

Abstract
Objectives
Approximately 50% of serous epithelial ovarian cancers (EOC) contain molecular defects in homologous recombination (HR) DNA repair pathways. Poly(ADP-ribose) polymerase inhibitors (PARPi) have efficacy in HR-deficient, but not HR-proficient, EOC tumors as a single agent. Our goal was to determine whether the histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA), can sensitize HR-proficient ovarian cancer cells to the PARPi AZD-2281 (olaparib).
Methods
Ovarian cancer cell lines (SKOV-3, OVCAR-8, NCI/ADR-Res, UWB1.289 BRCA1null and UWB1.289 + BRCA1 wild-type) were treated with saline vehicle, olaparib, SAHA or olaparib/SAHA. Sulforhodamine B (SRB) assessed cytotoxicity and immunofluorescence and Western blot assays assessed markers of apoptosis (cleaved PARP) and DNA damage (pH2AX and RAD51). Drug effects were also tested in SKOV-3 xenografts in Nude mice. Affymetrix microarray experiments were performed in vehicle and SAHA-treated SKOV-3 cells.
Results
In a microarray analysis, SAHA induced coordinated down-regulation of HR pathway genes, including RAD51 and BRCA1. Nuclear co-expression of RAD51 and pH2AX, a marker of efficient HR repair, was reduced approximately 40% by SAHA treatment alone and combined with olaparib. SAHA combined with olaparib induced apoptosis and pH2AX expression to a greater extent than either drug alone. Olaparib reduced cell viability at increasing concentrations and SAHA enhanced these effects in 4 of 5 cell lines, including BRCA1 null and wild-type cells, in vitro and in SKOV-3 xenografts in vivo.
Conclusions
These results provide preclinical rationale for targeting DNA damage response pathways by combining small molecule PARPi with HDACi as a mechanism for reducing HR efficiency in ovarian cancer.
Keywords: vorinostat (SAHA), olaparib (Ola), histone deacetylase inhibitors (HDACi), poly (ADP-ribose) polymerase inhibitors (PARPi), phosphorylated gamma histone H2AX (pH2AX), ovarian cancer
INTRODUCTION
Serous epithelial ovarian cancer (EOC) is characterized by frequent genetic and epigenetic alterations in gene members of the homologous recombination (HR) DNA repair pathway [1, 2]. Approximately 50% of high grade serous EOC harbor molecular alterations in the HR pathway, which include: germline and somatic BRCA1/2 mutations, in 15% and 6–7% of cases respectively; hypermethylation of BRCA1 and RAD51C; and mutations in Fanconi anemia genes, PTEN, ATM and ATR [1, 2]. Exploiting these defects in DNA damage response and repair mechanisms, HR-deficient EOC tumors are highly sensitive to poly-ADP ribose polymerase inhibitor (PARPi) therapy in the presence [3–5] and absence of BRCA1/2 mutations [6, 7] in clinical trials. PARPi are a novel class of anticancer agents that stimulate synthetic lethality via DNA damage induction [8, 9]. Inhibition of PARP-1 and PARP-2, which play a prominent role in base excision repair, results in single-strand DNA breaks (SSBs) [10]. The accumulation of unrepaired SSBs generates double-strand breaks (DSBs) at stalled DNA replication forks during S phase [11, 12]. Such lesions are particularly lethal in HR-deficient cells because replication fork-associated DSBs are predominantly repaired by HR [12, 13], and unrepaired DSBs ultimately trigger apoptosis [14].
Of the PARPi, olaparib has been the most widely studied and is currently in the most advanced stage of clinical development [3, 4, 6, 7, 15]. Despite these encouraging results, EOC tumors with an intact HR pathway (approximately 50% of all cases) do not respond well to PARPi and may not benefit from treatment with this novel class of drugs [3–5, 7]. A combination of PARPi with agents that inhibit HR could be an effective strategy for expanding the use of PARPi to HR-proficient EOC tumors.
We have previously shown that histone deacetylase inhibitors (HDACi) alter DNA damage response and sensitize ovarian cancer cells to the effects of DNA-damaging drugs such as cisplatin [16]. HDAC proteins play an important role in DNA damage response and repair [17], and HDACi are known to impair HR in cancer cells through reduced expression of critical genes such as BRCA1 and RAD51 [18–20]. Suberoylanilide hydroxamic acid (SAHA), or vorinostat, is an inhibitor of classes I, II, and IV HDACs that is currently approved as single-agent therapy for refractory cutaneous T-cell lymphoma [21, 22]. In this study, we hypothesized that SAHA alters the expression of HR pathway genes in ovarian cancer cells and thus enhances the anti-tumor activity of olaparib in HR-proficient tumors.
Here, we demonstrated that SAHA treatment led to coordinated downregulation of HR pathway genes, including RAD51 and BRCA1. Consistent with this finding, the established marker of efficient HR repair, nuclear co-expression of RAD51 with pH2AX in response to DNA damage, was reduced by SAHA alone and in combination with olaparib. Furthermore, SAHA combined with olaparib induced robust and prolonged activation of pH2AX, indicative of deficient DNA repair and associated with apoptosis. SAHA also enhanced the cytotoxic effects of olaparib in ovarian cancer cells in vitro and in vivo. Our results indicate that targeting DNA damage response pathways with PARPi and HDACi combination therapy is a novel strategy with the potential to be effective in both HR-deficient and -proficient EOC tumors.
MATERIALS AND METHODS
Cell culture and compounds
The epithelial ovarian cancer cell lines SKOV-3, UWB1.289+BRCA1 wild-type BRCA1 wild type (Brca1 WT) and UWB1.289 BRCA1 null (Brca1 Null) cell lines (American Type Culture Collection, Manassas, VA), OVCAR-8 and NCI/ADR-RES (National Cancer Institute, Bethesda, MD) were maintained in culture as previously described [23, 24]. The SKOV-3, OVCAR-8 and NCI/ADR-RES cell lines are represented in the National Cancer Institute 60 (NCI 60) Cancer Panel [25, 26], and the BRCA1 WT and BRCA null cell lines have been well described [27]. Cell lines were authenticated in December 2013 by the Vanderbilt VANTAGE Genomics Core using the GenePrint 10 kit (Promega, Madison, WI). Gene loci profiles were verified using, where applicable, NCI 60 (25, 26), COGCELL, and ATCC reference databases. All cell lines used tested negative for mycoplasma. Cells were treated with 0.01% DMSO as vehicle control; vorinostat (SAHA) (synthesized at the Broad Institute, Cambridge, MA); AZD-2281 (olaparib) (Astra Zeneca Pharmaceuticals, Wilmington, DE); and the combination of SAHA and olaparib.
Design of Microarray Experiment, RNA isolation and Affymetrix GeneChip Hybridization
SKOV-3 ovarian cancer cells were treated with 1μM SAHA or vehicle (0.01% DMSO) for 6 or 24 hours, harvested and processed for RNA isolation. Data for both time-points for SAHA and vehicle-treated cells were obtained in duplicate. Total RNA was isolated using the Qiagen RNeasy Kit (Qiagen, Valencia, CA) according to manufacturer’s instructions. cDNA synthesis and hybridization on oligonucleotide microarrays (U133 Plus 2.0 Array GeneChip, Affymetrix, Inc., Santa Clara, CA) containing approximately 54,700 transcripts were carried out using standard protocols. Microarray experiments were performed at the Dana Farber Cancer Institute Microarray facility.
Bioinformatic Analysis and Statistics
Raw gene expression data (Affymetrix .CEL files) were imported in BRB-ArrayTools Version: 4.2.0 - Beta_2 (Biometrics Research Branch, National Cancer Institute) and were processed using Robust Multi-Array (RMA) analysis. In order to assess whether the gene expression profiles of vehicle and SAHA treated SKOV-3 cells were enriched for specific pathways or functional groups of genes, we performed gene set analysis (GSA) as described by Efron and Tibshirani [28]. GSA is an evolution of the previously reported Gene Set Enrichment Analysis (GSEA) and was performed using the Gene Set Analysis Tool of the Biometric Research Branch (BRB) Array Tools software. Gene Ontology assignments, Biocarta and KEGG pathways, and annotated gene sets from The Broad Institute Molecular Signatures Database (MSigDB) were analyzed. The Efron-Tibshirani “maxmean” test was applied to identify gene sets at a GSA P<0.05 significance level. Comparison of individual genes between SAHA and vehicle treated cells was performed using the Class Comparison Between Groups of Arrays Tool of BRB-ArrayTools. Parametric t-Test p values and false discovery rate (FRD) values were reported for each gene. Apart from the microarray experiments, statistical values shown represented mean + SE of at least 3 independent experiments with * p < 0.05. MIAME-compliant microarray data is publically available at the GEO repository (series entry GSE53603, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE53603).
Cell proliferation and cytotoxicity assays
Sulforhodamine B (SRB) assays were used to determine cell proliferation and cytotoxicity and were conducted as previously described [16]. Absorbance was measured at 510nm using a Spectramax M5 spectrophotometer (Molecular Devices, Sunnyvale, CA) in the High-Throughput Screening Core of the Vanderbilt Institute of Chemical Biology. The interaction between fixed ratios of SAHA and olaparib was measured with the Combination Index (CI) method [29]. A CI level of < 0.9, CI = 0.9–1.1 and CI > 1.1 indicates synergy, additivity and antagonism respectively, between drug combinations.
Immunofluorescence
After treatment with a clinically achievable dose of olaparib (10μM) [4], SAHA (1μM), or the combination, ovarian cancer cells were fixed, permeabilized and stained with anti-phospho H2AX (Ser139) (pH2AX) (Millipore, Billerica, MA), anti-RAD51 (Millipore), and anti-cleaved PARP (Cell Signaling Technology, Beverly, MA) as per established protocols [24, 30]. Binding of the primary antibodies was detected with Alexa Flour anti-mouse IgG 594 or Alexa Flour anti-rabbit IgG 488 secondary antibodies (Invitrogen, Carlsbad, CA), as appropriate. Images were acquired and analyzed as previously described [24].
Western Blotting
In cells treated with olaparib (10μM), SAHA (1μM), or the combination, whole cell protein isolation, hydrochloric acid extraction of histones, Western Blotting and signal detection were performed as described [23, 31]. Antibodies used were anti-RAD51 (Millipore), anti-BRCA1 (Millipore), anti-PARP (Cell Signaling Technology, Danvers, MA), anti-cleaved caspase-3 (Cell Signaling), and anti-phospho H2AX (Ser139) (Millipore, Billerica, MA). Loading controls were anti-histone H3 (Millipore) and β-actin (Sigma) for histones and total proteins, respectively.
Animals
Four to five-week-old female athymic Nude-Foxn1nu mice were purchased from Harlan Laboratories (Indianapolis, IN). For this subcutaneous xenograft model, 5 × 106 SKOV-3 tumor cells in a 200 μL of mixture of PBS and Matrigel (1:1 v/v) (BD Biosciences, San Jose, CA) were injected into the right flank of each mouse. After the tumors reached approximately 200mm3, we treated the mice in cohorts as follows: vehicle control (0.01% DMSO in PBS, n=5), olaparib (10mg/kg, n=5), SAHA (50mg/kg, n=5) and the olaparib/SAHA combination (n=5) via intraperitoneal injection for 3 weeks. Animals were examined biweekly for the effects of tumor burden and tumor growth, and tumor measurements were performed weekly. Tumor volume was calculated weekly from caliper measurements of the smallest (SD) and largest diameter (LD) using the formula: volume = [LD × SD2] × π/6 [31]. After 3 weeks of treatment and 24 h after the final dose of drug, the mice were euthanized according to protocol and necropsy was performed. Tumors were excised and processed as below. Experiments performed were approved by the Vanderbilt University Institutional Animal Use and Care Committee, and all animals were maintained in accordance to guidelines of the American Association of Laboratory Animal Care.
Immunohistochemistry
Tissue fixation, processing and sectioning methods of SKOV-3 xenografts have been previously described [16]. Hematoxylin and eosin staining for histology and immunostaining for cleaved caspase-3, mib-1/Ki67 and phospho H2AX (Ser139) were performed as described [16]. For assessment of cleaved caspase-3, mib-1/Ki67 and phospho H2AX (Ser139) staining in SKOV-3 xenografts, at least 1000 cells were counted in at least 5 independent fields under high power (x40).
Statistics
Unless otherwise specified, the Student’s t test was used for comparisons between groups for in vitro experiments. For in vivo mouse experiments and for immunohistochemical analysis of tumor tissue, we compared groups with a Mann-Whitney t test. In all cases, p < 0.05 was considered statistically significant.
RESULTS
SAHA downregulates homologous recombination (HR) pathway gene expression and reduces RAD51 foci formation in response to DNA damage in SKOV-3 ovarian cancer cells
Our group has previously shown that HDACi enhance the cytotoxic effects of DNA-damaging agents in ovarian cancer cells [16, 24]. Several lines of evidence suggest HDACi-induced defects in DNA damage response and repair processes contribute mechanistically to this sensitization. We have shown that enhanced cytotoxicity in ovarian cancer cells due to combined treatment is associated with persistent activation of pH2AX [16, 24], a marker of DNA damage that is directly linked to impaired DNA repair and apoptosis [14]. Other groups have also demonstrated that HDACi treatment reduces HR and RAD51 expression in response to DNA damage in multiple tumor cell types [18, 19].
Here, we assessed SAHA-induced changes in HR gene expression at a global/pathway and single gene level in SKOV-3 ovarian cancer cells. SKOV-3 cells have high basal expression of HR gene products and a functional HR pathway [32, 33], and hence are a suitable cell line model of HR-proficient EOC. RNA was isolated from SAHA and vehicle-treated cells, followed by Affymetrix oligonucleotide microarray hybridization. As shown in Figure 1A, fifteen HR pathway genes including BRCA1, BRCA2, RAD51 and CHEK1 were downregulated by SAHA treatment compared to vehicle-treated cells at 24 h (p and FDR level of 0.05). SAHA-induced downregulation of RAD51 and BRCA1 expression was confirmed by Western blot (Figure 1B). We also performed pathway analysis and assessed gene sets from Gene Ontology (GO), Biocarta, KEGG pathways and MSigDB (The Broad Institute Molecular Signatures Database) related to HR pathway. As shown in Table 1, HR pathway (as defined by GO, Biocarta, KEGG and MSigDB gene sets) was downregulated in SAHA-treated compared to vehicle-treated SKOV-3 cells.
Figure 1. SAHA down-regulates Homologous Recombination (HR) pathway genes in SKOV-3 cells.
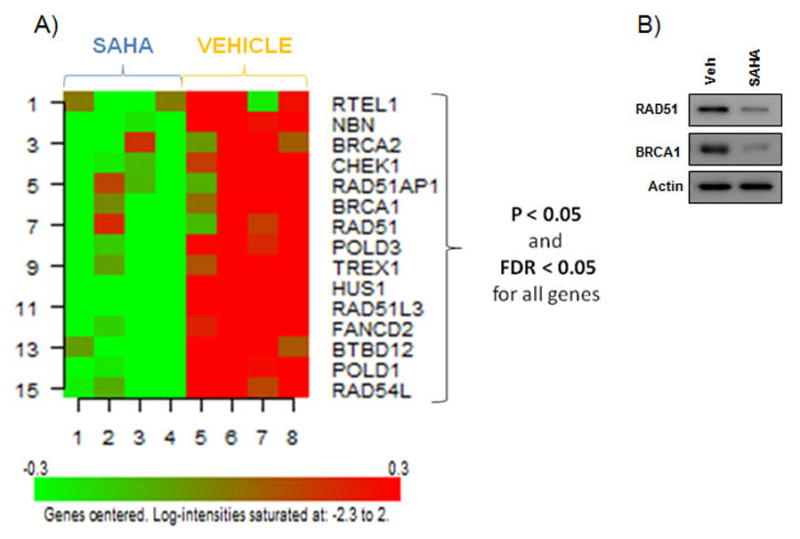
A) A heat map expression plot of 15 downregulated HR pathway genes in SKOV-3 cells after 24 h treatment with 1μM SAHA compared to vehicle (0.01% DMSO) treated controls. B) A representative Western blot showing 1μM SAHA-induced downregulation of RAD51 and BRCA1 expression after 24 h treatment. Actin was used as a loading control.
Table 1.
Homologous Recombination Pathway is downregulated in SAHA-treated SKOV-3 cells
| BIOCARTA pathways gene sets | GSA p(value) |
|---|---|
| 1. ATM Signaling Pathway | 0.025 |
| 2. Role of BRCA1, BRCA2 and ATR in Cancer Susceptibility | 0.015 |
| GO pathways gene sets | |
| 1. Double-strand break repair via homologous recombination | 0.08 |
| KEGG pathways gene sets | |
| 1. KEGG_Homologous_Recombination | 0.015 |
| BROAD MSig DATABASE gene sets | |
| 1. REACTOME_ Homologous_Recombination _Repair | 0.02 |
| 2. REACTOME_Double_Strand_Break_Repair | 0.01 |
Next, we explored a direct link between SAHA-mediated inhibition of expression of HR pathway genes and HR efficiency in response to DNA damage in olaparib-treated SKOV-3 cells. To determine whether SAHA-induced downregulation of RAD51 protein would affect recruitment of RAD51 to sites of double-strand DNA breaks, we used an IF assay measuring HR by the nuclear co-expression of RAD51 and pH2AX in response to DNA damage [34]. In SKOV-3 cells pre-treated for 6 h with the known inducer of double-strand DNA breaks, cisplatin (0.5 μM), there was no significant difference in the percentage of cells displaying co-expression of RAD51 and pH2AX with olaparib treatment compared to vehicle (Figure 2A and B). However, cells co-expressing RAD51 and pH2AX were reduced by approximately 40% when SAHA was used as a single agent or in combination with olaparib to chemically induce HR-deficiency (Figure 2A and B). Thus, HR efficiency in olaparib-treated HR-proficient cells was reduced by combination treatment with SAHA.
Figure 2. SAHA decreases formation of RAD51 foci in SKOV-3 cells in vitro.

Cells pre-treated with 0.5μM cisplatin for 6 h were then treated with vehicle (0.01% DMSO), olaparib (OLA) (10μM), SAHA (1μM) or the combination for a further 24 h. A) Representative images (40X) of IF staining for RAD51 (green) and pH2AX (red). DAPI-stained nuclei are in blue. B) The percentage of cells showing co-expression of RAD51 and pH2AX (>5 foci was considered positive staining) was calculated for each drug. Values are mean + SD for 3 independent experiments. At least 100 cells were counted (x40) for each drug treatment per experiment. * p < 0.01 compared to vehicle; b p < 0.01 relative to olaparib alone, both Student’s t test, Student’s t test.
SAHA combined with olaparib induces pH2AX activation and apoptosis more than olaparib alone in SKOV-3 cells
Having demonstrated functional inhibition of efficient HR by SAHA treatment alone and combined with olaparib in response to a DNA damage stimulus in SKOV-3 cells, we next determined whether SAHA could sensitize cells to the cytotoxic effects of olaparib. We have identified persistent pH2AX activation, which is associated with DNA damage-induced apoptosis [14], as a sensitive surrogate marker of cytotoxicity in ovarian cancer cells [16, 24]. Therefore, we examined whether pH2AX expression was enhanced by the combination of SAHA and olaparib compared to olaparib alone after 24 h treatment. First, we quantified drug effects on nuclear pH2AX expression by IF in SKOV-3 ovarian cancer cells (Figure 3A). Compared to vehicle-treated cells, olaparib significantly decreased the number of pH2AX-negative cells (less than 6 foci), and increased the number of cells displaying intermediate (between 6–20 foci) and high (greater than 20 foci, pan-nuclear staining where individual foci are not countable) pH2AX expression. Compared to olaparib and SAHA alone, combination treatment did not significantly reduce the number of pH2AX-negative cells, but did induce a robust increase in the proportion of cells displaying greater than 20 pH2AX foci. Second, we confirmed enhanced pH2AX expression in cells treated with SAHA/olaparib in Western blot analysis of purified histone extracts (Figure 3B).
Figure 3. Olaparib combined with SAHA increases SKOV-3 cell apoptosis and expression of the DNA damage mark pH2AX in vitro.
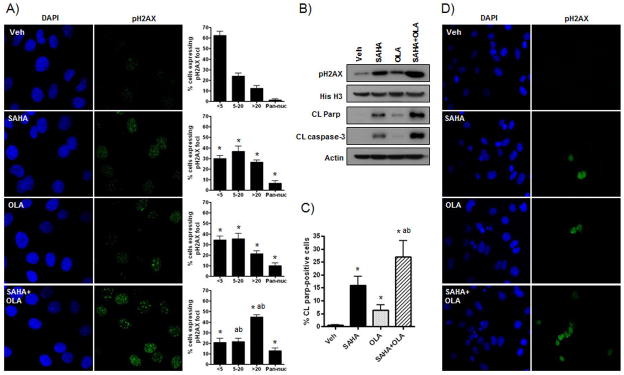
SKOV-3 ovarian cancer cells were treated with vehicle (0.01% DMSO), olaparib (OLA) (10μM), SAHA (1μM) or the combination for 24 h and evaluated by Western blot and IF staining for the expression of the DNA damage marker, pH2AX, and the apoptotic marks, cleaved PARP and cleaved caspase-3. A) IF nuclear stain for pH2AX (green). DAPI-stained nuclei are in blue. The number of cell nuclei displaying less than 6 foci (negative), between 6–20 foci, greater than 20 foci and diffuse pan-nuclear (Pan-nuc) staining for pH2AX foci was quantified and is presented in percentages (40X). B) Representative Western blot analysis of cleaved PARP and cleaved caspase 3 in whole cell lysates, and pH2AX in histone extracts. Loading controls were β-actin and histone H3, respectively. C) The percentage of cells showing cleaved PARP expression was calculated for each drug. D) Representative images for IF stain for cleaved PARP (green). DAPI-stained nuclei are in blue. Values in A) and C) are mean + SD for 5 and 3 independent experiments, respectively. At least 100 cells were counted (40X) for each drug treatment per experiment. * p < 0.01 compared to vehicle; a p < 0.01 relative to SAHA alone; b p < 0.01 relative to olaparib alone, all Student’s t test.
To assess induction of apoptosis associated with prolonged pH2AX activation [14], we measured the expression of the established apoptotic markers, cleaved PARP and cleaved caspase-3, in SKOV-3 cells treated with SAHA, olaparib and the combination at 24 h. Consistent with the pH2AX data, Western blot analysis of cleaved PARP and cleaved caspase-3 expression showed enhanced apoptosis with combined SAHA/olaparib treatment compared to olaparib alone (Figure 3B). These results were confirmed by IF analysis, where significant upregulation in the percentage of cells positive for cleaved PARP expression was observed for SAHA/olaparib treatment compared to single drug treatment (Figure 3C&D).
SAHA exhibits synergistic activity with olaparib in ovarian cancer cell lines in vitro
We then assessed the cytotoxic effects of olaparib (5–100 μM), SAHA (0.5–10 μM) and of the SAHA/olaparib combination in SRB growth assays. As shown in Figure 4A, SAHA reduced cell viability at increasing concentrations and enhanced the effects of olaparib in SKOV-3 cells following 72 h treatment. Combination Index analysis indicated that SAHA and olaparib strongly synergized in these experiments (Figure 4B). In addition, SAHA reduced cell viability and enhanced the effects of olaparib in the HR-deficient BRCA1 NULL (UWB1.289 BRCA1 null) and HR-proficient BRCA1 WT (UWB1.289+BRCA1 wild-type) cells (Figure 4C). Notably, BRCA1 null cells were considerably more sensitive to olaparib alone than BRCA1 WT cells, especially at lower concentrations. NCI/ADR-Res cells, which are resistant to many chemotherapeutic drugs, were exquisitely sensitive to SAHA as previously shown [24]. However, they did not respond to olaparib alone, and did not respond more to the olaparib/SAHA combination compared to SAHA alone (Figure 4D). Olaparib and SAHA activities were also synergistic in OVCAR-8, BRCA1 null and BRCA1 WT cells as demonstrated by CI levels below 0.9 in all cases (Figure 4B).
Figure 4. Olaparib reduces cell viability alone and combined with SAHA in ovarian cancer cells.
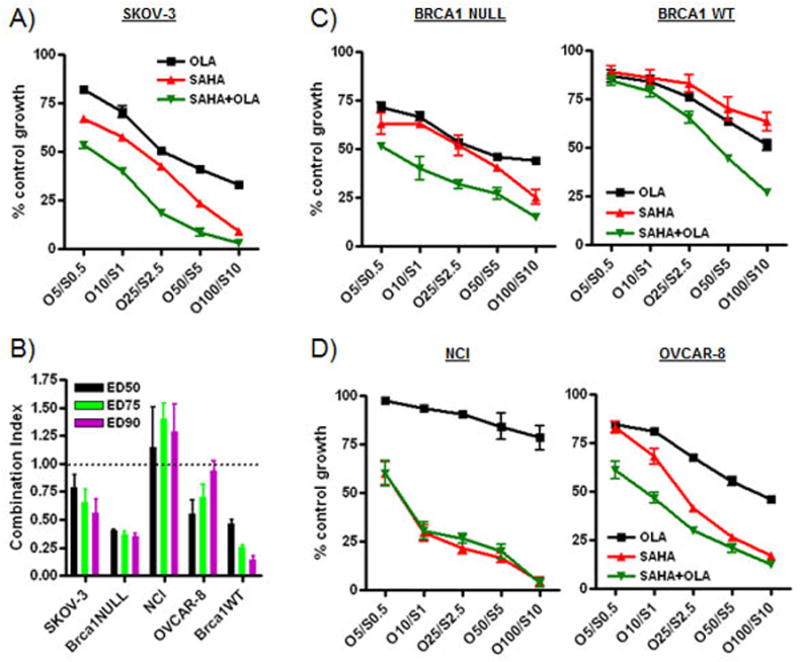
A,C,D) Ovarian cancer cells (SKOV-3, OVCAR-8, NCI/ADR-Res, UWB1.289 BRCA1 null and UWB1.289+BRCA1 wild-type) were treated with olaparib (OLA) (5–100μM), SAHA (0.5–10μM) or both. SRB assays were performed to assess cytotoxicity and cell viability after 72 h of treatment. Each dose was replicated 6 times. Values are shown as the mean + SD for three independent experiments. B) Combination results for the drug treatments are shown. A CI of < 1.0 is considered to be synergistic.
Combination treatment with SAHA and olaparib shows enhanced inhibitory effects on ovarian cancer tumor growth in vivo
Nude mice injected subcutaneously with SKOV-3 cells were treated with olaparib alone and in combination with SAHA for 3 weeks. Measurement of changes in tumor volume from the start to cessation of treatment showed that the rate of tumor growth was significantly slower in mice treated with the SAHA/olaparib combination compared to either vehicle (p=0.016, Mann-Whitney test) or olaparib alone (p=0.032, Mann-Whitney test) (Figure 5A). There was a trend in greater inhibition in tumor growth between SAHA and the combination that just failed to reach statistical significance (p=0.056).
Figure 5. SAHA inhibits tumor growth alone and in combination with OLA in vivo.

Nude mice injected SC with SKOV-3 cells were treated with olaparib (OLA) (10mg/kg thrice weekly), SAHA (50mg/kg twice thrice weekly), the OLA/SAHA combination at the same doses and schedule, and vehicle (0.01% DMSO thrice weekly) for 3 weeks following establishment of the tumors (approx 200mm3). 5 mice per treatment group were used. A) Tumor volume change from start of treatment period to sacrifice (%). In harvested tumors, the percentage of cells staining for B) pH2AX, C) cleaved caspase-3 and D) ki67/mib-1 by IHC was calculated. Values in B–D) are mean + SD for 3 independent experiments. At least 1000 cells were counted (40X) for each drug treatment per experiment. * p < 0.02 compared to vehicle; a p < 0.02 relative to SAHA alone; b p < 0.01 relative to olaparib alone, Mann-Whitney t test. E) High power (x40) images of H&E, pH2AX, cleaved caspase-3 and ki67/mib-1 staining in representative tumor sections for each drug treatment.
Harvested tumors were stained for pH2AX, cleaved caspase-3 and the proliferation marker ki67/mib-1. Consistent with our in vitro results, olaparib treatment alone significantly increased pH2AX expression relative to vehicle (p<0.001, Mann-Whitney t test) (Figure 5B), but did not significantly alter expression of cleaved caspase-3 (p=0.21, Mann-Whitney t test) (Figure 5C) or ki67/mib-1 (p=0.06, Mann-Whitney t test) (Figure 5E). However, the combination of SAHA and olaparib resulted in significantly increased cleaved caspase-3 (p<0.001, Mann-Whitney t test) and reduced ki67/mib-1 (p=0.002, Mann-Whitney t test) levels compared to olaparib alone, consistent with the enhanced inhibition of tumor growth observed. Representative images are shown in Figure 5E.
DISCUSSION
It is well known that PARPi are effective in HR-deficient, but not HR-proficient, EOC as single agents in clinical trials [3–5, 7]. Here, we have confirmed that the most frequently clinically used PARPi, olaparib [3, 4, 6, 7, 15], had minimal effects on cell growth and apoptosis in preclinical models of HR-proficient EOC. In these models, we tested the hypothesis that relative olaparib resistance could be overcome by co-treatment with the HDACi, SAHA. We found that SAHA induced coordinated downregulation of HR pathway genes, such as RAD51 and BRCA1, in SKOV-3 ovarian cancer cells. Moreover, there was synergistic enhancement of cytotoxicity in both HR-proficient (SKOV-3 and BRCA1 WT) and HR-deficient (BRCA1 NULL) cells with combination SAHA/olaparib treatment in vitro. Combination therapy also reduced tumor growth in SKOV-3 xenografts in vivo to a greater extent than olaparib alone. Enhanced cytotoxicity of the combination was associated with prolonged activation of pH2AX and reduced formation of RAD51 foci in response to DNA damage, both indicators of inefficient DNA repair mechanisms. However, since enhanced cytotoxicity was observed in both HR-proficient and HR-deficient cells, it is unclear whether SAHA-mediated disruption of efficient HR is a requirement for this synergy. Demonstrating a direct mechanistic link between SAHA-inhibited HR and sensitization to olaparib-mediated cytotoxicity will be the focus of future studies. Collectively, these preclinical studies provide a rationale for future investigation of PARPi/HDACi combination strategies to target DNA damage response pathways in selected ovarian tumors.
Our results indicate an association between SAHA-mediated inhibition of RAD51 expression and reduced formation of RAD51 foci following DNA damage. Other studies have shown similar findings of reduced HR and RAD51 expression by other HDACi [18, 19]. However, alternative non-transcriptional mechanisms of SAHA-mediated decreases in RAD51 foci have been proposed, such as reduced stabilization and chromatin association of RAD51 [34]. It is also possible that SAHA-induced changes in cell cycle may influence formation of RAD51 foci, although this was not observed in irradiated multiple myeloma cells [34]. The precise mechanism by which SAHA reduces RAD51 function during DNA damage responses in ovarian cancer cells will be the focus of future studies.
Although SAHA is FDA-approved as a single agent for the treatment of cutaneous T cell lymphoma (14, 15), its clinical effectiveness is limited by poor pharmacokinetics [35], and it has minimal therapeutic benefit as a single agent in a phase II ovarian cancer clinical trial [36]. SAHA inhibits both class I and II HDACs, and strong evidence suggest that the Class I HDACs such as HDAC3 play a critical role in DNA repair [37–39]. Therefore, more potent, isoform-targeted HDACi with improved pharmacokinetics may be required to more effectively treat ovarian cancer patients. Nevertheless, our findings provide proof-of-principle of the effectiveness of a strategy combining drugs which inhibit both single and double-strand DNA break repair. This concept is especially relevant to ovarian cancer, because approximately 50% of high grade serous EOC do not harbor genetic and epigenetic alterations of the HR DNA repair pathway [1, 2], the lack of which markedly decreases susceptibility to PARPi (7–9, 11).
In conclusion, the response of ovarian cancer cells to the PARPi olaparib is enhanced by co-treatment with the HDACi, SAHA in vitro and in vivo. The key sensor of DNA damage, pH2AX, is activated by combination treatment above that of either drug alone. Prolonged and robust activation of pH2AX and reduced HR function (formation of RAD51 foci) is associated with enhanced DNA damage-induced cell death with the combination treatment. Our preclinical studies indicate that responses to PARPi in HR-proficient ovarian epithelial tumors can be improved by combination therapy with HR-inhibiting drugs such as HDACi. Thus, we have identified a novel treatment strategy of combining PARPi with HDACi with the potential to improve responses of both HR-deficient and -proficient ovarian cancers to PARPi therapy.
RESEARCH HIGHLIGHTS.
SAHA downregulates homologous recombination pathway genes in ovarian cancer cells
Formation of RAD51 foci is inhibited by SAHA alone and combined with olaparib
Anti-tumor effects of olaparib in vitro and in vivo are enhanced by SAHA
Acknowledgments
We acknowledge the Vanderbilt University High-Throughput Screening Core and the Vanderbilt Immunohistochemistry Core. The Authors also thank Dr Edward Holson (Stanley Center for Psychiatric Research, The Broad Institute, Cambridge, MA) for providing SAHA.
Grant Support: 1K08CA148887-01 (DK) and the Department of Defense Ovarian Cancer Academy Award DOD W81XWH-10-1-0585/OC093426 (PAK).
Footnotes
CONFLICT OF INTEREST
The authors disclose no potential conflicts of interest.
This manuscript was presented in part as a poster at the Society of Gynecologic Oncology’s 43rd Annual Meeting on Women’s Cancer, Austin, TX, March, 2012.
References
- 1.Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alsop K, Fereday S, Meldrum C, Defazio A, Emmanuel C, George J, et al. BRCA Mutation Frequency and Patterns of Treatment Response in BRCA Mutation-Positive Women With Ovarian Cancer: A Report From the Australian Ovarian Cancer Study Group. J Clin Oncol. 2012;30:2654–63. doi: 10.1200/JCO.2011.39.8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376:245–51. doi: 10.1016/S0140-6736(10)60893-8. [DOI] [PubMed] [Google Scholar]
- 4.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 5.Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28:2512–9. doi: 10.1200/JCO.2009.26.9589. [DOI] [PubMed] [Google Scholar]
- 6.Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12:852–61. doi: 10.1016/S1470-2045(11)70214-5. [DOI] [PubMed] [Google Scholar]
- 7.Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366:1382–92. doi: 10.1056/NEJMoa1105535. [DOI] [PubMed] [Google Scholar]
- 8.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 9.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 10.Haince JF, Rouleau M, Hendzel MJ, Masson JY, Poirier GG. Targeting poly(ADP-ribosyl)ation: a promising approach in cancer therapy. Trends Mol Med. 2005;11(10):456–63. doi: 10.1016/j.molmed.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 11.Chen A. PARP inhibitors: its role in treatment of cancer. Chin J Cancer. 2011;30:463–71. doi: 10.5732/cjc.011.10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saleh-Gohari N, Bryant HE, Schultz N, Parker KM, Cassel TN, Helleday T. Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol Cell Biol. 2005;25:7158–69. doi: 10.1128/MCB.25.16.7158-7169.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arnaudeau C, Lundin C, Helleday T. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol. 2001;307:1235–45. doi: 10.1006/jmbi.2001.4564. [DOI] [PubMed] [Google Scholar]
- 14.Banath JP, Klokov D, MacPhail SH, Banuelos CA, Olive PL. Residual gammaH2AX foci as an indication of lethal DNA lesions. BMC Cancer. 2010;10:4. doi: 10.1186/1471-2407-10-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaye SB, Lubinski J, Matulonis U, Ang JE, Gourley C, Karlan BY, et al. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J Clin Oncol. 2011;30:372–9. doi: 10.1200/JCO.2011.36.9215. [DOI] [PubMed] [Google Scholar]
- 16.Wilson AJ, Lalani AS, Wass E, Saskowski J, Khabele D. Romidepsin (FK228) combined with cisplatin stimulates DNA damage-induced cell death in ovarian cancer. Gynecol Oncol. 2012;127:579–86. doi: 10.1016/j.ygyno.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 18.Koprinarova M, Botev P, Russev G. Histone deacetylase inhibitor sodium butyrate enhances cellular radiosensitivity by inhibiting both DNA nonhomologous end joining and homologous recombination. DNA Repair (Amst) 2011;10:970–7. doi: 10.1016/j.dnarep.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 19.Adimoolam S, Sirisawad M, Chen J, Thiemann P, Ford JM, Buggy JJ. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc Natl Acad Sci U S A. 2007;104:19482–7. doi: 10.1073/pnas.0707828104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ladd B, Ackroyd JJ, Hicks JK, Canman CE, Flanagan SA, Shewach DS. Inhibition of homologous recombination with vorinostat synergistically enhances ganciclovir cytotoxicity. DNA Repair (Amst) 2013;12:1114–21. doi: 10.1016/j.dnarep.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. The Oncologist. 2007;12:1247–52. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 22.Mann BS, Johnson JR, He K, Sridhara R, Abraham S, Booth BP, et al. Vorinostat for treatment of cutaneous manifestations of advanced primary cutaneous T-cell lymphoma. Clin Cancer Res. 2007;13:2318–22. doi: 10.1158/1078-0432.CCR-06-2672. [DOI] [PubMed] [Google Scholar]
- 23.Khabele D, Son DS, Parl AK, Goldberg GL, Augenlicht LH, Mariadason JM, et al. Drug-induced inactivation or gene silencing of class I histone deacetylases suppresses ovarian cancer cell growth: implications for therapy. Cancer Biol Ther. 2007;6:795–801. doi: 10.4161/cbt.6.5.4007. [DOI] [PubMed] [Google Scholar]
- 24.Wilson AJ, Holson E, Wagner F, Zhang YL, Fass DM, Haggarty SJ, et al. The DNA damage mark pH2AX differentiates the cytotoxic effects of small molecule HDAC inhibitors in ovarian cancer cells. Cancer Biol Ther. 2011;12:484–93. doi: 10.4161/cbt.12.6.15956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lorenzi PL, Reinhold WC, Varma S, Hutchinson AA, Pommier Y, Chanock SJ, et al. DNA fingerprinting of the NCI-60 cell line panel. Mol Cancer Ther. 2009;8:713–24. doi: 10.1158/1535-7163.MCT-08-0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scudiero DA, Monks A, Sausville EA. Cell line designation change: multidrug-resistant cell line in the NCI anticancer screen. J Nat Cancer Inst. 1998;90:862. doi: 10.1093/jnci/90.11.862. [DOI] [PubMed] [Google Scholar]
- 27.DelloRusso C, Welcsh PL, Wang W, Garcia RL, King MC, Swisher EM. Functional characterization of a novel BRCA1-null ovarian cancer cell line in response to ionizing radiation. Mol Cancer Re. 2007;5:35–45. doi: 10.1158/1541-7786.MCR-06-0234. [DOI] [PubMed] [Google Scholar]
- 28.Effron B, Tibshirani R. On testing the significance of sets of genes. Ann Appl Stat. 2007;1:107–29. [Google Scholar]
- 29.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 30.Wilson AJ, Arango D, Mariadason JM, Heerdt BG, Augenlicht LH. TR3/Nur77 in colon cancer cell apoptosis. Cancer Res. 2003;63:5401–7. [PubMed] [Google Scholar]
- 31.Wilson AJ, Byun DS, Nasser S, Murray LB, Ayyanar K, Arango D, et al. HDAC4 promotes growth of colon cancer cells via repression of p21. Mol Biol Cell. 2008;19:4062–75. doi: 10.1091/mbc.E08-02-0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rauh-Adelmann C, Lau KM, Sabeti N, Long JP, Mok SC, Ho SM. Altered expression of BRCA1, BRCA2, and a newly identified BRCA2 exon 12 deletion variant in malignant human ovarian, prostate, and breast cancer cell lines. Mol Carcinogenesis. 2000;28:236–46. doi: 10.1002/1098-2744(200008)28:4<236::aid-mc6>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 33.Zhou C, Huang P, Liu J. The carboxyl-terminal of BRCA1 is required for subnuclear assembly of RAD51 after treatment with cisplatin but not ionizing radiation in human breast and ovarian cancer cells. Biochem Biophys Res Comm. 2005;336:952–60. doi: 10.1016/j.bbrc.2005.08.197. [DOI] [PubMed] [Google Scholar]
- 34.Chen X, Wong P, Radany EH, Stark JM, Laulier C, Wong JY. Suberoylanilide hydroxamic acid as a radiosensitizer through modulation of RAD51 protein and inhibition of homology-directed repair in multiple myeloma. Mol Cancer Res. 2012;10:1052–64. doi: 10.1158/1541-7786.MCR-11-0587. [DOI] [PubMed] [Google Scholar]
- 35.Suzuki T, Miyata N. Non-hydroxamate histone deacetylase inhibitors. Curr Med Chem. 2005;12:2867–80. doi: 10.2174/092986705774454706. [DOI] [PubMed] [Google Scholar]
- 36.Modesitt SC, Sill M, Hoffman JS, Bender DP. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol. 2008;109:182–6. doi: 10.1016/j.ygyno.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 37.Bhaskara S, Chyla BJ, Amann JM, Knutson SK, Cortez D, Sun ZW, et al. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol Cell. 2008;30:61–72. doi: 10.1016/j.molcel.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bhaskara S, Knutson SK, Jiang G, Chandrasekharan MB, Wilson AJ, Zheng S, et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell. 2010;18:436–47. doi: 10.1016/j.ccr.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, et al. Chemical phylogenetics of histone deacetylases. Nat Chem Biol. 2010;6:238–43. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]