

Abstract
The role of ceramide neo-genesis in cellular stress response signaling is gaining increasing attention with recent progress in elucidating the novel roles and biochemical properties of the ceramide synthase (CerS) enzymes. Selective tissue and subcellular distribution of the six mammalian CerS isoforms, combined with distinct fatty acyl chain length substrate preferences, implicate differential functions of specific ceramide species in cellular signaling. We report here that ionizing radiation (IR) induces de novo synthesis of ceramide to influence HeLa cell apoptosis by specifically activating CerS isoforms 2, 5, and 6 that generate opposing anti- and pro-apoptotic ceramides in mitochondrial membranes. Overexpression of CerS2 resulted in partial protection from IR-induced apoptosis whereas overexpression of CerS5 increased apoptosis in HeLa cells. Knockdown studies determined that CerS2 is responsible for all observable IR-induced C24:0 CerS activity, and while CerS5 and CerS6 each confer ∼50% of the C16:0 CerS baseline synthetic activity, both are required for IR-induced activity. Additionally, co-immunoprecipitation studies suggest that CerS2, 5, and 6 might exist as heterocomplexes in HeLa cells, providing further insight into regulation of CerS proteins. These data add to the growing body of evidence demonstrating interplay among the CerS proteins in a stress stimulus-, cell type- and subcellular compartment-specific manner.
Keywords: ceramide synthase, LASS, CerS, ionizing radiation, apoptosis, HeLa cell
1. Introduction
Diverse cellular and environmental stresses (e.g. chemotherapeutics [1], heat shock [2], ischemia-reperfusion [3], ultraviolet radiation [4], and ionizing radiation (IR) [5], to list a few) stimulate cells to generate ceramide, an established second messenger in apoptotic signaling pathways [6-8]. Ceramide (N-acyl-D-erythro-sphingosine) can be generated via two major pathways: by hydrolysis of sphingomyelin via sphingomyelinases, or by ceramide synthase (CerS)-mediated synthesis, either via de novo acylation of the sphingoid base sphinganine with fatty acyl-CoAs of varying chain length from C14 to C26 to yield (dihydro)ceramides, followed by oxidation of sphinganine to sphingosine to yield ceramide, or via a salvage (or recycling) pathway where ceramide is deacylated by ceramidases to form sphingosine, which is reutilized by CerS to re-generate ceramide [9]. The sphinganine analogue, fumonisin B1 (FB1), is a competitive inhibitor of CerS activity [10].
IR-induced CerS-mediated ceramide generation, and subsequent apoptosis, occurs in a cell-type specific manner. Unlike the fast generation of ceramide at the plasma membrane (seconds to minutes) via sphingomyelinases, engagement of CerS and ceramide neo-genesis is delayed (hours to days) in almost every system defined to date [1, 11]. Furthermore, it was recently found that IR activates CerS to generate ceramide de novo in C. elegans germ cell mitochondrial membranes [12], implicating involvement of ceramide in the commitment step of the mitochondrial death pathway. In mammals a pathway analogous to that in C. elegans, termed the mitochondrial death (also known as intrinsic) pathway is the main pathway for apoptotic death. In this pathway, signals, instigated by pro-apoptotic stimuli, converge on mitochondria to induce mitochondrial outer membrane permeabilization (MOMP), the commitment step in this apoptotic process. MOMP results in release of apoptogenic factors, such as cytochrome c, to trigger activation of caspases, key effector components of apoptosis [13, 14]. MOMP is regulated by pro- and anti-apoptotic B-cell lymphoma 2 (Bcl-2) family members [15]. Relevant to this study, Bcl-xL, an anti-apoptotic Bcl-2 protein, protects numerous cell types from apoptosis by preventing MOMP [16-18]. The biologic significance of CerS in mediating mammalian apoptosis has been confirmed in vivo, as CerS transactivates disease pathogenesis in several experimental models of human disease [19-24].
There are six identified mammalian CerS isoforms, termed CerS/LASS1-6, characterized by a conserved lag1 domain. Although these six CerS isoforms all have a similar Km value towards sphinganine in the low micromolar range [25], they differ in their fatty acyl-CoA substrate chain length preference. For example, CerS1 uses exclusively long chain C18:0-fatty acyl-CoA [26], CerS5 and CerS6 prefer C16:0 [27, 28], whereas CerS2 utilizes very-long-chain species such as C24:0 and C24:1 [25, 28]. CerS family members also exhibit different tissue-specific expression patterns. CerS1 is largely confined to tissues of the nervous system [28] and CerS3 expression is predominant in testis [29] and keratinocytes [30]. CerS2, CerS4, CerS5, and CerS6 appear to have a broader tissue distribution [27, 28, 31], which corresponds to evidence showing that C24:1-, C24:0-, and C16:0-ceramides are the most abundant species in cells of many tissues, including epithelial cells, fibroblasts, and cells of the immune system [32, 33]. The interplay between long chain C16:0-ceramide and very long chain C24:1- and C24:0-ceramides has come into recent spotlight regarding their roles in maintaining cellular homeostasis [34-36], as C16:0-ceramide species is most often pro-apoptotic, whereas C24-ceramides do not appear to display this propensity. There is also evidence of intracellular differences in CerS distribution. CerS activity was initially localized to the cytoplasmic leaflet of the endoplasmic reticulum [37, 38], but increasing evidence reveals that CerS have additional subcellular localizations, such as perinuclear membranes [39] and the mitochondria-associated-membrane (MAM) [40], where distinct ceramides may confer discrete, and at times even opposing, signaling endpoints, including apoptosis and cell survival [39, 41, 42].
The mitochondrial-associated-membrane (MAM) is a compartment comprising the physical interaction between the endoplasmic reticulum (ER) and mitochondria (reviewed in [43]), enriched in lipid synthetic and transfer proteins, including CerS [40]. Recently, it was shown that long-chain ceramide generated de novo is rapidly transferred from MAM to mitochondria, likely catalyzed by a not yet identified transfer protein, resulting in MOMP [44]. Our laboratory also recently identified the MAM fraction of HeLa human cervical carcinoma cells as the site of IR-induced CerS activity, and the mitochondria as the predominant site of IR-induced ceramide elevation (Lee and Kolesnick, submitted). Based on this information, here we report that IR induces de novo ceramide synthesis to influence HeLa cell apoptosis, specifically activating CerS2, 5, and 6 in the MAM, generating opposing anti- and pro-apoptotic mitochondrial ceramides.
2. Materials and Methods
2.1. Cell culture, transfection, FB1-treatment, and irradiation
HeLa cells were cultured in low glucose Dulbecco's Modified Eagle's Medium (DMEM; Gibco BRL) supplemented with 10% fetal bovine serum (FBS), penicillin (50 units/ml), streptomycin (50 μg/ml) and 2 mM glutamine. Cells were transfected using Fugene 6 transfection reagent (Roche) in antibiotic-free culture media according to manufacturer's protocol. Medium containing DNA and reagent was replaced 6 h after transfection with complete culture medium. FB1 (Biomol) was solubilized in 1× PBS at a concentration of 5 mM and added to cells at final concentration of 75 μM. Note commercially available FB1 is a biologic product isolated from Fusarium verticillioides and Fusarium proliferatum that displays batch-to-batch variation. Hence, effectiveness of each batch must be tested empirically. Irradiation was carried out at 22°C using a Cs-137 irradiator (Shepherd Mark-I, model 68, SN 643) at a dose rate of 240 cGy/min.
2.2. Cloning pCMV2B-CerS1, 2, 5, and 6, pcDNA3-HA-CerS2, and pCMV3B-CerS6
We cloned full length human CerS1, 2, 5 and 6 into the pCMV2B plasmid vector (Stratagene) as described previously [25]. We cloned full length human CerS2 into the pcDNA3 plasmid vector (Invitrogen), containing an N-terminal HA tag, using human liver tissue library (Clontech). The genes were inserted using the following primers flanked with HinDIII and EcoRI restriction sites (Gene Link, Inc.): 5′ggaattcctccagaccttgtatgattac′3 and 5′cgaagcttgggagcggggtagttccttggc′3. The PCR products and pcDNA3-HA were digested with EcoR1-HinDIII, ligated, transformed into E.coli DH5α (Invitrogen), and directly sequenced. Full length human CerS6 was inserted into the pCMV3B plasmid vector (Stratagene), containing an N-terminal myc tag, by BamHI and EcoRI restriction digest of pCMV2B-CerS6 plasmid, subsequent ligation of the CerS6 insert into BamHI and EcoRI-digested pCMV3B, transformation, and direct sequencing.
2.3. Cloning pSUPER-CerS2, 5 and 6
Sense and antisense shRNA constructs flanked by HindIII restriction enzyme sites for each CerS isoform were annealed and ligated into the pSUPER expression vector (OligoEngine, Seattle, WA) as follows:
CerS2 (NM_181746)
sense 5′gatccccggatatcccatacagagcattcaagagatgctctgtatgggatatccttttta; antisense 5′agcttaaaaaggatatcccatacagagcatctcttgaatgctctgtatgggatatccggg.
CerS5 (NM_147190.2)
sense 5′gatccccgactgcaaggcactgaggattcaagagatcctcagtgccttgcagtcttttta; antisense 5′agcttaaaaagactgcaaggcactgaggatctcttgaatcctcagtgccttgcagtcggg.
CerS6 (NM_203463.1)
sense 5′gatcccctgaactgcttctggtcttattcaagagataagaccagaagcagttcattttta; antisense 5′agcttaaaaatgaactgcttctggtcttatctcttgaataagaccagaagcagttcaggg.
Vectors were transformed into E.coli DH5α (Invitrogen) and inserts verified by direct sequencing. shRNA expression vectors were screened for gene specific mRNA knockdown by real-time qPCR, and for protein knockdown by western blotting for CerS2 and CerS6.
2.4. Reverse transcriptase (RT)-PCR
Total RNA was isolated from cultured HeLa cells using an RNeasy Mini Kit (Qiagen), converted to cDNA using SuperScript® III First-Strand Synthesis System (Invitrogen), and PCR was performed using Fast Start Taq DNA Polymerase Kit (Roche) according to manufacturers' protocols. PCR conditions were as follows: initial denaturation for 4 min at 95°C, followed by 30 cycles of 30 s at 95°C, 30 s at 55°C, and 1 min at 72°C. A final elongation step followed for 10 min at 72°C. PCR products were separated by electrophoresis in 1% agarose gels. Primers are as follows: CerS1-5′cgtcgcggcctggctgagcacg and 5′gccgatggtaggagccgccgc. CerS2-5′ctccagaccttgtatgattact and 5′ggccacatggtggatgatctg; CerS3 - 5′ggactggcaaagaagtg and 5′cagggtgttacaggtct; CerS4- 5′ggaaccaggatcgacc and 5′ggactcgtagtatgtgg; CerS5-5′ggagggcctgtcaaagc and 5′cctagtcgtgtaacc; CerS6- 5′cgacaaagacgcaatcagg and 5′cggcaaacataacaaacagg; β-actin- 5′gctcgtcgtcgacaacggctc and 5′caaacatgatctgggtcatcttctc. Results were confirmed using previously published CerS1-6 primers [30].
2.5. Isolation of subcellular fractions from HeLa cells
All procedures were conducted at 4°C. For these studies, attached and floating cells were collected by centrifugation at 500×g for 10 min, washed twice, resuspended in SHE buffer [250 mM sucrose, 10 mM HEPES-KOH pH 7.4, 1 mM EGTA, protease inhibitor cocktail (Roche)] and homogenized twice by 20 strokes in a loose-fitting Dounce homogenizer. Cell debris and nuclei were pelleted by centrifugation at 800×g for 5 min. The post-nuclear supernatant was collected and centrifuged at 10,000×g for 10 min and the pellet containing heavy membrane fractions (P10) was resuspended in 1 ml SHE buffer. MAM-free mitochondria were isolated by Percoll gradient according to published methods [45], with slight modification. For these studies, P10 fractions were layered on top of 20 ml of medium consisting of 2.2 ml of 2.5 M sucrose, 6.55 ml of Percoll and 12.25 ml of 10 mM HEPES, pH 7.4, 1 mM EGTA, and centrifuged at 100,000×g for 1.5 h in a Beckman Ti-55.2 rotor. Two bands were recovered from the gradient; a lower band corresponding to mitochondria (Rf: 0.48) and an upper band containing MAM (Rf: 0.23) [45]. Mitochondria were collected using a Pasteur pipette, diluted 10-fold with SHE buffer and washed twice by centrifugation at 10,000×g for 10 min to remove the Percoll. To isolate the ER-enriched membrane fraction, the supernatant of the P10 fraction was centrifuged at 100,000×g for 1 h. Fractionation was monitored by immunoblotting using mouse monoclonal anti-COX II (mitochondrial marker) and rabbit polyclonal anti-Climp63 or anti-Calnexin (ER Markers).
2.6. CerS activity assay
For these studies, 75×106 cells were pelleted, washed once with cold PBS, and resuspended in 300 μl of homogenization buffer [25 mM HEPES pH 7.4, 5 mM EGTA, 50 mM NaF, protease inhibitor cocktail (Roche)]. Cells were disrupted on ice by sonication, and lysates centrifuged at 800×g for 5 min. The post-nuclear supernatant was centrifuged at 100,000×g for 1 h and the microsomal pellet (P100) was resuspended into 1 mL of homogenization buffer. To measure CerS activity in subcellular fractions, mitochondrial-enriched, ER-enriched, and MAM-enriched fractions were prepared as described in “Isolation of subcellular fractions from HeLa cells”. Membranes were prepared fresh daily.
CerS5 and CerS6 activities using palmitoyl-CoA were measured as previously described [5]. CerS2 activity using lignoceroyl-CoA was measured in 100 μg of HeLa cell homogenate incubated with [4,5-3H] sphinganine (specific activity of 15 Ci/mmol, American Radiolabeled Chemicals)/20 μM defatted-bovine serum albumin (Sigma), and 100 μM of lignoceroyl acyl-CoA (Avanti Polar Lipids, Alabaster, AL). The reaction was stopped after 30 min and lipids extracted and analyzed as above. Nonlinear fitting and Michaelis-Menten analyses were performed to determine Km and Vmax values using Prism 5.0b software.
2.7. Diacylglycerol Kinase (DGK) Assay
For ceramide measurement, cells were grown to confluency in 6-well tissue culture dishes. Floating cells were collected in chilled 13×100 mm glass tubes and collected by centrifugation at 500×g for 5 min. The pelleted cells were washed with cold 1× PBS and re-centrifuged. Adherent cells were lysed on ice for 10 min in 0.5 mL ice-cold methanol, collected with a rubber policeman, and added to the pelleted cells in glass tubes. Methanol lyses was repeated in tissue culture wells, and scraped cells added to the glass tubes. One ml of chloroform and 0.6 ml buffered saline solution [135 mM NaCI, 1.5 mM CaCI2, 0.5 mM MgCI2, 5.6 mM glucose, and 10 mM HEPES [pH 7.2]]/ 10 mM EDTA were added to tubes and lipids were extracted according to the DGK assay method [46].
2.8. Antibodies, Immunoprecipitation (IP), and Immunoblotting
Anti-FLAG (A8592, Sigma; 1:1000), anti-HA (H6908, Sigma; 1:1000), anti-myc (2278, Cell Signaling; 1:1000), anti-CerS2 (H00029956-M02, Abnova; 1:1000), anti-CerS6 (X2303P, Exalpha Biologicals; 1:200 and H00253782-M01, Abnova; 1:1000) were used as indicated for immunoblotting. Anti-FLAG M2-agarose beads (A2220, Sigma) were used to IP FLAG-CerS proteins, and anti-HA agarose beads (A-2095, Sigma) were used to IP HA-CerS2, according to manufacturer's instructions. Anti-CerS6 (H00253782-M01, Abnova; 1 μg) was used to IP CerS6 from pre-cleared cell lysate for 16 h on a roller apparatus at 4°C. Protein A/G beads (sc-2003, Santa Cruz) were then added and incubated for 2 h. Beads were washed extensively in 1× PBS and resuspended for SDS-PAGE analysis. Post-IP supernatant was analyzed for residual CerS6 using anti-CerS6 (X2303P, Exalpha Biologicals; 1:200). Proteins were separated on a 10-12% SDS-PAGE gel, transferred to 0.2 μm PVDF membrane (Biorad), and western blots carried out according to manufacturers' recommended protocols.
2.9. Apoptosis Quantification
bis-benzimide staining
Method was carried out as previously described [47].
Colorimetric assay for caspase-3
Cells seeded in 6 well plates at 25% confluency were transiently transfected with either 0.5 μg pCMV2B empty vector, pCMV2B-CerS5, pcDNA3.1-HA-Bcl-xL [48], or both pCMV2B-CerS5 and pcDNA3.1-HA-Bcl-xL, as described above and irradiated 24 h later. Cell lysate was prepared 48 h after IR and caspase activity measured as previously described [49, 50], with the following modification: in a 40 μl system, 50 μg HeLa lysate was incubated with 50 μM fluorogenic N-acetyl-Asp-Glu-Val-Asp-AFC (Ac-DEVD-AFC) substrate (Enzo Life Sciences).
2.10. Analysis of ceramides by LC-MS/MS
Analyses of the ceramides in MAM-free HeLa cell mitochondria, isolated as described above, were performed by combined LC-MS/MS. Lipid extraction and sample preparation were performed according to [51, 52]. The instrumentation employed was an API4000 Q-trap hybrid triple quadrupole linear ion-trap mass spectrometer (Applied Biosystems, Foster City, CA) equipped with a turboionspray ionization source interfaced with an automated Agilent 1100 series liquid chromatograph and autosampler (Agilent Technologies, Wilmington, DE). The sphingolipids were ionized via electrospray ionization (ESI) with detection via multiple reaction monitoring (MRM). Analysis of sphingoid bases and the molecular species of ceramides employed ESI in positive ions with MRM analysis, with C17-sphingosine and N-C17-ceramide as the internal standards. Details of the procedure are described in [51, 52].
2.11. Statistical Analysis
Statistical analysis was performed by unpaired, Student's t-test, using Graphpad Prism Version 5.0b software. For enzyme kinetic analysis, Vmax and Km values were derived by nonlinear fitting using Graphpad Prism, and differences in model parameters (Km, Vmax) between groups were compared using a z-test.
3. Results and Discussion
3.1. Ionizing radiation (IR) induces HeLa cell apoptosis via CerS-mediated ceramide generation
IR stimulates ceramide production in various cell types yielding apoptotic signaling, either by acid sphingomyelinase-mediated hydrolysis of sphingomyelin [53] or via CerS-mediated de novo generation [11]. We show here, in HeLa cells, that IR induction of apoptosis is dose-dependent, with as little as 5 Gy eliciting a significant apoptotic response (p<0.01 vs. 0 Gy each at all doses ≥5 Gy), increasing up to 20 Gy (Fig. 1A). Significant apoptosis is delayed, beginning 30 h after exposure, with levels increasing over time [see Figs. 3A,B, empty vector-transfected cells; note that control experiments showed no difference between empty vector-transfected and untransfected HeLa cells in response to IR (data not shown)]. Furthermore, pretreatment with the competitive CerS inhibitor FB1 (75 μM) reduced IR-induced apoptosis by 50%, indicating that HeLa cell apoptosis is a CerS-regulated process (p<0.02 vs. control).
Fig. 1. Ionizing radiation induces CerS-mediated apoptosis in HeLa cells.
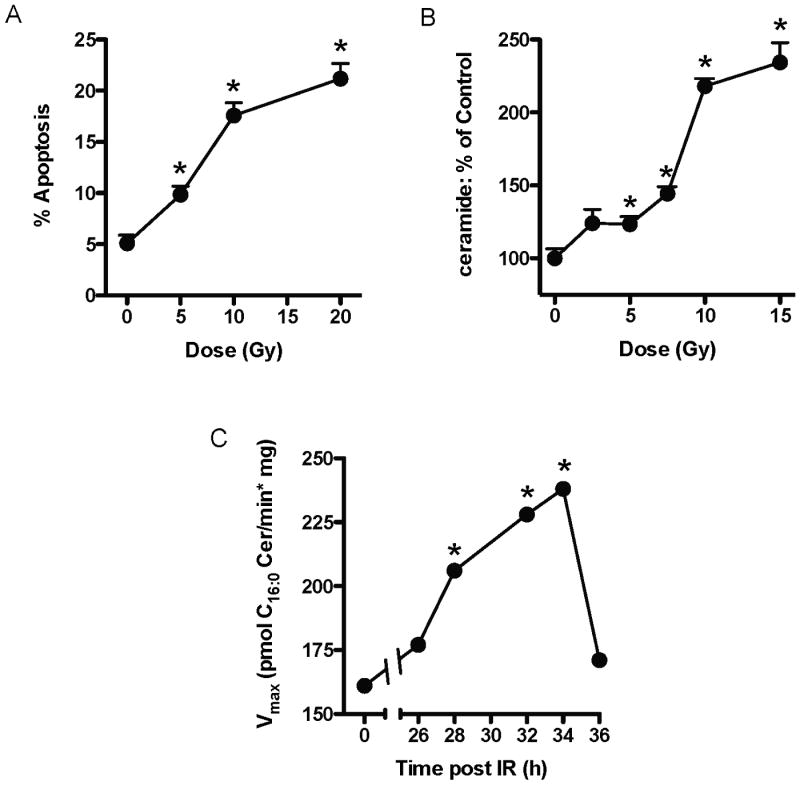
(A) Dose-dependent induction of apoptosis in response to IR. Morphologic changes of nuclear apoptosis were detected using the DNA-specific fluorochrome bis-benzimide. Data (mean±SEM) are from 1 of 3 similar experiments analyzing 400 cells per point. (p<0.01 vs. 0 Gy) (B) Dose response of ceramide generation measured 28 h after radiation. Cellular lipids were extracted and ceramide was quantified by the diacylglycerol kinase assay. Data (mean±SEM) are from 1 of 3 similar independent experiments performed in triplicate. (p<0.001 vs. 0 Gy) (C) Time-dependent CerS activation measured in post-nuclear extracts prepared from HeLa cells upon exposure to 10 Gy. Vmax values are derived from full Michaelis-Menten kinetics analyses of palmitoyl-CoA-specific CerS activity for each time point. (p<0.05 vs. 0 h)
Fig. 3. CerS2 and CerS5 overexpression define opposing roles in radiation-induced mitochondrial apoptosis.

(A) CerS2 overexpression partially protects HeLa cells from time-dependent IR-induced apoptosis measured by bis-benzimide staining. Data (mean±SD) are representative of 3 similar independent experiments performed in triplicate analyzing >200 cells per point. (p<0.01 vs. vector 15 Gy). Overexpression of CerS5 significantly increased apoptosis after 10 Gy, quantified in (B) by bis-benzimide staining (p<0.05 vs. vector 10 Gy) and in (C) by caspase activation (*p<0.02 vs. vector IR). Data (mean±SEM) in (B) are from 1 of 4 independent studies performed in triplicate analyzing >400 cells per point. In (C), BcL-xL overexpression abrogates both CerS5 overexpression- and IR-induced caspase activity elevations. Data (mean±SEM) in (C) are from 2 independent studies performed in duplicate. (**p<0.001 vs. vector IR). Overexpression of each CerS gene and bcl-xL was confirmed by direct western blot on cell lysate.
IR-induced apoptosis was preceded by dose-dependent elevation in total cellular ceramide, with a maximal 2.34-fold increase 28 h after 15 Gy (Fig. 1B and not shown; p<0.001 vs. 0 Gy). Using palmitoyl-CoA and sphinganine substrates, we determined timing of IR-induced increase in CerS activity in post-nuclear supernatants prepared from HeLa cells at various time points after 10 Gy exposure. Fig. 1C plots Vmax values derived from full Michaelis-Menten kinetics performed at each time point, showing significant IR-induced activity beginning at 28 h following exposure, with no significant change in Km towards sphinganine for all timepoints (Km=3.4-5.8 μM). Vmax steadily increased to 1.5-fold over baseline until a zenith was reached at 34 h, before dropping to nearly baseline levels at 36 h (p<0.05 for each timepoint vs. 0 h). Timing of CerS activation, ceramide generation, and apoptosis in HeLa cells seen here is congruent with the delayed mode of CerS-mediated events observed in multiple model systems [1, 11].
3.2. CerS2, 5, and 6 can synthesize IR-induced mitochondrial ceramides in HeLa cells
Our laboratory previously identified mitochondria as the site of IR-induced ceramide elevation in HeLa cells (Lee and Kolesnick, submitted). Mass spectrometry analyses of mitochondrial ceramides revealed specific increases in C16:0-, C24:1-, and C24:0- ceramides at 33 h after 10 Gy (Fig. 2A). Other ceramide and dihydroceramide molecular species were present at either very low or undetectable levels. To determine candidate CerS isoforms capable of generating these ceramide species, we performed RT-PCR on total mRNA isolated from HeLa cells using two separate sets of CerS homolog-specific primers, revealing the presence of CerS1, 2, 5, and 6 transcripts (Fig. 2B). These data were confirmed by real-time qPCR (data not shown). Prior published work points to CerS2 as the isoform responsible for generating very long chain C24:1- and C24:0-ceramides [25, 28], whereas CerS5 and CerS6 generate C16:0-ceramide [27, 28].
Fig. 2. Ionizing radiation-induced mitochondrial ceramide elevations via CerS2, 5, and 6.

(A) Ceramide molecular species were analyzed by mass spectrometry in mitochondria purified from control and irradiated (10 Gy) HeLa cells 33 h following exposure. Data (mean±SEM) are derived from biological duplicates, and IR-responsive ceramide species were confirmed by 2 additional independent studies. (B) RT-PCR products using CerS-specific primers on total mRNA isolated from HeLa cells were resolved on a 1% agarose gel to reveal transcripts encoding CerS1, 2, 5, and 6. These results were confirmed using a second set of homolog-specific primers as well as by real-time qPCR analyses.
3.3. Overexpression of certain HeLa cell CerS isoforms influences IR-induced mitochondrial apoptotic outcome
To study effects of CerS protein overexpression on HeLa cell apoptosis, we cloned N-terminally FLAG-tagged full-length CerS1, 5, and 6 constructs into the pCMV2B mammalian expression vector, as described in Materials and Methods. We also generated N-terminally HA-tagged full-length CerS2 and cloned it into the pcDNA3 mammalian expression vector. Cells overexpressing either CerS1, 2, 5, or 6 for 24 h were exposed to IR (0 or 15 Gy), and apoptosis was quantified by bis-benzimide staining and compared to empty vector-transfected cells. Overexpression was confirmed in cell lysates by western blot using anti-FLAG and anti-HA antibodies (data not shown).
Although CerS1 and CerS6 overexpression yielded no significant differences in IR-induced apoptosis compared to empty vector-transfected cells (data not shown), CerS2 and CerS5 overexpression significantly altered apoptosis. CerS2 overexpression yielded 42.8% protection at 36 h after 15 Gy (Fig. 3A; p=0.001 vs. vector 15 Gy). Conversely, CerS5 overexpression significantly increased apoptosis in a time- and dose-dependent manner compared to empty vector-transfected controls (Fig. 3B and data not shown), elevating apoptosis levels 1.8-fold at both 36 h and 42 h after 10 Gy (p<0.05 each vs. vector 10 Gy). IR-induced, CerS5-enhanced, apoptosis was confirmed by a second method of apoptosis quantification, caspase activity (*; p<0.02 vs. vector IR) (Fig. 3C).
Co-overexpression of HA-Bcl-xL with FLAG-CerS5 abrogated both the IR- and CerS5 overexpression-induced effects on apoptosis, reducing caspase activity in cell lysates to almost the unirradiated empty vector-transfected cell background level. (**; p<0.001 vs. vector IR) (Fig. 3C). These data indicate that both these IR- and CerS5-induced cellular events occur via the mitochondrial apoptotic pathway in HeLa cells. Overexpression of HA-Bcl-xL and FLAG-CerS5 were confirmed in cell lysates by western blot using anti-HA and anti-FLAG antibodies, respectively (Fig. 3C).
We hypothesized that isoform-specific knockdown of CerS would differentially affect IR-induced apoptosis in HeLa cells, i.e. CerS2 knockdown might enhance apoptosis, while CerS5 and CerS6 knockdown might decrease apoptosis compared to normal irradiated HeLa cells. For these studies, we attempted knockdown of CerS2, 5, and 6 via either transfecting cells with CerS isoform-specific antisense oligonucleotides or with vectors producing CerS isoform-specific shRNA. Unfortunately, knocking down de novo ceramide synthesis, even partially, for the length of time required by our apoptosis studies (>60 h) was cytotoxic and apoptosis could not be accurately quantified in CerS knockdown cells by bis-benzimide staining, caspase activity, or Annexin-V staining assays (data not shown).
Our unpublished findings confirm recent reports that loss of CerS2 triggers drastic changes in sphingolipid metabolism and cellular homeostasis, namely a counterregulatory upregulation of C16:0-ceramide synthesis occurred via a CerS-independent mechanism [34-36]. Based on multiple isoform-specific features, Laviad et al. described CerS2 as having likeness to a “housekeeping” gene, synthesizing essential lipids for resting cell function [31]. Additionally, decreasing cellular C16:0-ceramide via siRNA-mediated knockdown of CerS6 in human head and neck squamous cell carcinoma cells triggered the ER stress response, resulting in cell death [42]. Collectively, these studies imply that cell-wide loss of CerS isoforms, and corresponding ceramides, disrupts the balance between C16:0-, and very long chain C24:1- and C24:0-ceramide levels necessary for normal cell function.
3.4. CerS2 is responsible for all IR-induced lignoceroyl-CoA-dependent CerS activity in HeLa cells
CerS2 appears to be the predominant CerS isoform in HeLa cells (Fig. 2B) generating C24:0-ceramide [25], one of the mitochondrial ceramide species upregulated post IR (Fig. 2A). We recently identified the MAM as the site of IR-induced CerS activation (Lee and Kolesnick, submitted), and studied here whether CerS2 resides in this subcellular compartment. We purified fractions enriched in endoplasmic reticulum (ER), mitochondria (MT), and MAM from HeLa cell homogenate and detected CerS2 by direct western blot with anti-CerS2 antibody. CerS2 resides predominantly in the ER-enriched fraction, but is also present at lower levels the MAM-enriched fraction (Fig. 4A). Michaelis-Menten kinetics analyses on homogenate prepared from empty vector-transfected HeLa cells 32 h after exposure to 15 Gy revealed a significant 2.1-fold increase in lignoceroyl-CoA-dependent Vmax compared to unirradiated cells (p<0.05 vs. vector), with no significant change in Km towards CoA (Fig. 4B). Transfection with a CerS2-specific shRNA-producing vector decreased baseline Vmax activity by 62% and obliterated IR-induced CerS activity, indicating that CerS2 is required for all IR-induced lignoceroyl-CoA-dependent activity in HeLa cells. The ability of this shRNA-producing vector to knock down CerS2 mRNA levels in a homolog-specific manner was confirmed by real-time qPCR (Supplementary Table 1). These studies were performed at a time prior to substantial shRNA-induced cytotoxicity, as described in section 3.3.
Fig. 4. CerS2 confers all ionizing radiation-induced lignoceroyl-CoA-dependent CerS activity.
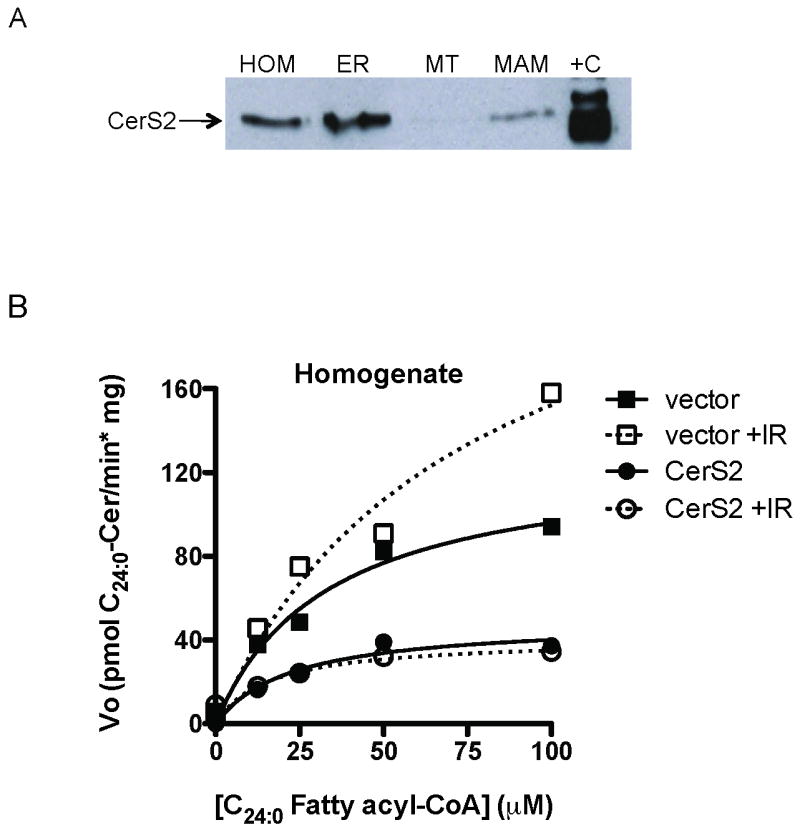
(A) Proteins from subcellular fractions purified from HeLa cells were analyzed by SDS-PAGE/western blot for the presence of CerS2. (B) Lignoceroyl-CoA-dependent CerS activity was measured in HeLa cell homogenate prepared from cells transfected with a CerS2-specific shRNA vector 32 h after exposure to 15 Gy. Data represent 1 of 2 similar experiments each. For each experiment, CerS isoform-specific mRNA down-regulation was confirmed by real-time qPCR.
3.5. CerS5 and CerS6 are both required for IR-induced palmitoyl-CoA-dependent CerS activity in HeLa cells
Mass spectrometry data showing a 47% increase in HeLa cell mitochondrial C16:0-ceramide after exposure to 10 Gy (Fig. 2A) lead us to hypothesize that CerS5 and CerS6 reside, at least in part, in the MAM compartment of HeLa cells. We confirmed the presence of endogenous CerS6 by quantitative IP western blotting in both ER- and MAM-enriched fractions (Fig. 5A). Unfortunately, we were not able to localize CerS5 in a similar manner, as we rigorously screened all CerS antibodies for isoform cross reactivity and determined that all commercially-available anti-CerS5 antibodies cross react with CerS6 (unpublished data).
Fig. 5. CerS5 and 6 are both required for ionizing radiation-induced palmitoyl-CoA-dependent CerS activity.
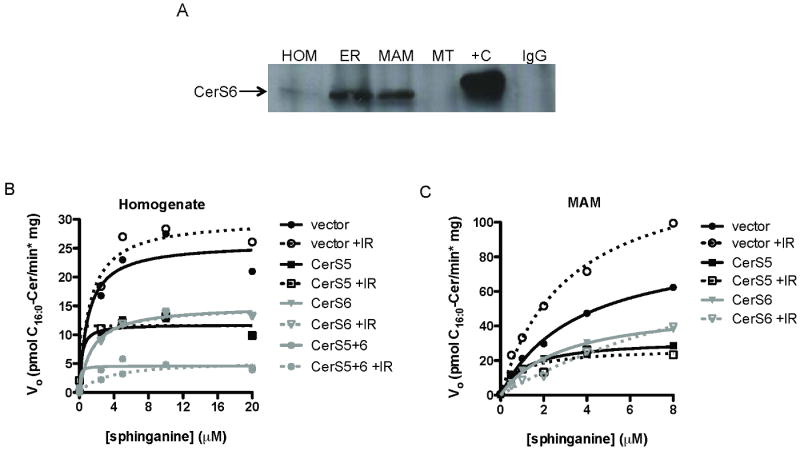
(A) Proteins from HeLa cell subcellular fractions were analyzed by quantitative IP western blotting for presence of CerS6, as detailed in Materials and Methods. (B) Kinetic analysis of palmitoyl-CoA-dependent CerS activity measured in homogenates prepared from unirradiated and irradiated (20 Gy) HeLa cells transfected with shRNA to either CerS5 or CerS6, or both. (C) Kinetics of palmitoyl-CoA-dependent CerS activity measured in MAM-enriched fractions purified from unirradiated and irradiated (10 Gy) HeLa cells transfected with shRNA to either CerS5 or CerS6. Data represent 1 of at least 2 similar experiments. For each experiment, CerS gene-specific mRNA down-regulation was confirmed by real-time qPCR.
Palmitoyl-CoA is the preferred fatty acyl-CoA substrate for both CerS5 and CerS6, and therefore the two isoforms cannot be distinguished using a palmitoyl-CoA-dependent activity assay. To identify which C16:0-ceramide-producing CerS isoform(s) is IR-inducible in HeLa cells, we knocked down either or both CerS5 and CerS6 by transfecting cells with isoform-specific, shRNA-producing vectors. Michaelis-Menten kinetics of palmitoyl-CoA-dependent CerS activities were performed on homogenates prepared from both unirradiated and irradiated (20 Gy) HeLa cells (Fig. 5B). Here, IR increased palmitoyl-CoA Vmax activity by ∼20% in empty vector-transfected cells (Km towards sphinganine ∼1 μM at 0 and 20 Gy). Knocking down CerS5 and 6 individually decreased resting Vmax by ∼50%, and simultaneous knockdown of both CerS5 and 6 decreased resting palmitoyl-CoA-dependent CerS activity by >80% compared to unirradiated empty vector-transfected cells, with no significant changes to Km value towards sphinganine. Furthermore, knocking down CerS5 and CerS6, either individually or concomitantly, obliterated all IR-induced palmitoyl-CoA activity (Fig. 5B).
We performed Michaelis-Menten kinetics analyses on the MAM fraction purified from these cells to determine the effects of CerS5 and 6 knockdown in this site of enriched IR-induced CerS activity. Palmitoyl-CoA-dependent CerS Vmax activity increased significantly by 1.51-fold in the MAM-enriched fraction prepared from irradiated (10 Gy) cells compared to unirradiated vector-transfected HeLa cells (p<0.001 vs. vector), with no significant change in Km towards sphinganine (Fig. 5C). Additionally, we observed no IR-induced palmitoyl-CoA-dependent CerS activity in the ER-, and little IR-induced activity in the MT-enriched fractions of these cells (data not shown), consistent with the notion that CerS exist in multiple subcellular locations where they are activated by distinct stimuli to elicit cellular responses [39, 40]. MAM purified from CerS5 or CerS6 knockdown cells, like the homogenate, exhibited decreased baseline activity (∼50% Vmax each) and no IR-induced activity (Fig. 5C). Furthermore, simultaneous knockdown of both CerS5 and 6 abrogated almost all baseline CerS activity in the MAM-enriched fraction (data not shown). Together, Figs. 5B and 5C suggest that both CerS5 and CerS6 are requisite for IR-induced palmitoyl-CoA-dependent CerS activity.
3.6. CerS2, CerS5, and CerS6 form heterocomplexes in HeLa cells
Evidence of CerS interplay with regulatory proteins has provided insight into the biologic role(s) of mammalian CerS isoforms [11, 31, 54]. Based on results in Fig. 5C, we hypothesized that the CerS proteins might interact with one another, at least in the context of IR-induced activation. Thirty-two hours after exposure to 0 or 10 Gy, we immunoprecipitated FLAG-CerS2 from HeLa cell lysate prepared from cells co-overexpressing myc-CerS6 and by SDS-PAGE/western blotting determined that CerS2 co-immunoprecipitated with CerS6, regardless of IR exposure (Fig. 6A). Similar experiments were performed with lysate prepared from unirradiated and irradiated cells co-overexpressing FLAG-CerS5 and myc-CerS6 (Fig. 6B) and FLAG-CerS5 and HA-CerS2 (Fig. 6C). CerS5 was shown to co-IP with CerS6 (Fig. 6B) and CerS2 (Fig. 6C) in both unirradiated and irradiated cells. Binding of each CerS combination was confirmed with the reverse IP/western blot antibody combinations, and correct overexpression of tagged CerS proteins was confirmed in all lysates by direct western blot (data not shown). These data suggest that physical interplay might exist among the CerS proteins.
Fig. 6. CerS2, 5, and 6 form heterocomplexes.
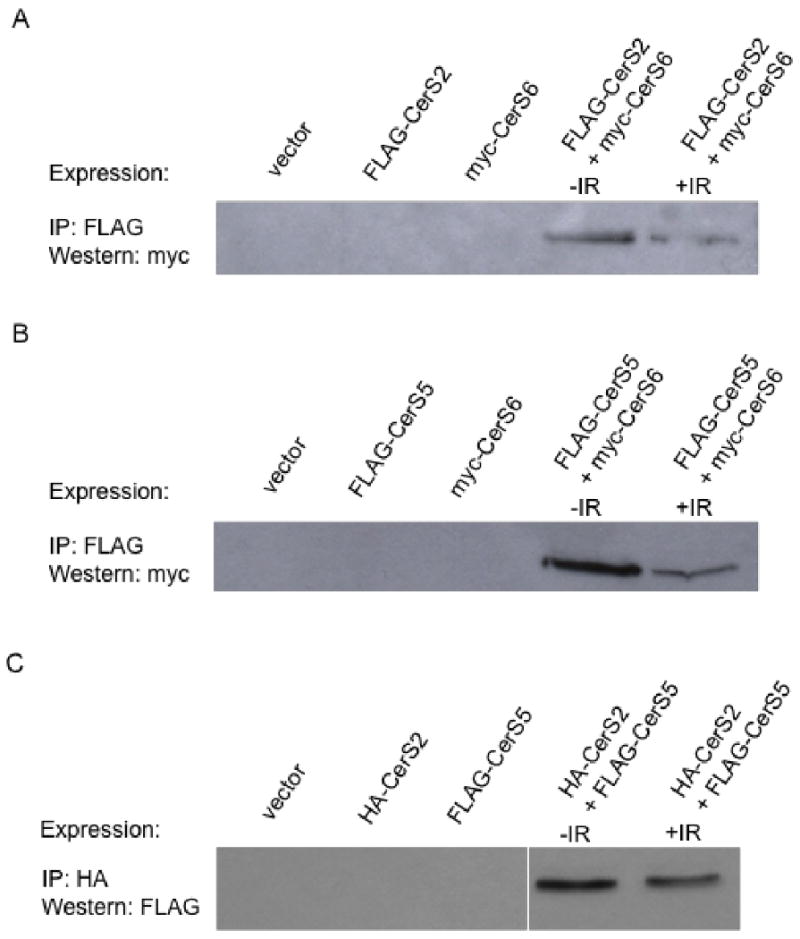
(A) Lysate was prepared from HeLa cells overexpressing FLAG-CerS2 and myc-CerS6 32 h after exposure to 0 or 10 Gy. FLAG-CerS2 was immunoprecipitated from lysate with anti-FLAG conjugated agarose beads and co-immunoprecipitation of myc-CerS6 determined by SDS-PAGE/western blotting with anti-myc monoclonal antibody. Similar experiments were performed with unirradiated and irradiated cells overexpressing (B) FLAG-CerS5 and myc-CerS6 and (C) HA-CerS2 and FLAG-CerS5, where FLAG-CerS5 and HA-CerS2 were immunoprecipitated using anti-FLAG and anti-HA conjugated agarose beads, respectively. Co-immunoprecipitation of myc-CerS6 and FLAG-CerS5 were determined with (B) anti-myc and (C) anti-FLAG western blots, respectively. Western blots represent 1 of 2 similar experiments, and binding was confirmed with reverse IP/western blot antibody combinations. Overexpression of tagged CerS proteins was confirmed in all lysates by direct western blot.
We do not know the biologic purpose of these CerS complexes, but hypothesize that their interaction plays a regulatory role in IR-induced activation. The requirement for CerS6 in IR-induced palmitoyl-CoA-dependent activity (Figs. 5B and 5C), despite the fact that overexpression did not increase IR-induced apoptosis as did CerS5 overexpression, suggests this physical interaction with CerS5 is crucial for stress-induced activation. Perhaps endogenous CerS6 may already be functioning at full capacity for induction of IR-induced apoptosis in HeLa cells, and CerS5 is limiting. In this scenario, only enhancing CerS5 expression, and not CerS6 expression, will impact IR-induced C16:0-ceramide generation, and subsequent apoptosis.
In fact, we show that all three IR-inducible HeLa CerS isoforms may coexist as a complex, raising the possibility that the combined activation of these three CerS isoforms discriminates outcome, determined by a balance between pro- and anti-apoptotic ceramide species. In this regard, C16:0-ceramide has been implicated in regulating apoptosis via both the acid sphingomyelinase- and CerS-mediated pathways, in a cell type-dependent manner [39, 47, 55, 56], in most instances promoting apoptosis, and studies show cellular homeostasis maintained by a balance between C16:0- and very-long-chain C24:1- and C24:0-ceramides [31, 34-36]. Consistent with these observations, our overexpression studies indicate that IR-induced C16:0-ceramide generation via CerS5 is pro-apoptotic (Figs. 3B and 3C), and IR-induced C24:1- and C24:0-ceramides via CerS2 are pro-survival (Fig. 3A). Perhaps these CerS2-CerS5-CerS6 complexes in MAM regulate generation of these ceramides of opposing function in response to IR, which when transferred to mitochondria serve to regulate commitment to MOMP. Further biochemical studies are required to determine the true role of the CerS heterocomplexes.
Our results provide insight into the biological differences between and interplay among the CerS isoforms. Collectively, our data suggest that upon IR stress, HeLa cells activate specific CerS isoforms to generate distinct ceramide species that confer opposing signaling outcomes, ceramides that are subsequently transferred to mitochondria to regulate apoptosis induction. The distinctive properties of the HeLa cell CerS isoforms in the context of IR-induced activation and apoptosis adds to the growing potential of manipulating CerS proteins and ceramide levels to impact the therapeutic index.
Supplementary Material
Acknowledgments
This research was funded by NIH grants CA105125 (A. Haimovitz-Friedman) and CA85704 (R. Kolesnick) and Virginia and D.K. Ludwig Fund for Cancer Research (Z. Fuks). Thanks to the University of Chicago Department of Medicine for the support to the Lipidomics facility.
The abbreviations used are
- CerS
ceramide synthase
- IR
ionizing radiation
- Bcl-2
B-cell lymphoma 2
- MOMP
mitochondrial outer membrane permeabilization
- MAM
mitochondria-associated-membrane
- ER
endoplasmic reticulum
- FB1
fumonisin B1
- shRNA
short hairpin RNA
- MT
mitochondria
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bose R, Verheij M, Haimovitz-Friedman A, Scotto K, Fuks Z, Kolesnick R. Cell. 1995;82:405–414. doi: 10.1016/0092-8674(95)90429-8. [DOI] [PubMed] [Google Scholar]
- 2.Jenkins GM, Cowart LA, Signorelli P, Pettus BJ, Chalfant CE, Hannun YA. J Biol Chem. 2002;277:42572–42578. doi: 10.1074/jbc.M207346200. [DOI] [PubMed] [Google Scholar]
- 3.Yu J, Novgorodov SA, Chudakova D, Zhu H, Bielawska A, Bielawski J, Obeid LM, Kindy MS, Gudz TI. J Biol Chem. 2007;282:25940–25949. doi: 10.1074/jbc.M701812200. [DOI] [PubMed] [Google Scholar]
- 4.Rotolo JA, Zhang J, Donepudi M, Lee H, Fuks Z, Kolesnick R. J Biol Chem. 2005;280:26425–26434. doi: 10.1074/jbc.M414569200. [DOI] [PubMed] [Google Scholar]
- 5.Truman JP, Gueven N, Lavin M, Leibel S, Kolesnick R, Fuks Z, Haimovitz-Friedman A. J Biol Chem. 2005;280:23262–23272. doi: 10.1074/jbc.M503701200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Obeid LM, Linardic CM, Karolak LA, Hannun YA. Science. 1993;259:1769–1771. doi: 10.1126/science.8456305. [DOI] [PubMed] [Google Scholar]
- 7.Dbaibo GS. Biochem Soc Trans. 1997;25:557–561. doi: 10.1042/bst0250557. [DOI] [PubMed] [Google Scholar]
- 8.Pettus BJ, Chalfant CE, Hannun YA. Biochim Biophys Acta. 2002;1585:114–125. doi: 10.1016/s1388-1981(02)00331-1. [DOI] [PubMed] [Google Scholar]
- 9.Hannun YA, Obeid LM. Nature Reviews. Molecular Cell Biology. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 10.Merrill AH, Jr, Liotta DC, Riley RT. Trends Cell Biol. 1996;6:218–223. doi: 10.1016/0962-8924(96)10021-0. [DOI] [PubMed] [Google Scholar]
- 11.Liao WC, Haimovitz-Friedman A, Persaud RS, McLoughlin M, Ehleiter D, Zhang N, Gatei M, Lavin M, Kolesnick R, Fuks Z. J Biol Chem. 1999;274:17908–17917. doi: 10.1074/jbc.274.25.17908. [DOI] [PubMed] [Google Scholar]
- 12.Deng X, Yin X, Allan R, Lu DD, Maurer CW, Haimovitz-Friedman A, Fuks Z, Shaham S, Kolesnick R. Science. 2008;322:110–115. doi: 10.1126/science.1158111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gross A, McDonnell JM, Korsmeyer SJ. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 14.Green DR, Kroemer G. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 15.Jourdain A, Martinou JC. Int J Biochem Cell Biol. 2009;41:1884–1889. doi: 10.1016/j.biocel.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 16.Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S, et al. Science. 1995;267:1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- 17.Hon H, Rucker EB, 3rd, Hennighausen L, Jacob J. J Immunol. 2004;173:4425–4432. doi: 10.4049/jimmunol.173.7.4425. [DOI] [PubMed] [Google Scholar]
- 18.Breckenridge DG, Xue D. Curr Opin Cell Biol. 2004;16:647–652. doi: 10.1016/j.ceb.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 19.Hwang D, Popat R, Bragdon C, O'Donnell KE, Sonis ST. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:321–329. doi: 10.1016/j.tripleo.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 20.Petrache I, Natarajan V, Zhen L, Medler TR, Richter AT, Cho C, Hubbard WC, Berdyshev EV, Tuder RM. Nat Med. 2005;11:491–498. doi: 10.1038/nm1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cuzzocrea S, Di Paola R, Genovese T, Mazzon E, Esposito E, Crisafulli C, Bramanti P, Salvemini D. J Pharmacol Exp Ther. 2008;327:45–57. doi: 10.1124/jpet.108.139808. [DOI] [PubMed] [Google Scholar]
- 22.Masini E, Giannini L, Nistri S, Cinci L, Mastroianni R, Xu W, Comhair SA, Li D, Cuzzocrea S, Matuschak GM, Salvemini D. J Pharmacol Exp Ther. 2008;324:548–557. doi: 10.1124/jpet.107.131565. [DOI] [PubMed] [Google Scholar]
- 23.Uehara K, Miura S, Takeuchi T, Taki T, Nakashita M, Adachi M, Inamura T, Ogawa T, Akiba Y, Suzuki H, Nagata H, Ishii H. J Pharmacol Exp Ther. 2003;305:232–239. doi: 10.1124/jpet.102.045195. [DOI] [PubMed] [Google Scholar]
- 24.Ch'ang HJ, Maj JG, Paris F, Xing HR, Zhang J, Truman JP, Cardon-Cardo C, Haimovitz-Friedman A, Kolesnick R, Fuks Z. Nat Med. 2005;11:484–490. doi: 10.1038/nm1237. [DOI] [PubMed] [Google Scholar]
- 25.Lahiri S, Lee H, Mesicek J, Fuks Z, Haimovitz-Friedman A, Kolesnick RN, Futerman AH. FEBS Lett. 2007;581:5289–5294. doi: 10.1016/j.febslet.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 26.Venkataraman K, Riebeling C, Bodennec J, Riezman H, Allegood JC, Sullards MC, Merrill AH, Jr, Futerman AH. J Biol Chem. 2002;277:35642–35649. doi: 10.1074/jbc.M205211200. [DOI] [PubMed] [Google Scholar]
- 27.Riebeling C, Allegood JC, Wang E, Merrill AH, Jr, Futerman AH. J Biol Chem. 2003;278:43452–43459. doi: 10.1074/jbc.M307104200. [DOI] [PubMed] [Google Scholar]
- 28.Mizutani Y, Kihara A, Igarashi Y. Biochem J. 2005;390:263–271. doi: 10.1042/BJ20050291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mizutani Y, Kihara A, Igarashi Y. Biochem J. 2006;398:531–538. doi: 10.1042/BJ20060379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mizutani Y, Kihara A, Chiba H, Tojo H, Igarashi Y. J Lipid Res. 2008;49:2356–2364. doi: 10.1194/jlr.M800158-JLR200. [DOI] [PubMed] [Google Scholar]
- 31.Laviad EL, Albee L, Pankova-Kholmyansky I, Epstein S, Park H, Merrill AH, Jr, Futerman AH. J Biol Chem. 2008;283:5677–5684. doi: 10.1074/jbc.M707386200. [DOI] [PubMed] [Google Scholar]
- 32.Pettus BJ, Bielawska A, Kroesen BJ, Moeller PD, Szulc ZM, Hannun YA, Busman M. Rapid Commun Mass Spectrom. 2003;17:1203–1211. doi: 10.1002/rcm.1043. [DOI] [PubMed] [Google Scholar]
- 33.Pettus BJ, Baes M, Busman M, Hannun YA, Van Veldhoven PP. Rapid Commun Mass Spectrom. 2004;18:1569–1574. doi: 10.1002/rcm.1520. [DOI] [PubMed] [Google Scholar]
- 34.Pewzner-Jung Y, Park H, Laviad EL, Silva LC, Lahiri S, Stiban J, Erez-Roman R, Brugger B, Sachsenheimer T, Wieland F, Prieto M, Merrill AH, Jr, Futerman AH. J Biol Chem. 2010;285:10902–10910. doi: 10.1074/jbc.M109.077594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spassieva SD, Mullen TD, Townsend DM, Obeid LM. Biochem J. 2009;424:273–283. doi: 10.1042/BJ20090699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pewzner-Jung Y, Brenner O, Braun S, Laviad EL, Ben-Dor S, Feldmesser E, Horn-Saban S, Amann-Zalcenstein D, Raanan C, Berkutzki T, Erez-Roman R, Ben-David O, Levy M, Holzman D, Park H, Nyska A, Merrill AH, Jr, Futerman AH. J Biol Chem. 2010;285:10911–10923. doi: 10.1074/jbc.M109.077610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mandon EC, Ehses I, Rother J, van Echten G, Sandhoff K. J Biol Chem. 1992;267:11144–11148. [PubMed] [Google Scholar]
- 38.Hirschberg K, Rodger J, Futerman AH. Biochem J. 1993;290(Pt 3):751–757. doi: 10.1042/bj2900751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.White-Gilbertson S, Mullen T, Senkal C, Lu P, Ogretmen B, Obeid L, Voelkel-Johnson C. Oncogene. 2009;28:1132–1141. doi: 10.1038/onc.2008.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bionda C, Portoukalian J, Schmitt D, Rodriguez-Lafrasse C, Ardail D. Biochem J. 2004;382:527–533. doi: 10.1042/BJ20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koybasi S, Senkal CE, Sundararaj K, Spassieva S, Bielawski J, Osta W, Day TA, Jiang JC, Jazwinski SM, Hannun YA, Obeid LM, Ogretmen B. J Biol Chem. 2004;279:44311–44319. doi: 10.1074/jbc.M406920200. [DOI] [PubMed] [Google Scholar]
- 42.Senkal CE, Ponnusamy S, Bielawski J, Hannun YA, Ogretmen B. FASEB J. 2010;24:296–308. doi: 10.1096/fj.09-135087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hayashi T, Rizzuto R, Hajnoczky G, Su TP. Trends Cell Biol. 2009;19:81–88. doi: 10.1016/j.tcb.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stiban J, Caputo L, Colombini M. J Lipid Res. 2008;49:625–634. doi: 10.1194/jlr.M700480-JLR200. [DOI] [PubMed] [Google Scholar]
- 45.Vance JE. J Biol Chem. 1990;265:7248–7256. [PubMed] [Google Scholar]
- 46.Bose R, Kolesnick R. Methods Enzymol. 2000;322:373–378. doi: 10.1016/s0076-6879(00)22034-x. [DOI] [PubMed] [Google Scholar]
- 47.Cremesti A, Paris F, Grassme H, Holler N, Tschopp J, Fuks Z, Gulbins E, Kolesnick R. J Biol Chem. 2001;276:23954–23961. doi: 10.1074/jbc.M101866200. [DOI] [PubMed] [Google Scholar]
- 48.Gao Z, Shao Y, Jiang X. J Biol Chem. 2005;280:38271–38275. doi: 10.1074/jbc.M506488200. [DOI] [PubMed] [Google Scholar]
- 49.Shao Y, Gao Z, Marks PA, Jiang X. Proc Natl Acad Sci U S A. 2004;101:18030–18035. doi: 10.1073/pnas.0408345102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pan W, da Graca LS, Shao Y, Yin Q, Wu H, Jiang X. J Biol Chem. 2009;284:6946–6954. doi: 10.1074/jbc.M805801200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Berdyshev EV, Gorshkova IA, Usatyuk P, Zhao Y, Saatian B, Hubbard W, Natarajan V. Cell Signal. 2006;18:1779–1792. doi: 10.1016/j.cellsig.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 52.Berdyshev EV, Gorshkova I, Skobeleva A, Bittman R, Lu X, Dudek SM, Mirzapoiazova T, Garcia JG, Natarajan V. J Biol Chem. 2009;284:5467–5477. doi: 10.1074/jbc.M805186200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garcia-Barros M, Paris F, Cordon-Cardo C, Lyden D, Rafii S, Haimovitz-Friedman A, Fuks Z, Kolesnick R. Science. 2003;300:1155–1159. doi: 10.1126/science.1082504. [DOI] [PubMed] [Google Scholar]
- 54.Vallee B, Riezman H. EMBO J. 2005;24:730–741. doi: 10.1038/sj.emboj.7600562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taha TA, Mullen TD, Obeid LM. Biochim Biophys Acta. 2006;1758:2027–2036. doi: 10.1016/j.bbamem.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Renert AF, Leprince P, Dieu M, Renaut J, Raes M, Bours V, Chapelle JP, Piette J, Merville MP, Fillet M. J Proteome Res. 2009;8:4810–4822. doi: 10.1021/pr9005316. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.