

Abstract
Traumatic brain injury (TBI) is a cause of death and disability and can lead to tauopathy-related dementia at an early age. Pathologically, TBI results in axonal injury that is coupled to tau hyperphosphorylation, leading to microtubule instability and tau-mediated neurodegeneration. This suggests that the forms of this protein might serve as neuroinjury-related biomarkers for diagnosis of injury severity and prognosis of the neurological damage prior to clinical expression. We initially determined whether we could detect tau in body fluids using a highly sensitive assay. We used a novel immunoassay, enhanced immunoassay using multi-arrayed fiberoptics (EIMAF) either alone or in combination with rolling circle amplification (a-EIMAF) for the detection of total (T) and phosphorylated (P) tau proteins from brains and biofluids (blood, CSF) of rodents following controlled cortical impact (CCI) and human patients post severe TBI (sTBI). This assay technology for tau is the most sensitive to date with a detection limit of approximately 100 ag/mL for either T-tau and P-tau. In the rodent models, T-tau and P-tau levels in brain and blood increased following CCI during the acute phase and remained high during the chronic phase (30 d). In human CSF samples, T-tau and P-tau increased during the sampling period (5–6 d). T-tau and P-tau in human serum rose during the acute phase and decreased during the chronic stage but was still detectable beyond six months post sTBI. Thus, EIMAF has the potential for assessing both the severity of the proximal injury and the prognosis using easily accessible samples.
Key words: : brain and biofluids, EIMAF, rolling circle amplification, severe traumatic brain injury, total and phosphorylated tau
Introduction
Traumatic brain injury (TBI) is one of the leading causes of death and disability among all traumas, and an increasing body of literature implicates TBI as an independent risk factor for developing Alzheimer's disease (AD).1–9 The incidence of TBI in the United States is comparable to that of stroke but affects younger people, thus resulting in a greater healthcare burden.10
TBI covers a wide range of injuries, from mild to moderate and severe. Factors that influence the neuropathology—such as the number of repeated impacts, types and extent of injury, and regions of the brain where the trauma occurs—have not yet been clearly elucidated. Although most cases of TBI are mild or moderate, most of the TBI animal model systems and studies have focused on severe TBI (sTBI). Even so, reliable predictors of sTBI outcome, particularly during the early stages following neurotrauma, have not been established and are being sought. This emphasizes the need to identify and characterize reliable neurological and biochemical TBI biomarkers for diagnosis and prognosis.
Currently, a TBI patient is evaluated only by clinical assessment and neuroimaging, which have their own limitations in predicting the functional impairments associated with the chronic conditions that accompany a significant TBI. Historically, classification of TBI severity has been based on a Glasgow Coma Scale (GCS) score. However, this widely-used clinical neurological score may be influenced by unrelated factors, such a patient's consumption of drugs or alcohol, prescribed medications, and other extra-cerebral injuries. Therefore, establishing a complementary approach of patient evaluation using neurological assessment in combination with biochemical biomarkers will reliably and objectively determine the severity of a TBI, which can then guide treatment regimens.11–20
Tau is a microtubule-associated protein localized mainly in neuronal cells and functions as a major structural element in the axonal cytoskeleton. Total tau (T-tau) is abundant in the central nervous system (CNS), and in particular, in unmyelinated axons and cortical interneurons.21,22 Under normal circumstances, the phosphorylation of tau (P-tau) is responsible for regulating its biological activity. However, excessive tau phosphorylation (i.e., hyperphosphorylation) is associated with several neurodegenerative diseases and are referred to as tauopathies.23–25 For example, one of the hallmarks of AD is the presence of neurofibrillary tangles (NFTs) that are composed of P-tau that forms paired helical filaments (PHFs), and also includes increased T-tau and P-tau in the cerebrospinal fluid (CSF).22,26,27 Pathological phosphorylation of tau has been found at a number of sites including Thr-181, Ser-198, Ser-199, Ser-202, Thr-205, Thr-231, Ser-356, Ser-404 and Ser-422, which are phosphorylated by casein kinases, cyclic adenosine monophosphate (cAMP)-dependent protein kinase, glycogen synthase kinase-3β (GSK-3β), cAMP-dependent protein kinase, cyclin-dependent kinase 5, and tau-tubulin kinases (TTBK).28–32
The analysis of P-tau is crucial in the diagnosis of AD.33 However, the significance of P-tau levels following TBI is unclear. Rodent TBI models do not produce NFTs post-injury. However, since tau levels may markedly influence the pathophysiology of TBI both in the acute and chronic phases, tau may still be an informative biomarker for TBI. Increased tau levels have been reported in the CSF following TBI and also show promise as a specific serum biomarker in both human patients and experimental models.13,15,16,34–36 Although there is a rapid rise in tau protein levels in the CSF post-TBI,13,34 the peak and temporal progression of serum tau levels have not been adequately evaluated.15,16
Tau-associated neuropathology, particularly the presence of NFTs, has been reported in the brains of athletes who have played contact sports (boxers, football and ice hockey players, wrestlers) and who sustained concussions during their career. This pathological condition has been termed chronic traumatic encephalopathy (CTE).37–42 Common symptoms in CTE include memory loss, Parkinson-like movements, dementia, aggression, confusion, and depression.38,41, 43–46 Although the majority of CTE cases display widespread NFTs, in contrast to AD, amyloid-β (Aβ) pathology is less frequent.42,47
Reports on the time course of T-tau and P-tau levels following TBI are limited and include those of Gabbita and colleagues48 and Liliang and colleagues.49 We previously described the development of an assay termed SOFIA (surround optical fiber immunoassay) for the detection of the abnormal prion protein in prion diseased animals and humans.50–54 As a result of our continued efforts to develop advanced biomarker assay technologies from readily accessible samples, we have changed the term SOFIA to EIMAF (enhanced immunoassay using multi-arrayed fiberoptics).
Here, we report the use of EIMAF with and without pre-assay target amplification for T-tau and P-tau detection in both rodent and human CNS tissue and/or biofluid samples (blood, CSF) following sTBI. Based on these findings from rodent TBI models and clinical samples obtained from individuals with TBI, we believe that the EIMAF technology for determining T-tau and P-tau levels provide the degree of sensitivity needed for assessing whether measuring tau levels provides a prognostic biomarker for patient recovery and/or development of a tauopathy.
Methods
Controlled cortical impact (CCI)
Brain trauma in rats and mice was produced using an electromagnetic contusion device (Myneurolab, St. Louis, MO). Adult male (280–300 g) Sprague-Dawley rats (Harlan, Indianapolis, IN) or C57Bl/6J mice (∼30 g; Jackson Labs, Bar Harbor, ME) were anesthetized with 4% isoflurane in a carrier gas of oxygen (0.8 L/min) and maintained in 2.5% isoflurane as anesthesia in the same carrier gas. Core body temperature was monitored continuously and maintained at 37±1° C. Animals were placed onto a stereotactic apparatus (David Kopf, Tujinga, CA) in a prone position and secured by ear and incisor bars. Following a midline cranial incision and reflection of the soft tissues, a unilateral craniotomy (ipsilateral to site of impact; 4 mm and 7 mm diameter for mice and rats, respectively) was performed adjacent to the central suture, midway between bregma and lambda. The dura mater was kept intact over the cortex.
Brain trauma was produced by impacting the right (ipsilateral) cortex with an aluminum impactor tip (housed in a pneumatic cylinder; 3.5 mm and 5 mm diameter for mice and rats, respectively) at an impact velocity of 4.5 msec with a 1.5 mm (mice) or 2.5 mm (rats) depth and 150 msec dwell time. The craniotomy was covered with a plastic plate that was cemented (Grip Cement, Dentsply, York, PA) to the skull. Sham-injured control animals were subjected to the same surgical procedures but did not receive the impact injury.
Animals were monitored and recovery from anesthesia was confirmed when they regained their ability to right themselves and ambulate. Appropriate pre- and post-injury management was preformed to minimize pain and discomfort and to insure compliance with guidelines set forth by the State University of New York (SUNY) Downstate Medical Center Institutional Animal Care and Use Committee (protocol #'s 08-477-10 and 13-10382) and the National Institutes of Health (NIH) guidelines detailed in the Guide for the Care and Use of Animals.
Brain tissue and blood samples were harvested from mice and rats at selected times after the CCI. At each time point, mice and rats were anesthetized with isoflurane before sample collection. Blood was collected in non-heparinized tubes from tail veins or by cardiac exsanguination. Following centrifugation, serum was obtained, stored at −80o C, and diluted (1:20) prior to use. Brains were removed immediately following cervical dislocation and stored at −80o C. In addition to these samples, frozen brains and blood from 8-month-old JNPL3 (P301L) and T-tau knockout (TauKO) mice were generously supplied by Dr. Karen Duff (Columbia University Medical Center, New York, NY).
Human samples
Samples were derived from six patients with blunt trauma to the head and with a GCS score <8 enrolled in a sTBI study where CSF was collected from adult subjects presenting to the emergency department of Ben Taub General Hospital, Baylor College of Medicine, Houston, Texas. The study protocol was approved by the Baylor College of Medicine (IRB # H-13606). CSF was collected until a ventriculostomy catheter was no longer clinically indicated. CSF samples (10 mL), with a collection time not exceeding 1 h, were diverted to 15 mL conical polypropylene centrifuge tubes (BD Falcon) and centrifuged at 4,000×g at room temperature for 5 min to remove loose cells and debris. One mL aliquots of the cleared supernatants were pipetted into cryogenic tubes, snap-frozen and stored at −80°C.
Control CSF samples were purchased from Bioreclamation, Inc. Archived TBI CSF samples also were assessed. Archived serum samples from sTBI subjects and non-TBI controls were collected at the University of Pittsburgh Medical Center (IRB #'s PRO08020342, IRB0308021).
Enrolled subjects in this cohort also sustained blunt trauma to the head and had an admission GCS score <8. Initial blood samples were collected during the acute stage (<3 d post-injury) and also at approximately one month, three months, and six months post-TBI. Non-TBI control sera were collected only once per subject using similar procedures. After collection, blood samples were allowed to coagulate for 30-60 min at room temperature before centrifugation at 2,500×g for 10 min. Supernatants (serum) were collected, aliquoted, snap-frozen and stored at −80°C. Prior to analysis, serum and CSF were diluted 1:20 and 1:100, respectively.
Human CSF and serum samples were transferred to SUNY Downstate for use in this study with NIH clinical exemption 4 (IRB # 00003624) from Federal regulations.
Preparation of tissue extracts
Soluble tissue extracts were generated by homogenization of rodent brain tissue in 1x lysis buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EGTA, 5 mM EDTA, 1% Triton X-100, 1 mM Na vanadate, 1 mM dithiothreitol, and 100×Halt protease inhibitor (Fisher Scientific) on ice. Extracts were transferred to 1.5 mL microcentrifuge tubes, microfuged at 10,000×g for 15 min at 4° C and the supernatants (soluble proteins) were collected and stored at −80° C until analysis. Protein concentrations were determined using a micro BCA protein assay kit (Fisher Scientific, Morristown, NJ).
EIMAF and a-EIMAF
The anti-tau monoclonal antibodies (Mabs) used were previously described55 and epitopes are indicated in italics below. For EIMAF coupled to rolling circle amplification (a-EIMAF), high-binding 96-well microtiter plates (Costar) were coated with capture Mab at 6 μg/mL final concentration (Mab DA31 [aa150-190] for T-tau and Mabs CP13 [pSer-202] rodents samples or RZ3 [pThr-231] human samples for P-tau). Following an overnight incubation at 40o C, unoccupied binding sites were blocked for 1 h with casein. A 100 μl aliquot of diluted brain or blood (serum at 1:20 dilution is used to avoid matrix effects) sample was added, incubated and followed by the addition of a biotinylated detection Mab DA9 (aa102-140) (100 μL at 4 μg/mL final Mab concentration).
Five 10-min washes with phosphate-buffered saline containing 0.2% Tween-20 (PBST) were followed with the addition of 100 μL of streptavidin (5 μg/mL) per well and incubation for 1 h at 37o C. A biotinylated prostate-specific DNA primer (5′-TTTTTTTGTCCGTGCTAGAAGGAAA-CAGTTAC-3′) (100 μl at 4 μg/mL) was added for 1 h at 37o C. Following the addition of a T4-DNA ligase-pretreated prostate DNA template (1 mg/ mL), RCA was initiated by adding 100 μl of reaction mixture consisting of: φ29 DNA polymerase reaction buffer, bovine serum albumin, nucleotide triphosphates supplemented with dUTP-Texas Red, and φ29 DNA polymerase.56 Incubation for several hours is followed by PBST washes, addition of 1N NaOH, neutralization with 1 M Tris-HCl, pH 7.5, heat treatment (100o C for 15 min) and fluorescence analysis using surround optical fluorescence detection.
For direct, non-amplified detection and relative quantitation of tau, EIMAF was performed as detailed previously and briefly described here.50 For direct EIMAF, tissue homogenates or biofluids were added to the capture Ab followed by the biotinylated detection Mab DA9. Following a 1 h incubation, streptavidin conjugated to Rhodamine Red X (1:1000; Invitrogen, Norwalk, CT) was added and incubated for 1 h. The wells were washed with TBS containing Tween-20, treated with NaOH and neutralized. A 90 μL sample was drawn up into a 100 μL Microcap (Drummond Scientific, Broomall, PA) micro-capillary tube which was then inserted into a specifically designed tube sample holder for laser excitation and emission quantitation. Each EIMAF and a-EIMAF sample was tested in triplicate and, depending on available sample volumes, duplicated in independent experiments. Although we have found that a-EIMAF is not required for all serum samples, it is required for many of them. Therefore, our standard protocol is to assay all non-CNS samples directly by a-EIMAF.
Immunoblotting and ELISA
For capture enzyme-linked immunosorbent assay (ELISA), 96-well microtiter plates were coated with 100 μL of purified capture antibody (Mab DA31 for T-tau, Mab CP13 for rodent P-tau, Mab RZ3 for human P-tau) at a concentration of 5 μg/mL in PBS. After overnight incubation at 4° C the wells were washed twice with PBST and blocked by adding 200 μL 1% non-fat dry milk in PBS, pH 7.4. A 100-μL aliquot of PBS-diluted antigen was incubated at 37o C for 1 h followed by four washes with PBST. Biotinylated DA9 (100 μL at 1 μg/mL) was added and incubated for 1 h at room temperature (RT). Wells were washed four times with PBST followed by the addition of 100 μL of streptavidin conjugated to alkaline phosphatase and incubation at RT for 60 min. This was followed by the addition of 100 μL para-Nitrophenylphosphate substrate, incubation at 37o C for 60 min and optical density readings at 405 nm.
For immunoblotting analysis, soluble protein fractions (25 μg per lane) from the brain tissue extracts were separated using sodium dodecyl sulfate polyacrylamide gel electrophoresis (10% resolving gels). Equivalent sample volumes were loaded on each lane. Proteins were electrotransferred to nitrocellulose membranes and the blots were blocked using 5% non-fat dry milk in PBST for 30 min at RT. Blots were incubated with primary Ab (1:1000 dilution of Mab DA9 conditioned media for T-tau and Mab CP13 for P-tau) for 2 h at 37o C, washed three times in PBST (15 min each) followed by goat anti-mouse immunoglobulin G (IgG; Fab specific) conjugated with alkaline phosphatase (1:2000; Sigma-Aldrich Corporation, St. Louis, MO) in 1% non-fat dry milk - PBST for 2 h. Following three PBST washes (15 min each), the blots were developed in substrate buffer (100 mM Tris-HCl, pH 9.5, 100 mM NaCl, and 5 mM MgCl2) containing nitro blue tetrazolium (0.33 mg/mL)/5-bromo-4-chloro-3-indolyl-phosphate (0.165 mg/mL). All data shown are representative of three separate experiments. Quantification of tau proteins was performed by densitometric analysis using NIH Image J software. All data shown are representative of multiple independent experiments.
Results
The sensitivity of EIMAF for the detection of T-tau was determined by performing assays on serial dilutions of recombinant human Tau-441 (rTau; EMD Millipore, Billerica, MA; Fig. 1). Measuring rTau without pre-assay signal amplification by RCA, we reliably and repeatedly detected rTau down to <100 fg/mL. However, using RCA to increase the target signal detected by EIMAF (i.e., a-EIMAF) assay sensitivity was increased by approximately 3 logs to <100 ag/mL. The data presented in this figure also was used as a standard curve for quantifying T-tau levels in the experimental samples being assayed to calibrate the voltage readings expressed by EIMAF and a-EIMAF. To our knowledge, this is the most sensitive assay for T-tau detection to date.
FIG. 1.
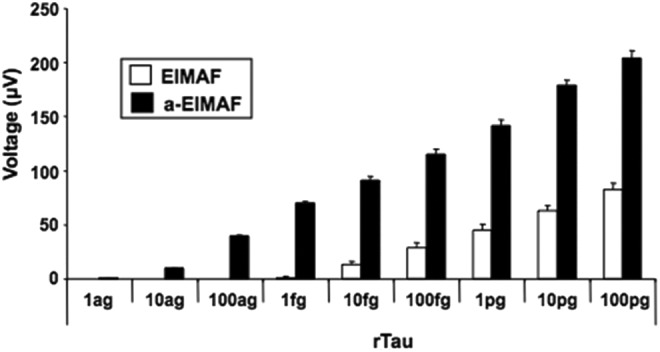
Sensitivity limits of enhanced immunoassay using multi-arrayed fiberoptics alone (EIMAF) and EIMAF coupled to rolling circle amplification (a-EIMAF) for rTau.
Similarly, for studies on EIMAF and a-EIMAF, sensitivity and dynamic range were determined for P-tau using, as a surrogate target, rTau phosphorylated by either TTBK-1 (Fig. 2A) or GSK-3β (Fig. 2B; SignalChem, Richmond, British Columbia). TTBK-1, a serine/threonine/tyrosine kinase belonging to the casein kinase 1 superfamily, phosphorylates at AD-related sites (Ser-198, Ser-199, Ser-202 and Ser-422) and also is associated with tau aggregation.30 GSK-3β is a physiological serine/threonine kinase for tau that targets numerous tau phosphorylation sites identified in NFT and other tau-positive inclusions. Phosphorylation of tau at Thr-231 by GSK-3β plays a critical role in regulating the ability of tau to bind to and stabilize microtubules.
FIG. 2.
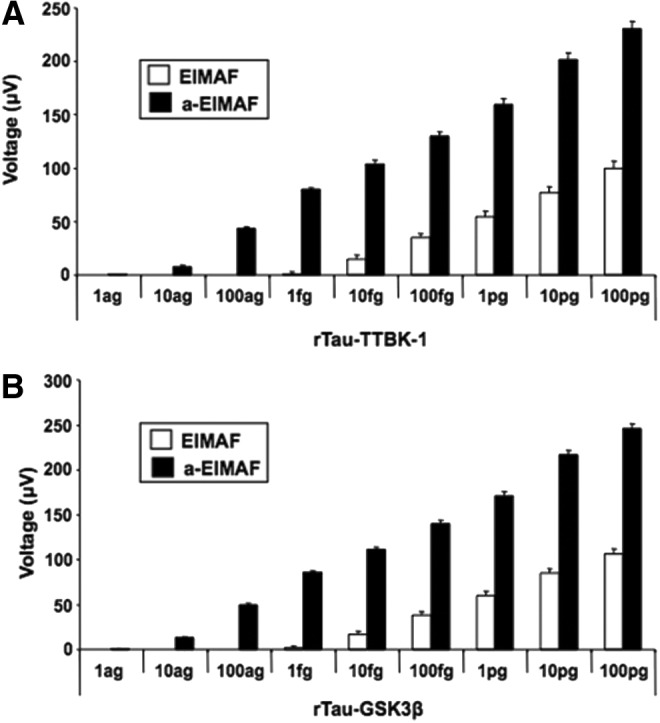
Sensitivity limits of enhanced immunoassay using multi-arrayed fiberoptics alone (EIMAF) and EIMAF coupled to rolling circle amplification (a-EIMAF) for P-tau using rTau-TTBK-1 with Mab CP13 (A) and rTau-GSK3β with Mab RZ3 (B).
In addition, Rankin and colleagues57 reported that the major sites phosphorylated by GSK-3β include Thr-205, Ser-214, Thr-231, Ser-262, Ser-356, Ser-400, Ser-404, and Ser-409. Our results support these findings in that we found CP13 (pSer-202) had greater reactivity for rTau-TTBK-1 than for rTau-GSK-3β, and, conversely, RZ3 (pThr-231) had greater reactivity for rTau-GSK-3β than for rTau-TTBK-1 (data not shown). Accordingly, we focused our efforts only on the more highly reactive combinations. The sensitivity and dynamic range of EIMAF and a-EIMAF for P-tau detection with CP13 (using rTau-TTBK-1; Fig. 2A) and RZ3 (using rTau-GSK-3β; Fig. 2B) are similar to the results obtained for T-tau detection using rTau (Fig. 1) and includes the approximate 3-log increase in detection with EIMAF following amplification.
We compared the sensitivity of EIMAF with sandwich ELISA (Fig. 3) using, as a source of T-tau and P-tau, serial 10-fold dilutions (10−4 - 10−12) of brain lysates from 8 month old JNPL3 (P301L) mice which exhibit extensive tauopathy and TauKO mice as negative controls.58,59 As anticipated, T-tau and P-tau were not detected in brain homogenates from TauKO mice by either sandwich ELISA or EIMAF demonstrating the high specificity of these immunoassays (Fig. 3). Both T-tau and P-tau were readily detectable by both sandwich ELISA and EIMAF. Both T-tau and P-tau were detected in the 10−4 dilution by sandwich ELISA, but not at 10−5 and 10−6 dilutions of T-tau and P-tau, respectively.
FIG. 3.

Enhanced immunoassay using multi-arrayed fiberoptics (EIMAF; ––) vs. sandwich enzyme-linked immunosorbent assay (ELISA; – –) for T-tau (●, ■) and P-tau (○, □) detection sensitivity. Detection of T-tau and P-tau was performed using serial dilutions of JNPL3 (P301L; ●, ○) and T-tau knockout (TauKO; ■, □) mouse brain homogenates. EIMAF has a fivefold to sixfold more sensitive detection limit than sandwich ELISA.
In contrast, a different profile was observed when EIMAF was used for T-tau and P-tau detection of the same brain lysates (Fig. 3). Detection of T-tau and P-tau by EIMAF in JNPL3 (P301L) brain remained constant from a 10−4 to 10−8 brain lysate dilution for P-tau and to a 10−9 brain lysate dilution for T-tau. P-tau and T-tau were not detected by EIMAF at a 10−11 and 10−12 brain lysate dilution, respectively. Taken together, these studies demonstrate that EIMAF has a sensitivity that is approximately 5–6 logs greater than sandwich ELISA with an additional ∼3 logs when a-EIMAF is used.
To further assess assay utilization, sensitivity and specificity, we compared the detection of T-tau in serum from 8 month old P301L mice and TauKO mice (data not shown). With use of a 1:20 serum dilution, T-tau from P301L mice was readily detectable by a-EIMAF but not by sandwich ELISA or EIMAF alone. As expected, diluted serum from TauKO mice gave readings similar to PBS background controls for all three assays. These studies demonstrate the utility of a-EIMAF for biomarker detection when the concentration of the analyte is too low for detection by conventional assays.
Using a-EIMAF, we monitored the levels of T-tau and P-tau in rat serum over a 30-d time period post-CCI (Fig. 4). At each time point, serum was collected from separate groups each consisting of five rats and serum samples were analyzed individually. We observed that there was a baseline level of T-tau even in naïve rat serum. The levels of T-tau began to increase gradually by Day 2 and continued until Day 3, after which it remained constant for the remainder of the time course. In contrast, P-tau levels were very low in naïve rat serum and there was a dramatic increase from baseline by Day 3 post-CCI that continued on an upward trend until Day 30.
FIG. 4.
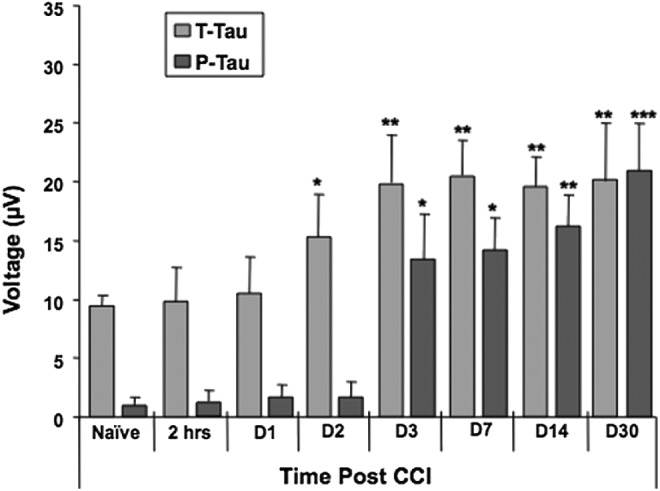
Detection of tau by enhanced immunoassay using multi-arrayed fiberoptics coupled to rolling circle amplification (a-EIMAF) in rat serum following severe traumatic brain injury. Adult male Sprague-Dawley rats were subjected to controlled cortical impact and blood was collected at various time points as indicated. The levels of T-tau and P-tau in serum was determined by a-EIMAF. Statistical analysis (t-test) was based on comparison to naïve rats. *p<0.001; **p<0.0001; ***p<0.00001.
Mice also were subjected to CCI and followed by a-EIMAF over a 7-d period during which changes in serum levels of T-tau and P-tau were determined (Fig. 5). The control (naïve, sham) and CCI groups each consisted of five mice. At each time point blood was collected, processed, and analyzed individually from each mouse prior to sacrifice. In contrast to the rat model, the T-tau levels in mouse serum increased at Day 1 post-CCI, the first time point selected, and showed continued increases at Days 3 and 7 in comparison to sham mice. Only values for Day 1 sham mice are plotted in Figure 5 since the values in sham mice at Days 1, 3, and 7 were all similar. In the CCI group, P-tau levels also followed the same profile increasing continuously from Day 1 to Day 7 (all statistically significant when compared with Day 1 sham). However, the increases in P-tau were more dramatic than T-tau over the 7-d period. Although it is currently unclear why P-tau levels showed a greater increase than T-tau in mice, this may be due to a difference in epitope accessibility with the P-tau epitope being more detectable in serum. However, this does not seem to be an issue with the rat model study described above (Fig. 5).
FIG. 5.
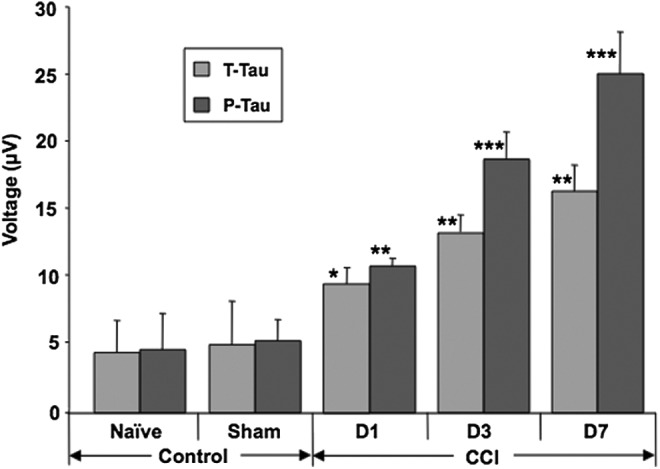
Detection of tau by enhanced immunoassay using multi-arrayed fiberoptics coupled to rolling circle amplification (a-EIMAF) in mouse serum following severe traumatic brain injury. At Days 1 (D1), 3 (D3), and 7 (D7) post–controlled cortical impact (CCI) or sham treatment, blood was collected and the serum was assayed for T-tau and P-tau by a-EIMAF. The levels of T-tau and P-tau in the sham-treated mice at D1 (shown), D3, and D7 did not change significantly. Statistical analysis (t-test) was based on comparison to sham-treated mice. *p<0.01; **p<0.001; ***p<0.00001.
To try to address this issue found in serum, Western blotting for T-tau and P-tau was performed on the mouse brain lysates (Fig. 6). Since Western blotting was performed on denatured proteins, the influence of tau conformation is eliminated. The immunoblotting data demonstrated that following CCI, increases in T-tau levels continued from Day 1 to Day 7 while increases in P-tau followed the T-tau increase and were observed at Days 3 and 7. However, P-tau levels did not exceed T-tau levels at any time point. Taken together, these results suggest that the discrepancy in data for the non-denatured T-tau and P-tau in murine serum described above (Fig. 5) is likely due to conformational differences resulting in altered epitope availability.
FIG. 6.
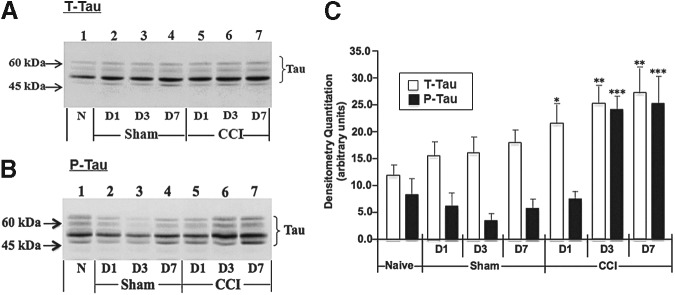
Western blotting of mouse brain lysates for tau. At Days 1, 3, and 7 post– controlled cortical impact (CCI) or sham treatment, soluble fractions (25 μg) of ipsilateral cortex from CD-1 mouse brain lysates were electrophoresed and Western blotted for T-tau with Mab DA9 (A) and P-tau with Mab CP13 (B). Western blotting of naïve (N) mouse brain lysate also was performed. Shown are representative Western blots. Densitometric quantitation (C) of the most intensely immunostained T-tau (white bars) and P-tau (black bars) bands was performed using Image J software. Statistical analysis (t-test) compares densitometry of CCI vs. sham-treatment. *p<0.01; **p<0.001; ***p<0.00001.
As a next step, the utility of EIMAF for detection of tau in human biofluids was investigated. First, timed acute phase human CSF samples collected from six sTBI patients were analyzed. T-tau and P-tau levels in diluted (1:100) CSF samples were measured using EIMAF or sandwich ELISA for comparison (Fig. 7). While T-tau and P-tau were readily detectable in the human CSF samples using EIMAF (Fig. 7A), sandwich ELISA (Fig. 7B) lacked the requisite sensitivity. Figure 7 presents the data obtained using serial CSF samples from six sTBI subjects extending from Days 0–6 after injury, in comparison with four non-TBI control CSF samples. In all six sTBI subjects, the T-tau and P-tau levels in the first available sample were higher than in the non-TBI controls (Fig. 7A). Interestingly, CSF T-tau levels either remained unchanged or rose during this time course with T-tau concentrations ranging from 13.3 to 20.2 ng/mL, exemplifying patient to patient variability. On the other hand, CSF P-tau levels for all six sTBI subjects exhibited an increase over the time course examined, from the first sample on Day 0 or 1 until the last sample collected on Days 5 or 6 (Fig. 7A), with the range of P-tau concentrations 7.1–19.8 ng/mL.
FIG. 7.

Detection of T-tau and P-tau in human cerebrospinal fluid (1:100 dilution) during the acute and chronic phases post severe brain injury using enhanced immunoassay using multi-arrayed fiberoptics coupled to rolling circle amplification (A) and enzyme-linked immunosorbent assay (B).
We next analyzed tau levels in a series of human serum samples taken from four sTBI patients (Patients 650, 671, 860, 921) during either the acute stage (≤3 d) or at three time points more remote from injury (≥21 d, approximately at one, three, and six months post-TBI; Fig. 8). In a group of non-TBI control subjects (Patients 130–133), serum T-tau levels were about threefold to fourfold higher than P-tau levels (∼144–158 fg/mL T-tau vs. ∼38–40 fg/mL P-tau). For sTBI subjects, although the actual T-tau and P-tau levels varied between the patients, the data profiles from each of the four patients produced similar findings: 1) serum levels of T-tau and P-tau during the acute phase (first serum samples Day 1–3) post-TBI rose dramatically (∼375–405 fg/mL T-tau; ∼380–410 fg/mL P-tau), compared with the levels in the non-TBI control sera; 2) P-tau levels became comparable to the T-tau levels in the samples during the acute phase of TBI; and 3) during the chronic stage (approximately one, three, and six months post-injury), the T-tau and P-tau levels declined steadily at similar but not identical rates for two of the patients (Patients 671, 860), slower for Patient 650 and slowest for Patient 921 (Fig. 8). Of note, even at the more remote stage (approximately three and six months post-injury), T-tau, and importantly P-tau, levels are still detectable in serum using a-EIMAF and remain significantly higher than the control values. Additional samples will be used to confirm these findings.
FIG. 8.

Detection of T-tau and P-tau in human serum post severe traumatic brain injury (sTBI) by enhanced immunoassay using multi-arrayed fiberoptics coupled to rolling circle amplification (a-EIMAF). Human serum samples were diluted 1:20 in phosphate-buffered saline and assayed by a-EIMAF for T-tau (Mab DA31) and P-tau (Mab RZ3) in combination with biotinylated Mab DA9 for detection. Serum levels of both T-tau and P-tau are elevated in the acute phase (range, Days 1–3), compared with control serum. In the chronic serum samples (range, Days 27–190), the majority of T-tau returned to control levels. In contrast, P-tau levels in most sTBI serum samples remained higher than control levels.
Discussion
In many instances, surviving TBI victims experience cognitive dysfunction throughout their life coupled with a diminished quality of life. The initial impact from TBI results in many cellular and biochemical changes, which exemplifies the complex pathophysiology, resulting in a disease process that increases and prolongs injury severity. The epidemiologic evidence implicates TBI as a probable risk factor for AD. This suggests that TBI can initiate mechanistic events leading to neurodegenerative changes. Axonal injury, observed in many sTBI patients, results in accumulation of Aβ peptides and NFTs, the main component of which is the hyperphosphorylated, insoluble and filamentous P-tau.60–64
Tau inclusions are composed of aggregated tau protein that is abnormally phosphorylated and/or hyperphosphorylated. Aggregated tau is resistant to dephosphorylation and the extent of tau aggregation corresponds to the degree of neuronal loss and neuron toxicity. Further hyperphosphorylated tau is resistant to proteolysis, fails to bind to microtubules and accumulates in neurons resulting in tau toxicity. Neurodegenerative disorders with tau inclusions within both glial and neuronal cell types are referred to as tauopathies.65 In addition to AD, these include frontotemporal lobar degeneration, progressive supranuclear palsy, Pick's disease, some prion diseases, amyotrophic lateral sclerosis/parkinsonism-dementia complex, CTE, and some genetic forms of Parkinson's disease.65–68 The fact that the tau inclusions are localized in the brain regions whose functions are compromised suggests that these inclusions are partially responsible for the neuropathogenesis of these disorders. This is strengthened by studies demonstrating that progression and duration of AD is correlated with NFT formation.69,70
In this report, we describe an ultrasensitive immunoassay technology (EIMAF) and its modification (a-EIMAF) that, for the first time, documents changes in T-tau and P-tau in two rodent models and human biofluid samples following sTBI. We have found that serum T-tau and P-tau levels generally increase during the acute stage of sTBI in rodent serum (from Days 2 to 30 for T-tau and from Days 3 to 7 for P-tau in rats and Days 1 through 7 in mice; Fig. 4 and Fig. 5). During the subacute/chronic state (Days 14–30), the increased levels of both T-tau and P-tau are maintained in the rat model (Fig. 4). In studies of human biofluid samples following sTBI, the elevated T-tau levels are generally sustained while elevated P-tau levels actually increase during the acute stage of sTBI (Days 0 to 6 post-injury) in human CSF (Fig. 8). Further, in human TBI, the serum samples from the acute phase of injury have the most elevated T-tau and P-tau levels (Fig. 8). However, T-tau levels appear to return to normal by about one month post-injury, while P-tau levels, though reduced, are still substantially higher than control levels even at six months post-injury (Fig. 8).
NFTs and CSF tau are commonly increased by a factor of 3 to 4 in AD.22,26,27 Following TBI, not only do the tau levels in the CSF increase but changes of tau levels in the serum suggest its use as a specific serum biomarker in humans and experimental models.35,36,49,71 Analysis of rat serum T-tau protein following TBI demonstrated that although tau levels in serum increased as a function of severity and time at 1 h and 6 h after TBI, the serum T-tau may not be suitable as a marker 24 h after injury.49 Consistent with the increase in T-tau, additional reports analyzing the biomarkers cleaved (c)-tau,48 S100β and neuron specific enolase (NSE),72,73 also reported increased levels within 6 h after TBI in rats. The levels of brain biomarkers found in serum is influenced by the integrity of the blood-brain barrier (BBB) integrity and 6 h after TBI the integrity of BBB would re-established resulting in a decrease in the serum tau levels.48 We did not observe a similar time course profile for serum T-tau and P-tau in our studies.
The evaluation of the neurological damage that results from TBI is a continuing challenge. Techniques used to assess brain trauma include neuropsychological assessments and neuroimaging, which partially lend themselves to subjective interpretation of the data. The use of biochemical methods for the detection of protein biomarkers can offer a more objective analysis of brain injury and be a valuable asset. For example, the capability of detecting biomarkers in biofluids such as blood or urine for evaluating the extent of brain damage presents a less stressful and minimally invasive procedure with reduced costs. Not only can the extent of injury be determined, but these same protein biomarkers also may be useful for monitoring the effectiveness of therapeutic interventions. In addition, the detection and analysis of protein biomarkers would complement the more subjective GCS score that may not be accurate in certain individuals, such as children. Some of the protein biomarkers that have been used are S100β, NSE, glial fibrillary acidic protein (and its breakdown products), ubiquitin carboxyl-terminal esterase L1, and C-tau.18,74–82 However, there are issues in the use of these proteins for assessing TBI, which includes sensitivity and specificity, use in adult versus pediatric patients, lack of correlation between the values in blood and CSF, and lack of correlation with the different levels of TBI severity.
In a recent study, the concentrations of plasma T-tau and serum S-100B and NSE were determined in professional ice hockey players who experienced concussions during the game.71 The goal of this study was to determine whether blood biomarkers could be used as a guide for acute diagnosis of concussion and injury recovery. Only T-tau was found to be significantly higher in the post-concussive player samples, compared with pre-season samples, in spite of the high degree of overlap in the range of T-tau values between the two sample groups. T-tau exhibited a biphasic increase following injury and had the greatest diagnostic accuracy when correlated with post-concussive symptoms over time. Further, the T-tau levels were highest during the first hour after concussion, which is similar to our results, but the actual T-tau levels reported (1–100 pg/mL) were greater than those of our study. Whether this is due to the differences in the samples analyzed, assay platform or reagents used is not clear. The levels of S-100B and NSE increased after a game in which no concussion occurred but T-tau levels were unaffected. Previous studies on serum tau levels after mild or sTBI reported a high degree of variability in the serum tau levels of patients using an sandwich ELISA platform.15,35
Epidemiological evidence, which includes the appearance of fibrillary Aβ plaques in the brain several years following a single sTBI, suggests that TBI may be an risk factor for the development of AD and may accelerate the pathophysiological processes leading to AD.3,7–9,83 Although there exists a causal connection, there are clinical and histopathological differences between AD and TBI,8 including the distribution of P-tau immunoreactive NFTs, suggesting that the neurophysiological and neuropathological mechanisms leading to the increased risk for neurodegenerative diseases following TBI are highly complex.
Tau pathology and NFTs also were observed in patients who suffered a single sTBI one to 47 years previously.84 The process of delayed NFT formation in human TBI remains to be explained. Immediately following sTBI, T-tau and P-tau were found to accumulate in both neuronal cell bodies and axons although without clear NFT pathology.85,86 In addition, NFTs were not found in TBI patients who died within 4 weeks of injury,85 suggesting that the mechanisms leading to NFT and/or CTE pathology requires a prolonged time post-injury to develop.
CTE is a clinical entity consisting of tau pathology—in particular, the accumulation of NFTs—in the brains of athletes who have been involved in contact sports (professional boxers, American football or ice hockey players, wrestlers) and who sustained several concussions during their career.37–42 Symptoms of CTE include memory loss, Parkinson-like movements, and dementia.38,41,43–45 In CTE, the vast majority of cases display extensive NFTs while Aβ pathology is much less frequently observed.42
Although repeated concussions/mild TBI should be regarded very seriously, the number of individuals examined is still low and the incidence of CTE, its risk factors, and the contribution of injury severity (mild/moderate/severe) and number of impacts has yet to be fully characterized.
Whether the patients in our study, whose P-tau levels remained high at the later time points during the chronic stage, are at a higher risk of developing a neurodegenerative disease is unknown at this time. Ideally, biomarker levels should closely correlate with a biological or pathogenic process87 or have use as a surrogate end-point. The correlation between early tau detection and the later development of neurodegenerative disease remains to be established since the variability in the TBI models do not allow straightforward extrapolation of the experimental results into clinical practice. Instead, the available evidence suggests that tau levels could be used as injury markers. In future studies, correlation of levels of various tau species in CSF, and/or serum with advanced neuroimaging such as diffuse tensor imaging or positron emission tomography combined with enhanced analytical tools may improve diagnosis/prognosis. Collectively, biomarker analysis and the pharmacological tools could provide crucial information on the importance of tau and Aβ peptides in the pathophysiology and long-term consequences of TBI.
TBI has been associated with hyperphosphorylation of the tau protein and the resulting P-tau is influential in the development of neurodegeneration. To define the precise relationship between TBI and tau levels in brain tissue, CSF and/or blood and clinical disease remains an important scientific challenge due to the association between TBI and the risk of developing neurodegenerative disease. Available experimental and clinical evidence suggests a complex relationship between increased tau protein release and NFT formation following TBI. Rodent studies have provided important mechanistic information and shed light into many aspects of tau expression following TBI although without consistently mirroring the histopathological findings observed in humans. TBI seems likely to accelerate the process leading to tauopathies, although the mechanisms involved and their relationship to the acute injury cascade are unclear. The increased use of tau as a biomarker in the clinical setting should enhance our understanding of TBI severity, recovery, therapeutic efficacy and the link between TBI and neurodegeneration.
In conclusion, we have developed tau-specific assay conditions that, in combination with the EIMAF technology, provides the sensitivity required to use the tau protein as a biomarker. In both experimental animal models and human samples the tau protein was detectable in CSF and blood following neurotrauma. Although these preliminary results are promising, validation of the assay will require analysis of larger sample sets. Follow-up studies also will allow discrimination of injury severities and help define parameters needed for accurate diagnosis and prognosis. All in all, the improved detection limits for tau demonstrated in this study using readily accessible samples provided by this fluorescence-based immunoassay system will greatly enhance future studies in this area. Further, this approach can easily be adapted to include additional biomarkers generating biomarker signature profiles that provide greater accuracy in TBI diagnosis/prognosis.
Acknowledgments
We acknowledge the technical support of Zhihui Yang (University of Florida) for clinical sample organization.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Chiu W.T., Huang S.J., Tsai S.H., Lin J-W., Tsai M-D., and Lin T-J. (2007). The impact of time, legislation, and geography on the epidemiology of traumatic brain injury. J. Clin. Neurosci. 14, 930–935 [DOI] [PubMed] [Google Scholar]
- 2.Clinton J., Ambler M.W., and Roberts G.W. (1991). Post-traumatic Alzheimer's disease: preponderance of a single plaque type. Neuropathol. Appl. Neurobiol. 17, 69–74 [DOI] [PubMed] [Google Scholar]
- 3.Mortimer J.A., Van Duijn C.M., Chandra V., Fratiglioni L., Graves A.B., Heyman A., Jorm A.F., Kokmen E., Rocca W.A., Shalat S.L., Soininen H., and Hofman A. (1991). Head trauma as a risk factor for Alzheimer's disease: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int. J. Epidemiol. 20, S28–S35 [DOI] [PubMed] [Google Scholar]
- 4.Breteler M.M., Claus J.J., Van Duijn C.M., Launer L.J., and Hofman A. (1992). Epidemiology of Alzheimer's disease. Epidemiol Rev 14, 59–82 [DOI] [PubMed] [Google Scholar]
- 5.Mayeux R., Ottman R., Tang M.X., Noboa-Bauza L., Marder K., Gurland B., and Stem Y. (1993). Genetic susceptibility and head injury as risk factors for Alzheimer's disease among community-dwelling elderly persons and their first-degree relatives. Ann. Neurol. 33, 494–501 [DOI] [PubMed] [Google Scholar]
- 6.Guo Z., Cupples L.A., Kurz A., Auerbach S.H., Volicer L., Chui H., Green R.C., Sadovnick A.D., Duara R., DeCarll C., Johnson K., Go R.C., Growdon J.H., Haines J.L., Kukull W.A., and Farrer L.A. (2000). Head injury and the risk of AD in the MIRAGE study. Neurology 54, 1316–1323 [DOI] [PubMed] [Google Scholar]
- 7.Plassman B.L., Havlik R.J., Steffens D.C., Helms M.J., Newman T.N., Drosdick D., Philips C., Gau B.A., Welsh-Bohmer K.A., Burke J.R., Guralnik J., and Breitner J.C.S. (2000). Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology 55, 1158–1166 [DOI] [PubMed] [Google Scholar]
- 8.Johnson V.E., Stewart W., and Smith D.H. (2010). Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer's disease? Nat. Rev. Neurosci. 11, 361–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Magnoni S., and Brody D.L. (2010). New perspectives on amyloid-beta dynamics after acute brain injury: moving between experimental approaches and studies in the human brain. Arch. Neurol. 67, 1068–1073 [DOI] [PubMed] [Google Scholar]
- 10.Li L.M., Menon D.K., and Janowitz T. (2014). Cross-sectional analysis of data from the U.S. clinical trials database reveals poor translational clinical trial effort for traumatic brain injury, compared with stroke. PLoS One 9, e84336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raabe A., Grolms C., Sorge O., Zimmermann M., and Seifert V., 1999. Serum S-100B protein in severe head injury. Neurosurgery 45, 477–483 [DOI] [PubMed] [Google Scholar]
- 12.Woertgen C., Rothoerl R., Metz C., and Brawanski A. (1999). Comparison of clinical, radiologic, and serum marker as prognostic factors after severe head injury. J. Trauma 47, 1126–1130 [DOI] [PubMed] [Google Scholar]
- 13.Franz G., Beer R., Kampfl A., Engelhardt K., Schmutzhard E., Ulmer H., and Delsenhammer F. (2003). Amyloid beta 1–42 and tau in cerebrospinal fluid after severe traumatic brain injury. Neurology 60, 1457–1461 [DOI] [PubMed] [Google Scholar]
- 14.Vos P.E., Lamers K.J., Hendriks J.C., van Haaren M., Beems T., Zimmerman C., van Geel W., de Reus H., Blert J., and Verbeek M.M. (2004). Glial and neuronal proteins in serum predict outcome after severe traumatic brain injury. Neurology 62, 1303–1310 [DOI] [PubMed] [Google Scholar]
- 15.Bulut M., Koksal O., Dogan S., Bolca N., Ozguc H., Korfali E., Ilcolet Y.O., and Parkiak M. (2006). Tau protein as a serum marker of brain damage in mild traumatic brain injury: preliminary results. Adv. Ther. 23, 12–22 [DOI] [PubMed] [Google Scholar]
- 16.Kavalci C., Pekdemir M., Durukan P., Ilhan N., Yildiz M., Serhatlioglu S., and Seckin D. (2007). The value of serum tau protein for the diagnosis of intracranial injury in minor head trauma. Am. J. Emerg. Med. 25, 391–395 [DOI] [PubMed] [Google Scholar]
- 17.Korfias S., Stranjalis G., Boviatsis E., Psachoulia C., Jullien G., Gregson B., Mendelow A.D., and Sakas D.E. (2007). Serum S-100B protein monitoring in patients with severe traumatic brain injury. Intensive Care Med. 33, 255–260 [DOI] [PubMed] [Google Scholar]
- 18.Papa L., Akinyi L., Liu M.C., Pineda J.A., Tepas J.J., III, Oli M.W., Zheng W., Robinson G., Robicsek S.A., Gabrielli A., Heaton S.C., Hannay H.J., Demery J.A., Brophy G.M., Layon J., Robertson C., Hayes R.L., and Wang K.K.W. (2010). Ubiquitin C-terminal hydrolase is a novel biomarker in humans for severe traumatic brain injury. Crit. Care Med. 38, 138–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mondello S., Robicsek S.A., Gabrielli A., Brophy G.M., Papa L., Tepas J., Robertson C., Buki A., Scharf D., Jixiang M., Akinyi L., Muller U., Wang K.K.W., and Hayes R.L. (2010). AlphaII-spectrin breakdown products (SBDPs): diagnosis and outcome in severe traumatic brain injury patients. J. Neurotrauma 27, 1203–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mondello S., Jeromin A., Buki A., Bullock R., Czeiter E., Kovacs N., Barzo P., Schmid K., Tortella F., Wang K.K., and Hayes R.L. (2012). Glial neuronal ratio: a novel index for differentiating injury type in patients with severe traumatic brain injury. J. Neurotrauma 29, 1096–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trojanowski J.Q., Schuck T., Schmidt M.L., and Lee V.M. (1989). Distribution of tau proteins in the normal human central and peripheral nervous system. J. Histochem. Cytochem. 37, 209–215 [DOI] [PubMed] [Google Scholar]
- 22.Sivanandam T.M. and Thakur M.K. (2012). Traumatic brain injury: a risk factor for Alzheimer's disease. Neurosci. Biobehav. Rev. 36, 1376–1381 [DOI] [PubMed] [Google Scholar]
- 23.Alonso A., Zaidi T., Novak M., Grundke-Iqbal I., and Iqbal K. (2001). Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. U.S.A. 98, 6923–6928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feijoo C., Campbell D.G., Jakes R., Goedert M., and Cuenda A. (2005). Evidence that phosphorylation of the microtubule-associated protein Tau by SAPK4/p38delta at Thr50 promotes microtubule assembly. J. Cell Sci. 118, 397–408 [DOI] [PubMed] [Google Scholar]
- 25.Morris M., Maeda S., Vossel K., and Mucke L. (2011). The many faces of tau. Neuron 70, 410–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blennow K. and Hampel H. (2003). CSF markers for incipient Alzheimer's disease. Lancet Neurol. 2, 605–613 [DOI] [PubMed] [Google Scholar]
- 27.Selkoe D. J. and Schenk D. (2003). Alzheimer's disease: molecular understanding predicts amyloid-based therapeutics. Ann. Rev. Pharmacol. Toxicol. 43, 545–584 [DOI] [PubMed] [Google Scholar]
- 28.Goedert M., Jakes R., Crowther R.A., Cohen P., Vanmechelen E., Vandermeeren M., and Cras P. (1994). Epitope mapping of monoclonal antibodies to the paired helical filaments of Alzheimer's disease: identification of phosphorylation sites in tau protein. Biochem. J. 301, 871–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davies P., 2000. Characterization and use of monoclonal antibodies to tau and paired helical filament tau. Methods Mol. Med. 32, 361–373 [DOI] [PubMed] [Google Scholar]
- 30.Sato S., Cerny R.L., Buescher J.L., and Ikezu T. (2006). Tau-tubulin kinase 1 (TTBK1), a neuron-specific tau kinase candidate, is involved in tau phosphorylation and aggregation. J. Neurochem. 9, 1573–1584 [DOI] [PubMed] [Google Scholar]
- 31.Hanger D.P., Byers H.L., Wray S., Leung K-Y., Saxton M.J., Seereeram A., Reynolds C.H., Ward M.A., and Anderton B.H. (2007). Novel phosphorylation sites in tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis. J. Biochem. 282, 23645–23654 [DOI] [PubMed] [Google Scholar]
- 32.Wang J-Z., Grundke-Iqbal I., and Iqbal K. (2007). Kinases and phosphatases and tau sites Involved in Alzheimer neurofibrillary degeneration. Eur. J. Neurosci. 25, 59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mattson N., Zegers I., Andreasson U., Bjerke M., Blankenstein M.A., Bowser R., Carrillo M.C., Gobomd J., Heath T., Jenkins R., Jeromin A., Kaplow J., Kidd D., Laterza O.F., Lockhart A., Lunn M.P., Martone R.L., Mills K., Pannee J., Ratcliffe M., Shaw L.M., Simon A.J., Soares H., Teunissen C.E., Verbeek M.M., Umek R.M., Vanderstichele H., Zetterberg H., Blennow K., and Portelius E. (2012). Reference measurement procedures for Alzheimer's disease cerebrospinal fluid biomarkers: definitions and approaches with focus on amyloid beta 42. Biomark. Med. 6, 409–417 [DOI] [PubMed] [Google Scholar]
- 34.Ost M., Nylen K., Csajbok L., Ohrfelt A.O., Tullberg M., Wikkelso C., Nellgard P., Rosengren L., Blennow K., and Nellgard B. (2006). Initial CSF total tau correlates with 1-year outcome in patients with traumatic brain injury. Neurology 67, 1600–1604 [DOI] [PubMed] [Google Scholar]
- 35.Liliang P.C., Liang C.L., Weng H.C., Lu K., Wang K.W., Chen H.J., and Chuang J.H. (2010). Tau proteins in serum predict outcome after severe traumatic brain injury. J. Surg. Res. 160, 302–307 [DOI] [PubMed] [Google Scholar]
- 36.Rostami E., Davidsson J., Ng K.C., Lu J., Gyorgy A., Walker J., Wingo D., Plantman S., Bellander B., Agoston D.V., and Risling M. (2012). A model for mild traumatic brain injury that induces limited transient memory impairment and increased levels of axon related serum biomarkers. Front. Neurol. 3, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Corsellis J.A., Bruton C.J., and Freeman-Browne D. (1973). The aftermath of boxing. Psychol. Med. 3, 270–303 [DOI] [PubMed] [Google Scholar]
- 38.Roberts G.W., Allsop D., and Bruton C. (1990). The occult aftermath of boxing. J Neurol Neurosurg. Psychiatry 53, 373–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dale G.E., Leigh P.N., Luthert P., Anderton B.H., and Roberts G.W. (1991). Neurofibrillary tangles in dementia pugilistica are ubiquitinated. J. Neurol. Neurosurg. Psychiatry 54, 116–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geddes J.F., Vowles G.H., Nicoll J.A., and Revesz T. (1999). Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol. 98, 171–178 [DOI] [PubMed] [Google Scholar]
- 41.McKee A.C., Cantu R.C., Nowinski C.J., Hedley-Whyte E.T., Gavett B.E., Budson A.E., Santini V.E., Lee H.S., Kubilus C.A., and Stern R.A. (2009). Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J. Neuropathol. Exp. Neurol. 68, 709–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McKee A.C., Stein T.D., Nowinski C.J., Stern R.A., Daneshvar D.H., Alvarez V.E., Lee H.S., Hall G., Wojtowicz S.M., Baugh C.M., Riley D.O., Kubilus C.A., Cormier K.A., Jacobs M.A., Martin B.R., Abraham C.R., Ikezu T., Reichard R.R., Wolozin B.L., Budson A.E., Goldstein L.E., Kowalk N.W., and Cantu R.C. (2013). The spectrum of disease in chronic traumatic encephalopathy. Brain 136, 43–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jordan B.D., Kanik A.B., Horwich M.S., Sweeney D., Relkin N.R., Petito C.K., and Gandy S. (1995). Apolipoprotein E epsilon 4 and fatal cerebral amyloid angiopathy associated with dementia pugilistica. Ann. Neurol. 38, 698–699 [DOI] [PubMed] [Google Scholar]
- 44.McKenzie K.J., McLellan D.R., Gentleman S.M., Maxwell W.L., Gennarelli T.A., and Graham D.I. (1996). Is beta-APP a marker of axonal damage in short-surviving head injury? Acta Neuropathol. 92, 608–613 [DOI] [PubMed] [Google Scholar]
- 45.Nowak L.A., Smith G.G., and Reyes P.F. (2009). Dementia in a retired world boxing champion: case report and literature review. Clin. Neuropathol. 28, 275–280 [PubMed] [Google Scholar]
- 46.Omalu B.I., Fitzsimmons R.P., Hammers J., and Bailes J. (2010). Chronic traumatic encephalopathy in a professional american wrestler. J. Forensic Nurs. 6, 130–136 [DOI] [PubMed] [Google Scholar]
- 47.Smith D.H., Johnson V.E., and Stewart W. (2013). Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat. Rev. Neurol. 9, 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gabbita S.P., Scheff S.W., Menard R.M., Roberts K., Fugaccia I., and Zemlan F.P. (2005). Cleaved-tau: a biomarker of neuronal damage after traumatic brain injury. J. Neurotrauma 22, 83–94 [DOI] [PubMed] [Google Scholar]
- 49.Liliang P.C., Liang C.L., Lu K., Wang K.W., Weng H.C., Hsieh C.H., Tsai Y-D., and Chen H-J. (2010). Relationship between injury severity and serum tau protein levels in traumatic brain injured rats. Resuscitation 81, 1205–1208 [DOI] [PubMed] [Google Scholar]
- 50.Chang B., Gray P., Piltch M., Bulgin M.S., Sorensen-Melson S., Miller M.W., Davies P., Brown D.R., Coughlin D.R., and Rubenstein R. (2009). Surround optical fiber immunoassay (SOFIA): more than an ultra-sensitive assay for PrP detection. J. Virol. Methods 159, 15–22 [DOI] [PubMed] [Google Scholar]
- 51.Rubenstein R., Chang B., Gray P., Piltch M., Bulgin M.S., Sorensen-Melson S., and Miller M.W. (2010). A novel method for preclinical detection of PrPSc in blood. J. Gen. Virol. 91, 1883–1892 [DOI] [PubMed] [Google Scholar]
- 52.Rubenstein R., Chang B., Gray P., Piltch M., Bulgin M., Sorensen-Melson S., and Miller M.W. (2011). Prion disease detection, PMCA kinetics, and IgG in urine from naturally/experimentally infected scrapie sheep and preclinical/clinical CWD deer. J. Virol. 85, 9031–9038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rubenstein R., Bulgin M.S., Chang B., Sorensen-Melson S., Petersen R.B., and LaFauci G. (2012). PrPSc detection and infectivity in semen from scrapie-infected sheep. J. Gen. Virol. 93, 1375–1383 [DOI] [PubMed] [Google Scholar]
- 54.Rubenstein R. and Chang B. (2013). Re-Assessment of PrPSc distribution in sporadic and variant CJD. PLoS ONE 8, e66352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Acker C.M., Forest S.K., Zinkowski R., Davies P., and d'Abramo C. (2013). Sensitive quantitative assays for tau and phosphor-tau in transgenic mouse models. Neurobiol. Aging 34, 338–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schweitzer B., Wiltshire S., Lambert J., O'Malley S., Kukanskia K., Zhu Z., Kingsmore S.F., Lizardi P.M., and Ward D.C. (2000). Immunoassays with rolling circle DNA amplification: a versatile platform for ultrasensitive antigen detection. Proc. Natl. Acad. Sci. U.S.A. 97, 10113–10119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rankin C.A., Sun Q., and Gamblin T.C. (2007). Tau phosphorylation by GSK-3β promotes tangle-like filament morphology. Mol. Neurodegen. 2, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lewis J., McGowan E., Rockwood J., Melrose H., Nacharaju P., Van Slegtenhorst M., Gwinn-Hardy K., Murphy M.P., Baker M., Yu X., Duff K., Hardy J., Corral A., Lin W-L., Yen S-H., Dickson D.W., Davies P., and Hutton M. (2000). Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 25, 402–405 [DOI] [PubMed] [Google Scholar]
- 59.Dawson H.N., Ferreira A., Eyster M.V., Ghoshal N., Binder L.I., and Vitek M.P. (2001). Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J. Cell Sci. 114, 1179–1187 [DOI] [PubMed] [Google Scholar]
- 60.Grundke-Iqbal I., Iqbal K., Quinlan M., Tung Y.C., Zaidi M.S., and Wisniewski H.M. (1986). Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 261, 6084–6089 [PubMed] [Google Scholar]
- 61.Nukina N. and Ihara Y. (1986). One of the antigenic determinants of paired helical filaments is related to tau protein. J. Biochem. 99, 1541–1544 [DOI] [PubMed] [Google Scholar]
- 62.Wood J.G., Mirra S.S., Pollock N.J., and Binder L.I. (1986). Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau. Proc. Natl. Acad. Sci. U.S.A. 83, 4040–4043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kondo J., Honda T., Mori H., Hamada Y., Miura R., Ogawara M., and Ihara Y. (1988). The carboxyl third of tau is tightly bound to paired helical filaments. Neuron 1, 827–834 [DOI] [PubMed] [Google Scholar]
- 64.Lee V.M., Balin B.J., Otvos L., Jr., and Trojanowski J.Q. (1991). A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science 251, 675–678 [DOI] [PubMed] [Google Scholar]
- 65.Lee V.M., Goedert M., and Trojanowski J.Q. (2001). Neurodegenerative tauopathies. Ann. Rev. Neurosci. 24, 1121–1159 [DOI] [PubMed] [Google Scholar]
- 66.Omalu B., Bailes J., Hamilton R.L., Kamboh M.I., Hammers J., Case M., and Fitzsimmons R. (2011). Emerging histomorphologic phenotypes of chronic traumatic encephalopathy in American athletes. Neurosurgery 69, 173–183 [DOI] [PubMed] [Google Scholar]
- 67.Rajput A., Dickson D.W., Robinson C.A., Ross O.A., Dachsel J.C., Lincoln S.J., Cobb S.A., Rajput M.L., and Farrer M.J. (2006). Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology 67, 1506–1508 [DOI] [PubMed] [Google Scholar]
- 68.Santpere G. and Ferrer I. (2009). LRRK2 and neurodegeneration. Acta Neuropathol. 117, 227–246 [DOI] [PubMed] [Google Scholar]
- 69.Ihara Y. (2001). PHF and PHF-like fibrils–cause or consequence? Neurol. Aging 22, 123–126 [DOI] [PubMed] [Google Scholar]
- 70.Giannakopoulos P., Herrmann F.R., Bussiere T., Bouras C., Kovari E., Perl D.P., Morrison J.H., Gold G., and Hof P.R. (2003). Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology 60, 1495–1500 [DOI] [PubMed] [Google Scholar]
- 71.Shahim P., Tegner Y., Wilson D.H., Randall J., Skillback T., Pazooki D., Kallberg B., Blennow K., and Zetterberg H. (2014). Blood biomarkers for brain injury in concussed professional ice hockey players. JAMA Neurol. 71, 684–692 [DOI] [PubMed] [Google Scholar]
- 72.Woertgen C., Rothoerl R.D., and Brawanski A. (2001). Neuron-specific enolase serum levels after controlled cortical impact injury in the rat. J. Neurotrauma 18, 569–573 [DOI] [PubMed] [Google Scholar]
- 73.Woertgen C., Rothoerl R.D., Wiesmann M., Missler U., and Brawanski A. (2002). Glial and neuronal serum markers after controlled cortical impact injury in the rat. Acta Neurochir. Suppl 81, 205–207 [DOI] [PubMed] [Google Scholar]
- 74.Rothermundt M., Peters M., Prehn J.H., and Arolt V. (2003). S100B in brain damage and neurodegeneration. Microsc. Res. Tech. 60, 614–632 [DOI] [PubMed] [Google Scholar]
- 75.Ross S.A., Cunningham R.T., Johnston C.F., and Rowlands B.J. (1996). NSE as an aid to outcome prediction in head injury. Br. J. Neurosurg. 10, 471–476 [DOI] [PubMed] [Google Scholar]
- 76.Pelinka L.E., Kroepfl A., Schmidhammer R., Krenn M., Buchinger W., Redl H., and Raabe A. (2004). Glial fibrillary acidic protein in serum after traumatic brain injury and multiple trauma. J. Trauma 57, 1006–1012 [DOI] [PubMed] [Google Scholar]
- 77.Honda M., Tsuruta R., Kaneko T., Kasaoka S., Yagi T., Todani M., Fujita M., Izumi T., and Meekawa T. (2010). Serum glial fibrillary acidic protein is a highly specific biomarker for traumatic brain injury in humans compared with S-100B and neuron-specific enolase. J. Trauma 69, 104–109 [DOI] [PubMed] [Google Scholar]
- 78.Papa L., Lewis L.M., Falk J.L., Zhang Z., Silvestri S., Giordano P., Brophy G.M., Demery J.A., Dixit N.K., Ferguson I., Liu M.C., Mo J., Akinyi L., Schmid K., Mondello S., Robertson C.S., Tortella F.C., Hayes R.L., and Wang K.K.W. (2012a). Elevated levels of serum glial fibrillary acidic protein breakdown products in mild and moderate traumatic brain injury are associated with intracranial lesions and neurosurgical intervention. Ann. Emerg. Med. 59, 471–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Okonkwo D.O., Yue J.K., Puccio A.M., Panczykowski D.M., Inoue T., McMahon P.J., Sorani M.D., Yuh E.L., Lingsma H.F., Maas A.I.R., Valadka A.B., Manley G.T., and Transforming Research and Clinical Knowledge in Traumatic Brain Injury investigators including, Casey S.S., Cheong M., Cooper S.R., Dams-O'Connor K., Gordon W.A., Hricik A.J., Hochberger K., Menon D.K., Mukherjee P., Sinha T.K., Schnyer D.M., and Vassar M.J. (2013). GFAP-BDP as an acute diagnostic marker in traumatic brain injury: eesults from the prospective transforming research and clinical knowledge in traumatic brain injury study. J. Neurotrauma 30, 1490–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Papa L., Lewis L.M., Silvestri S., Falk J.L., Giordano P., Brophy G.M., Demery J.A., Liu M.C., Mo J., Akinyi L., Mondello S., Schmid K., Robertson C., Tortella F.C., Hayes R.L., and Wang K.K.W. (2012b). Serum levels of ubiquitin C-terminal hydrolase distinguish mild traumatic brain injury from trauma controls and are elevated in mild and moderate traumatic brain injury patients with intracranial lesions and neurosurgical intervention. J. Trauma Acute Care Surg. 72, 1335–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Diaz-Arrastia R., Wang K.K.W., Papa L., Sorani M.D., Yue J.K., Puccio A.M., McMahon P.J., Inoue T., Yuh E.L., Lingsma H.F., Maas A.I.R., Valadka A.B., Okonkwo D.O., Manley G.T., and the TRACK-TBI Investigators including Casey S.S., Cheong M., Cooper S.R., Dams-O'Connor K., Gordon W.A., Hricik A.J., Menon D.K., Mukherjee P., Schnyer D.M., Sinha T.K., and Vassar M.J. (2014). Acute biomarkers of traumatic brain injury: relationship between plasma levels of ubiquitin C-terminal hydrolase-L1 (UCH-L1) and glial fibrillary acidic protein (GFAP). J. Neurotrauma 31, 19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ma M., Lindsell C.J., Rosenberry C.M., Shaw G.J., and Zemlan F.P. (2008). Serum cleaved tau does not predict postconcussion syndrome after mild traumatic brain injury. Am. J. Emerg. Med. 26, 763–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fleminger S., Oliver D.L., Lovestone S., Rabe-Hesketh S., and Giora A. (2003). Head injury as a risk factor for Alzheimer's disease: the evidence 10 years on; a partial replication. J. Neurol. Neurosurg. Psychiatry 74, 857–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Johnson V.E., Stewart W., and Smith D.H. (2012). Widespread tau and amyloid-Beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. 22, 142–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Smith C., Graham D.I., Murray L.S., and Nicoll J.A. (2003). Tau immunohistochemistry in acute brain injury. Neuropathol. Appl. Neurobiol. 29, 496–502 [DOI] [PubMed] [Google Scholar]
- 86.Uryu K., Chen X.H., Martinez D., Browne K.D., Johnson V.E., Graham D.I., Lee V.M-Y., Trojanowski J.Q., and Smith D.H. (2007). Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp. Neurol. 208, 185–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Czeiter E., Mondello S., Kovacs N., Sandor J., Gabrielli A., Schmid K., Tortella F., Wang K.K.W., Hayes R.L., Barzo P., Ezer E., Doczi T., and Buki A. (2012). Brain injury biomarkers may improve the predictive power of the IMPACT outcome calculator. J. Neurotrauma 29, 177–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]