotheses were difficult to test in previous studies due to the need to look at two distant point mutations (one germline and one somatic), but we attempted to evaluate these hypotheses in the context of our data. UPD is usually very large, covering many megabases including all the germline and somatic mutations within JAK2. It is biologically very unlikely that a JAK2 germline variant influences UPD events in cis, which is caused by mitotic recombination with a breakpoint that may be megabases away from JAK2; therefore, it is reasonable to assume that UPD events occur randomly in each of the two homologous chromosomes. If the fertile ground hypothesis is correct and if UPD occurs immediately after a somatic V617F mutation, UPD would increase the allelic fraction of any chromosome with V617F mutations and not favor germline risk variants. Therefore, our analysis renders some levels of support to the hypermutability hypothesis; however, we caution that this simplified scenario ignores the time-course relationship between distinct somatic mutations. In reality, UPD may occur after preferential gain of chromosomes carrying both germline risk alleles and somatic V617F mutations, so we still cannot exclude the possibility of the fertile ground hypothesis on V617F.
Taken together, our results demonstrate that germline risk variants at the JAK2 locus predispose to somatic mutations within JAK2, and that allelic dosage can be further increased by a serial subclonal expansion of allele-specific UPD, implicating convergent mechanisms of somatic mutations that impact the genetic load of gain-of-function mutations contributing to the observed clonal expansion and pathogenesis of PV. Compared to many genetic studies where germline events change protein-coding sequences or regulate gene expression, our study thus represents one of the first examples in the literature where germline variants confer disease susceptibility through a series of convergent somatic events.
Supplementary Material
Acknowledgments
We gratefully thank all the patients and their families who were enrolled in this study, as well as all the control subjects who donated blood samples to the Children’s Hospital of Philadelphia (CHOP) for genetic research purposes. We thank the technical staff at the Center for Applied Genomics at CHOP for producing the genotypes used for analysis and the nursing, medical assistant, and medical staff for their invaluable help with sample recruitments. The study was supported by grants from the 1P01CA108671-O1A2 (NCI) Myeloproliferative Disorders (MPD) Consortium and from the MPD Foundation to J.T.P.
Footnotes
Disclosure
The authors declare no conflicts of interest.
Contributor Information
Kai Wang, Center for Applied Genomics, Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania 19104, USA and Zilkha Neurogenetic Institute, Department of Psychiatry and Preventive Medicine, University of Southern California, Los Angeles, California 90089, USA
Sabina Swierczek, Department of Hematology and Genetics, University of Utah & ARUP Laboratories, Salt Lake City, Utah 84132, USA
Kimberly Hickman, Department of Hematology and Genetics, University of Utah & ARUP Laboratories, Salt Lake City, Utah 84132, USA
Josef T. Prchal, Department of Hematology and Genetics, University of Utah & ARUP Laboratories, Salt Lake City, Utah 84132, USA
Hakon Hakonarson, Center for Applied Genomics, the Abramson Research Center of the Joseph Stokes Jr. Research Institute, Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania 19104, USA and Department of Pediatrics, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania 19104, USA
References
- Campbell PJ. Somatic and germline genetics at the JAK2 locus. Nat Genet. 2009;41(4):385–386. doi: 10.1038/ng0409-385. [DOI] [PubMed] [Google Scholar]
- Campbell PJ, Baxter EJ, Beer PA, Scott LM, Bench AJ, Huntly BJ, Erber WN, Kusec R, Larsen TS, Giraudier S, Le Bousse-Kerdiles MC, Griesshammer M, Reilly JT, Cheung BY, Harrison CN, Green AR. Mutation of JAK2 in the myeloproliferative disorders: timing, clonality studies, cytogenetic associations, and role in leukemic transformation. Blood. 2006;108(10):3548–3555. doi: 10.1182/blood-2005-12-013748. [DOI] [PubMed] [Google Scholar]
- Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8(1):e1000294. doi: 10.1371/journal.pbio.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guglielmelli P, Biamonte F, Spolverini A, Pieri L, Isgro A, Antonioli E, Pancrazzi A, Bosi A, Barosi G, Vannucchi AM. Frequency and clinical correlates of JAK2 46/1 (GGCC) haplotype in primary myelofibrosis. Leukemia. 2010;24(8):1533–1537. doi: 10.1038/leu.2010.126. [DOI] [PubMed] [Google Scholar]
- Jelinek J, Oki Y, Gharibyan V, Bueso-Ramos C, Prchal JT, Verstovsek S, Beran M, Estey E, Kantarjian HM, Issa JP. JAK2 mutation 1849G>T is rare in acute leukemias but can be found in CMML, Philadelphia chromosome-negative CML, and megakaryocytic leukemia. Blood. 2005;106(10):3370–3373. doi: 10.1182/blood-2005-05-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones AV, Chase A, Silver RT, Oscier D, Zoi K, Wang YL, Cario H, Pahl HL, Collins A, Reiter A, Grand F, Cross NC. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat Genet. 2009;41(4):446–449. doi: 10.1038/ng.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpivaara O, Mukherjee S, Schram AM, Wadleigh M, Mullally A, Ebert BL, Bass A, Marubayashi S, Heguy A, Garcia-Manero G, Kantarjian H, Offit K, Stone RM, Gilliland DG, Klein RJ, Levine RL. A germline JAK2 SNP is associated with predisposition to the development of JAK2(V617F)-positive myeloproliferative neoplasms. Nat Genet. 2009;41(4):455–459. doi: 10.1038/ng.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kralovics R, Guan Y, Prchal JT. Acquired uniparental disomy of chromosome 9p is a frequent stem cell defect in polycythemia vera. Exp Hematol. 2002;30(3):229–236. doi: 10.1016/s0301-472x(01)00789-5. [DOI] [PubMed] [Google Scholar]
- Kralovics R, Stockton DW, Prchal JT. Clonal hematopoiesis in familial polycythemia vera suggests the involvement of multiple mutational events in the early pathogenesis of the disease. Blood. 2003;102(10):3793–3796. doi: 10.1182/blood-2003-03-0885. [DOI] [PubMed] [Google Scholar]
- Kralovics R, Teo SS, Li S, Theocharides A, Buser AS, Tichelli A, Skoda RC. Acquisition of the V617F mutation of JAK2 is a late genetic event in a subset of patients with myeloproliferative disorders. Blood. 2006;108(4):1377–1380. doi: 10.1182/blood-2005-11-009605. [DOI] [PubMed] [Google Scholar]
- Manolio TA, Brooks LD, Collins FS. A HapMap harvest of insights into the genetics of common disease. J Clin Invest. 2008;118(5):1590–1605. doi: 10.1172/JCI34772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussenzveig RH, Swierczek SI, Jelinek J, Gaikwad A, Liu E, Verstovsek S, Prchal JF, Prchal JT. Polycythemia vera is not initiated by JAK2V617F mutation. Exp Hematol. 2007;35(1):32–38. doi: 10.1016/j.exphem.2006.11.012. [DOI] [PubMed] [Google Scholar]
- Olcaydu D, Harutyunyan A, Jager R, Berg T, Gisslinger B, Pabinger I, Gisslinger H, Kralovics R. A common JAK2 haplotype confers susceptibility to myeloproliferative neoplasms. Nat Genet. 2009;41(4):450–454. doi: 10.1038/ng.341. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Santiago B, Malats N, Rothman N, Armengol L, Garcia-Closas M, Kogevinas M, Villa O, Hutchinson A, Earl J, Marenne G, Jacobs K, Rico D, Tardon A, Carrato A, Thomas G, Valencia A, Silverman D, Real FX, Chanock SJ, Perez-Jurado LA. Mosaic uniparental disomies and aneuploidies as large structural variants of the human genome. Am J Hum Genet. 2010;87(1):129–138. doi: 10.1016/j.ajhg.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott LM, Scott MA, Campbell PJ, Green AR. Progenitors homozygous for the V617F mutation occur in most patients with polycythemia vera, but not essential thrombocythemia. Blood. 2006;108(7):2435–2437. doi: 10.1182/blood-2006-04-018259. [DOI] [PubMed] [Google Scholar]
- Stephenson ML, Zamecnik PC. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc Natl Acad Sci U S A. 1978;75(1):285–288. doi: 10.1073/pnas.75.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swierczek SI, Agarwal N, Nussenzveig RH, Rothstein G, Wilson A, Artz A, Prchal JT. Hematopoiesis is not clonal in healthy elderly women. Blood. 2008;112(8):3186–3193. doi: 10.1182/blood-2008-03-143925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tefferi A, Thiele J, Vardiman JW. The 2008 World Health Organization classification system for myeloproliferative neoplasms: order out of chaos. Cancer. 2009;115(17):3842–3847. doi: 10.1002/cncr.24440. [DOI] [PubMed] [Google Scholar]
- Theocharides A, Boissinot M, Girodon F, Garand R, Teo SS, Lippert E, Talmant P, Tichelli A, Hermouet S, Skoda RC. Leukemic blasts in transformed JAK2-V617F-positive myeloproliferative disorders are frequently negative for the JAK2-V617F mutation. Blood. 2007;110(1):375–379. doi: 10.1182/blood-2006-12-062125. [DOI] [PubMed] [Google Scholar]
- Wang K, Dickson SP, Stolle CA, Krantz ID, Goldstein DB, Hakonarson H. Interpretation of association signals and identification of causal variants from genome-wide association studies. Am J Hum Genet. 2010;86(5):730–742. doi: 10.1016/j.ajhg.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SFA, Hakonarson H, Bucan M. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto G, Nannya Y, Kato M, Sanada M, Levine RL, Kawamata N, Hangaishi A, Kurokawa M, Chiba S, Gilliland DG, Koeffler HP, Ogawa S. Highly sensitive method for genomewide detection of allelic composition in nonpaired, primary tumor specimens by use of affymetrix single-nucleotide-polymorphism genotyping microarrays. Am J Hum Genet. 2007;81(1):114–126. doi: 10.1086/518809. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.