

Abstract
Endogenously produced carbon monoxide (CO) is commonly believed to be a ubiquitous second messenger involved in a wide range of physiological and pathological responses. The major evidence supporting this concept is that CO is produced endogenously via heme oxygenase-catalyzed breakdown of heme and that experimental exposure to CO alters tissue function. However, it remains to be conclusively demonstrated that there are specific receptors for CO and that endogenous CO production is sufficient to alter tissue function. Unlike other signaling molecules, CO is not significantly metabolized, and it is removed from cells solely via rapid diffusion into blood, which serves as a near infinite sink. This non-metabolizable nature of CO renders the physiology of this gas uniquely susceptible to quantitative modeling. This review analyzes each of the steps involved in CO signaling: 1) the background CO partial pressure (PCO) and the blood and tissue CO binding; 2) the affinity of the putative CO receptors; 3) the rate of endogenous tissue CO production; and 4) the tissue PCO that results from the balance between this endogenous CO production and diffusion to the blood sink. Because existing data demonstrate that virtually all endogenous CO production results from the routine “housekeeping” turnover of heme, only a small fraction can play a signaling role. The novel aspect of the present report is to demonstrate via physiological modeling that this small fraction of CO production is seemingly insufficient to raise intracellular PCO to the levels required for the conventional, specific messenger receptor activation. It is concluded that the many physiological alterations observed with exogenous CO administration are probably produced by the non-specific CO inhibition of cytochrome C oxidase activity, with release of reactive oxygen species (ROS) and that this ROS signaling pathway is a potential effector mechanism for endogenously produced CO.
Keywords: cytochrome, oxygenase, Krogh cylinder model, ROS, heme
Introduction
Scientific appreciation of the physiological implications of carbon monoxide (CO) has evolved from its recognition as an environmental toxin in 1857,1 to demonstration of its derivation from heme catabolism in 1952,2 to its present important status as an intracellular messenger that regulates hundreds of physiological and cytoprotective functions. CO is produced in animals exclusively by heme oxygenase (HO) catalysis of heme to biliverdin:3
| (1) |
In mammals, there are two forms of HO: an inducible form, HO-1 (also known as heat-shock protein 32), and a constitutive form, HO-2. A wide variety of stimuli induce HO-1, including oxidative stress, heat shock, ischemia, radiation, hypoxia, hyperoxia, and drugs.4–6 This amazing responsiveness to nearly every known cell-modifying signal has focused attention on the potential importance of HO in a wide range of clinical conditions. It is important to recognize that alterations in HO do not necessarily implicate CO as the HO effector, since HO has three other functions: removal of heme, release of Fe2+, and production of the anti-oxidant bilirubin from biliverdin, all of which are important cellular responses to stress and cell death.7 However, it is widely assumed that the primary effect of HO induction is the modification of cell function via production of CO, which serves as a second messenger. The focus of this review will be on the unique physiology required for CO to serve as the second messenger mediator of HO activity – a function invoked as a factor in a wide variety of clinical conditions. For example, a recent review of just the central nervous system conditions in which CO is implicated include Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, cerebral artery occlusion, and pain.8
The CO messenger literature is enormous, involving nearly every organ and regulatory system. There are a number of excellent recent reviews of this topic,5–13 with each review having a particular focus and emphasis. Seemingly lacking in these discussions is a critical examination of the unusual features of CO that complicate the ability of this gas to serve as a typical intracellular messenger. Most signaling molecules are produced solely for a signaling purpose and are then rapidly catabolized (eg, nitric oxide has a half-time of less than 2 seconds), and the background intracellular activity of the signaling molecule is maintained at a very low level. In contrast, there is minimal catabolism of CO, and endogenously produced CO is removed from the cell entirely via diffusion to the blood, where it is avidly bound by hemoglobin (Hb). Homeostasis of blood CO is established by pulmonary excretion, a relatively slow process with a half-time of approximately 4 hours (as discussed in the “Hb and Mb CO binding and pharmacokinetics” section). Thus, cells float in a sea of the messenger, which maintains a low, but not negligible-free CO concentration in the cell equal to that of blood, about 0.002 μM, or a CO partial pressure (PCO) of 0.0019 mmHg under normal conditions (Table 1). To provide a signal, CO has to be produced at a rate that appreciably raises the CO concentration over the basal level, ruling out the possibility that trivial CO production could serve as a messenger. The lack of catabolism of CO, its removal from the cell by diffusion to the blood, and its excretion solely via the lungs makes CO uniquely amenable to quantitative physiological modeling. For example, in contrast to other messengers, the total body production of CO is easily assessed (via breath measurements), and the intracellular PCO at baseline and following CO administration both can be determined, since intracellular PCO is in equilibrium with the blood PCO. Most important, the diffusion of CO between the cell and blood can be quantitatively modeled, making it possible to estimate the intracellular production rate required to maintain a given cellular PCO. In Table 1, we have summarized the fundamental physical chemical CO parameters (eg, solubility, diffusion coefficient) and the range of CO concentrations required for different biological effects.
Table 1.
Physiological CO parameters (at 37°C)
| Parameter | Value | Ref | Comments |
|---|---|---|---|
| Aqueous solubility (α) | At 37°C | ||
| mL STP/liter/mmHg | 0.024 | 68 | |
| μmole/liter/mmHg | 1.1 | ||
| Aqueous diffusion coefficient (D) | Interpolate between 30°C and 40°C | ||
| cm2/s | 3.26×10−5 | 108 | |
| Aqueous diffusivity (Kw = α D) | |||
| nmol/cm/mm Hg/s | 3.58×10−5 | ||
| Tissue CO diffusivity (K) | |||
| nmol/cm/mm Hg/s | 1.77×10−5 | 109 | From K for O2 divided by 1.32 |
| CO/O2 relative affinity (M) | |||
| Hemoglobin | 220 | 14 | |
| Myoglobin | 39 | 14 | |
| Cytochrome C oxidase | 2.5 | 29 | |
| Blood CO concentration | Normal human | ||
| % Carboxyhemoglobin | 0.8 | 18 | |
| PCO (mmHg) | 0.0019 | Estimated from M for hemoglobin | |
| End alveolar PCO (mmHg) | 0.0018 | 17 | |
| Free concentration (μM) | 0.002 | Estimated from PCO and α | |
| PCO for biological effect (mmHg) | |||
| Superoxide dismutase synthesis | 0.0076 | 42 | 1 h exposure |
| ROS-related cell death | 0.076 | 42 | 1 h exposure |
| Piglet pial arteriolar dilation | 0.09 | 60 | |
| Rat tail artery dilation | 10 | 62 |
Abbreviations: STP, standard temperature and pressure; ref, reference; ROS, reactive oxygen species; PCO, CO partial pressure; h, hour.
The fundamental (but rarely stated) hypothesis of CO signaling is that local tissue CO production raises the local cellular concentration to a level sufficiently above the background concentration to trigger the signaling, which is then terminated when the tissue production ceases and the CO diffuses to the blood sink. The crucial issue is whether the rate of tissue production is sufficient to reach signaling levels, given the rapid rate of diffusion between cells and the blood sink. In order to answer this question, the following items require careful consideration: 1) the background blood and tissue CO; 2) the CO affinity of the putative tissue CO signaling receptors; 3) the rate of endogenous tissue CO production; and 4) a quantitative analysis of the tissue CO concentration that is reached as a result of this CO production in the presence of the nearly infinite diffusion sink represented by the blood. This review will present what we believe to be the first detailed, quantitative discussions of each of these issues.
The “Hb and Mb CO binding and pharmacokinetics” section reviews the quantitative details of the binding of CO to Hb and myoglobin (Mb) and the pharmacokinetics of CO production, distribution, and excretion. This binding determines the steady state background CO concentration and the time dependence of its change when CO is exogenously administered.
Two fundamentally different receptor mechanisms have been proposed for CO second messenger signaling. The first, discussed in the “COX binding and ROS formation” section, is the CO binding to and inhibition of cellular cytochrome C oxidase (COX), a reaction which releases reactive oxygen species (ROS) that serve as a relatively non-specific effector. This type of “signaling” is unique to CO and raises the question of whether it is actually a physiological mechanism versus a non-specific toxic CO action. This issue is particularly important, because most recent research concerning the mode of action of inhaled CO invokes the mechanism of ROS formation secondary to COX inhibition. The second signaling mechanism is the classic second messenger type involving binding to a specific CO receptor which then triggers a unique cellular response. The “Specific CO sensors or receptors” section reviews the CO receptors that have been proposed, their affinity for CO, and the evidence supporting their second messenger function.
The “HO activity, heme substrate concentration, and the endogenous rate of CO tissue production” section reviews available information about the rate of endogenous tissue CO production that might serve a messenger function. There have been only a few, inexact measurements of this critical factor that determines the tissue CO concentration. In this review, it will be stressed that the vast majority of the total CO produced in the body results from the routine turnover of Hb and other tissue (eg, hepatic) hemes; only a very small fraction of the total CO endogenous production possibly could be designed to serve a specific CO signaling function.
The “Quantitative modeling of the steady state and time-dependent tissue CO in the presence of the blood sink” section describes a quantitative model of the steady state and time-dependent tissue CO concentrations that result from the balance between the endogenous tissue CO production and the rate of diffusion to the blood sink. This calculation is designed to mimic the physiological effect of local tissue HO activity. Although the model is a crude approximation of the complicated tissue–blood interaction, it provides the first analysis of the basic requirements necessary for a CO signaling mechanism, in particular, the large rates of local endogenous CO production required for signaling.
Finally, the “Discussion: does HO function via its release of CO?” section summarizes the results of this analysis and their implications for interpreting the function and importance of CO signaling. This analysis places limits on the possible second messenger role of CO and raises questions about the validity of some conventionally accepted views of CO signaling.
Hb and Mb CO binding and pharmacokinetics
Table 1 lists the important quantitative CO parameters that will be used in this review. The aqueous solubility (α) allows one to convert from the partial pressure in the gas phase (P) to the molar free concentration (c) in the solution with which the gas is in equilibrium:
| (2) |
The biologically important concentration of CO that drives reactions, receptor binding, or diffusion is c, the free (unbound) concentration of CO that is dissolved in the tissue fluid or plasma. The free concentration of CO is not directly measured, but rather the PCO is determined and is then used to calculate the free concentration via use of the solubility coefficient. Using an α =1.1 μmol/L/mmHg (Table 1), a 1 μM free solution CO concentration corresponds to a 0.9 mmHg (or 1,188 parts per million [ppm]) partial gas pressure.
The concept that CO serves a signaling function is largely derived from the observation that tissue function is altered by in vitro or in vivo exposure to CO. Such alterations could reflect a specific messenger function or a non-specific toxicity of this gas. To differentiate between these two possibilities, it is necessary to know the concentration of free CO that exists in tissue at baseline and following CO administration, values that are dependent on CO–O2 binding competition in Hb and tissue heme proteins. Classical CO toxicity is mediated via binding to Hb, with a resultant decreased O2 delivery to the tissues. This binding is quantitated by the fraction of CO-bound Hb (HbCO). Since standard whole-animal experimental CO administration is usually achieved via a short-period CO inhalation, it is important to understand the time dependence of the change in HbCO during and following this CO exposure, and its effect on tissue O2 delivery. In addition, since CO and O2 compete for COX binding, a decreased tissue oxygen partial pressure results in an increased CO–COX binding. The following analysis presents a detailed description of the complex non-linear relationship between HbCO versus PCO and . When analyzing this relationship, it is crucial to recognize that CO and O2 are competing for the same heme binding sites.
For Mb, where there is just one binding site and no significant cooperative binding (Hill coefficient =1), the equation for the fraction CO (MbCO) and O2 (MbO2) bound has the following standard competitive binding form:14
| (3) |
The binding is characterized by the two parameters, (P50)CO, the partial pressure for 50% binding of CO in the absence of O2, and MMb, which is the affinity of CO relative to O2:
| (4) |
The corresponding is MMb × (P50)CO. In the following calculations, values of (P50)CO =0.07 mmHg and MMb =39 will be used (Table 1).14
For Hb, where there is cooperative binding between the four heme sites, the fraction of CO (HbCO) and O2 (HbO2) bound can be approximately described by a variant of the Adair equation:14
| (5) |
where and MHb is the affinity of the heme subunit for CO relative to O2 and is defined by the Haldane relationship:
| (6) |
This equation assumes that CO and O2 compete for each of the four heme sites with the same relative affinity for MHb and that the cooperativity of binding is identical for O2 and CO. The following set of parameters will be used here:15,16 MHb =220; A1 =0.0218; A2 =0.000912; A3 =3.75×10−6; and A4 =2.47×10−6.
Although Equation 5 is not precise because the cooperativity of CO differs slightly from that of O2 and the Adair equation is not exact, even for pure oxygen,15,16 it is a quite good approximation and will be used in the following calculations to illustrate the physiologically important interactions between , PCO, and the fraction of Hb that is occupied by CO (HbCO) in human blood. Figure 1 shows a plot of Equations 3 and 5 for HbCO (red lines) and MbCO (black lines) as a function of the PCO for value of 95 (solid line), 50 (long-dashed line), and 20 mmHg (dot-dashed line). The competition between CO and O2 implies that, in the steady state, the PCO in the capillary (eg, tissue) is significantly less than the arterial PCO because the lower capillary leads to a greater HbCO affinity. Since the body is a closed system with most of the CO bound to Hb (see discussion of equation 10), the value of HbCO remains relatively constant as the blood moves from the artery to the capillaries. As illustrated in Figure 1, for a fixed HbCO of 0.2 (horizontal dashed line), as the decreases from 95 (artery) to 20 (vein) mmHg, the PCO falls from 0.112 to 0.047 mmHg.
Figure 1.
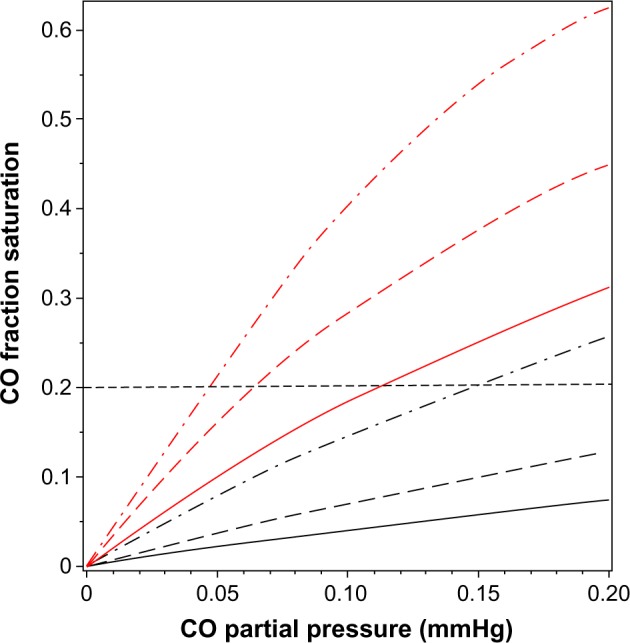
Plot of fraction of hemoglobin (red lines) and myoglobin (black lines) that is occupied by CO as a function of PCO, for a fixed of 95 mmHg (solid lines), 50 mmHg (long-dashed lines), and 20 mmHg (dot-dashed lines). The black, horizontal dashed line indicates that for a fixed HbCO fraction of 0.2, the equilibrium PCO is 0.112 mmHg for a of 95 mmHg, and decreases to a PCO of 0.047 mmHg when the decreases to 20 mmHg.
Abbreviations: , O2 partial pressure; Hb, hemoglobin; HbCO, CO-bound Hb; PCO, CO partial pressure.
Figure 2A shows the O2 Hb saturation (HbO2) as a function of in the presence of a PCO of 0, 0.1, 0.2, and 0.5 mmHg, as determined by Equation 5. The presence of CO not only reduces the value of HbO2, CO also changes the shape of the O2 dissociation curve, shifting it to the left. This shift is seen more clearly in Figure 2B, where the O2 saturation relative to the maximal O2 saturation at mmHg is plotted, instead of the absolute saturation, which is plotted in Figure 2A. The for 50% of maximum binding falls from a normal value of 26 mmHg in the absence of CO to 12.6 mmHg for a PCO of 0.5 mmHg. Thus, CO produces tissue anoxia not simply by displacing O2 from Hb but also because the tissue O2 must fall to lower values in order to release the O2 that has not been displaced.
Figure 2.

Fractional saturation of hemoglobin occupied by O2 as a function of the for PCO fixed at 0 mmHg (black line), 0.1 mmHg (red line), 0.2 mmHg (blue line), and 0.5 mmHg (green line). In (A), the absolute O2 saturation of the total hemoglobin is plotted, while in (B), the fraction saturated with O2 relative to the maximum O2 saturation at is plotted.
Abbreviations: , O2 partial pressure; PCO, CO partial pressure.
A critical concept central to this review is that the blood, which serves as a CO sink for all tissues, normally maintains a very low PCO. Venous blood PCO in humans can be estimated from the normal venous HbCO value of 0.8% and by applying Equation 6 to venous blood ( ; HbO2 =0.75 mmHg; MHb =220 mmHg):
| (7) |
Probably the best direct experimental measurement of human blood PCO is the breath holding end alveolar PCO value of 2.44 ppm (0.0018 mmHg) obtained by Furne et al.17 This value should be close to equilibrium with the average pulmonary capillary blood. These values are summarized in Table 1.
The classical approach to administering CO in experimental studies is via inspired air. To extrapolate from alterations in cell function observed in such studies to potential signaling mechanisms induced by endogenous CO production, it is necessary to know the cellular PCO that results from CO inhalation. Although the time-dependent pharmacokinetics are complicated, with a CO pulmonary diffusion limitation and varying rates of distribution to the different organs and tissues, a surprisingly simple expression derived by Coburn et al18 provides a good approximation. The long-time steady state (HbCO)ss can be obtained by application of the Haldane relation (Equation 6) to the pulmonary capillary blood:
| (8) |
where ICO is the inspired alveolar PCO, is the average pulmonary capillary , and HbO2 is the average pulmonary capillary HbO2 saturation. Using the approximate relations that HbCO + HbO ≈1 and for breathing room air:
| (9) |
That is, the steady state HbCO for an arbitrary value of inspired alveolar PCO (also known as ICO) can be estimated simply from the value of MHb (≈220). This represents the contribution from exogenous inspired CO. The total blood HbCO is the sum of this term plus the baseline endogenous HbCO. The time-dependent change in blood HbCO in response to inhaled CO (neglecting the endogenous CO production) is well approximated by a single exponential, based on the assumption that the total body CO volume of distribution can be represented by a well-mixed blood volume (Vb).18
| (10) |
where DL is the CO lung diffusivity, PAtm is the atmospheric pressure corrected for vapor pressure of water (713 mmHg), is alveolar ventilation, Vb is the effective blood volume, and [HbO2] is the average pulmonary capillary blood O2 concentration in mL STP/mL blood. Equations 9 and 10 provide good fits to the time-dependent experimental data in humans for inhaled CO ranging from 25 ppm (0.019 mmHg) to 500 ppm (0.38 mmHg), assuming that [HbO2] =0.2 and .19 The major simplifying assumption of this model is that the total body CO volume of distribution can be represented by an “equivalent” Vb. This explanation is a good approximation, because the vast majority of the total body CO binding is provided by Hb, eg, in humans there is roughly 800 g Hb, versus only 34.7 g Mb and much lower concentrations of other cellular hemes (eg, 0.78 g COX).20 Since Hb has the highest CO affinity (Table 1 and Figure 1), at least 94% of the total body CO is in the blood. A second assumption is that [HbO2] is constant during the entire CO inhalation time course. This is clearly incorrect if the CO concentration is high enough to saturate a significant fraction of Hb. Bruce and Bruce21 have described a much more detailed, multi-compartment, physiologically based pharmacokinetic model that, presumably, is more accurate. However, the experimental data are probably not good enough to distinguish them from the simple model described above.
From Equation 10, the time constant T for humans (DL ≈30 mL/min/mmHg; Vb ≈5.5 L; and ) is about 360 minutes, which is in good agreement with experimental data.19 It is of interest to see how this time constant scales for different animals, in particular the rat and mouse, which are the commonly used experimental models. Neglecting the 1/DL term (assume lung diffusion is infinite), which is only 30% of the term, the T for animal x relative to the human is given by:
| (11) |
Using values for the rat (Vb =0.07 g/g,22 and ) and the mouse (Vb =0.09 g/g,22 and , and (assuming dead space is 33% of tidal volume), the CO time constant is 70 minutes and 43 minutes for the rat and mouse, respectively. This value for the rat is in rough agreement with direct experimental measurements.24 A common experimental model is to expose mice to 250 ppm CO for a period of 1 hour, and from the above estimate of T, at the end of the hour, mice should have reached 75% of their steady state value.
COX binding and ROS formation
Although there are hundreds of heme proteins25 that could potentially be modified by CO, the only major proteins with well-characterized, relatively high affinity CO binding are Hb, Mb, cytochrome P450, and COX26 (refer to the “Specific CO sensors or receptors” section for a further discussion of other possible minor CO binding proteins). Of the four complexes in the electron transport chain, three of which contain heme proteins, only complex IV (otherwise known as COX) is significantly inhibited by low levels of CO.27 Historically, COX has been the major focus of studies of cellular CO toxicity. The quantitative aspects of the binding of CO to COX are of particular importance because this binding produces a non-specific asphyxiation of all cells. When investigating the actions of CO, it is essential to distinguish this general COX inhibition from the more directed, specific CO signaling mechanisms that are discussed in the “Specific CO sensors or receptors” section. This “toxicity” has recently taken on special importance because of a number of reports of alterations of physiological function induced by surprisingly low doses of CO (eg, 250 ppm for 1 hour, see Discussion section). These effects have been attributed to the formation of ROS produced by CO inhibition of COX, the last step in the electron transport chain,28 as opposed to an effect attributable to a conventional messenger function in which CO binds to a specific CO receptor.
As was the case for Hb and Mb, O2 and CO are competing for the same binding site in COX. Using purified COX, Petersen29 showed that CO behaves as a classical competitive enzyme inhibitor of O2, with the activity (V) characterized by the O2 Km and the CO inhibition constant (Ki):
| (12) |
The second equality is in the same form used to describe CO binding to Mb (Equation 3), with and MCOX = Km/Ki. The fraction of COX inhibited by the addition of a given PCO to a medium with a fixed is described by an equation similar to Equation 3, with (P50)CO equal to the Ki of CO:
| (13) |
It can be seen from Equation 13 that the fractional COX inhibition by CO is critically dependent on the at the COX binding site. At high PCO and , the Ki term in the denominator is negligible, and the fraction CO inhibition is characterized by MCOX.
The CO Ki of COX is about 0.3 μM (or 0.27 mmHg).29,30 Yoshikawa et al31 directly measured CO binding to purified anaerobic COX using infrared spectroscopy and found a Kd of 0.3 μM, identical to the CO Ki, as is predicted for a simple competitive inhibitor. Wohlrab and Ogunmola32 found a similar Kd of 0.46 μM (or 0.42 mmHg) for CO binding to COX in intact liver mitochondria. The corresponding Km (or Kd) of mitochondria for O2 is less certain and varies with the metabolic state, but is probably in the range of 0.1 to 0.8 μM.33,34 Petersen29 found a Km for O2 of 0.75 μM using the same purified COX enzyme assay used to determine the Ki of CO. Using this value of Km, the value of MCOX should be 0.75/0.3 or 2.5. These values of Km, Ki, and MCOX will be used throughout this report.
This apparently high CO–COX affinity of 0.3 μM (or 0.027 mmHg; or 35 ppm) does not, at first, seem consistent with the classic observation that CO has relatively minor effects on the rate of O2 consumption of whole organs.35,36 For example, Glabe et al,36 using isolated saline perfused rat hearts, measured the maximum O2 consumption and cellular adenosine triphosphate (ATP) and phosphocreatine using proton nuclear magnetic resonance and found no significant change in any of these variables, even for a saline PCO as high as a 100 mmHg (with a saline of 540 mmHg). Assuming that the at the COX binding site is 100 mmHg for this perfusion of 540 mmHg, and using Equation 13, one would predict that a PCO of 100 mmHg should produce about a 70% COX inhibition. However, this 70% COX inhibition would not be expected to have a major effect on organ oxidative metabolism because of the “branching” and “cushioning” phenomena originally described by Chance et al.37 Even though most of the COX is inhibited by CO, the remaining uninhibited COX can branch out and oxidize the neighboring CO-inhibited COX chains, leading to relatively normal ATP synthesis.33,37,38
A major focus of recent CO signaling research is the formation of ROS secondary to COX inhibition by CO. Zuckerbraun et al28 have shown that relatively low concentrations of CO (1 hour exposure to 250 ppm, or 0.19 mmHg) in a cell culture system produced a large increase in ROS formation, seemingly due to CO inhibition of COX. There is no evidence that other heme-containing enzymes that assist in the removal of ROS (eg, superoxide dismutase [SOD] or catalase) are inhibited by these CO concentrations. Normally, there is a small basal leak of ROS from the respiratory transport chain complexes I–III that feed into COX (complex IV), and this leak is markedly increased when COX is partially inhibited, presumably because of a blockade at the terminal step accompanying a buildup in the electron driving pressure.39 Nitric oxide (NO) also inhibits COX (in a somewhat different manner), but this inhibition is not thought to be a major NO signaling pathway, because NO–COX affinity is 50-fold less than the affinity of NO for guanylate cyclase.38
Although ROS production historically has been regarded as a pathologic process, it recently has been proposed that ROS can serve as an important signaling mechanism.40,41 If so, CO-induced COX inhibition should be considered to be a physiological process, with the resultant ROS formation being the best validated CO second messenger system. As discussed, large increases in ROS generation can occur with minimal changes in overall respiration, thus providing the putative messenger in the absence of cell asphyxiation. The “specificity” of this ROS release presumably would be determined by the cellular systems that are modified by increased ROS exposure. ROS release is remarkably sensitive to low CO exposures. Thom et al42 measured rates of cell death and the cellular levels of antioxidant enzymes such as SOD as a function of time after short exposures to CO in a cell culture system. Just a 1-hour exposure to 100 ppm CO (or 0.08 mmHg) resulted in a significantly increased ROS and an increase in cell death at 18 days. A 1-hour exposure to 10 ppm CO did not increase death rates, but was associated with a 3-fold increase in SOD levels, which were, presumably, a surrogate signal of increased ROS formation. This 10 ppm CO (0.008 mmHg) is only 4× the normal blood level and is commonly observed in smokers.43
The quantitative relationship between COX binding and ROS release is poorly understood. Not commonly considered is that competition between O2 and CO for COX (Equation 13) makes this relationship strongly dependent on the at the COX binding site. In the typical cell culture experiments in petri plates42 or 96-well tissue culture plates28, the effective cell is poorly characterized. Peng and Palsson44 have shown that these tissue cultures at high cell density may become oxygen limited, ie, the at the COX site may be close to 0 mmHg. Using the CO Ki of 0.27 mmHg and Equation 13, the maximum possible COX–CO (ie, within the limit as approaches 0 mmHg) is 2.7% for the 10 ppm CO (0.0076 mmHg) in the Thom et al42 experiment and 41% for the 250 ppm CO in the Zuckerbraun et al28 experiment. This last prediction is in good agreement with the 50% COX inhibition directly measured by Zuckerbraun et al28 at 250 ppm CO. Despite this 50% COX inhibition, Zuckerbraun et al28 found that there was no significant change in cellular ATP, consistent with the classic “branching” effect Chance et al37 described. If this quantitative analysis is correct, it indicates that very small fractional CO–COX binding (eg, 2.7%) can have significant physiological effects, presumably by producing a relatively large change in the rate of ROS production. It also implies that, specifically, it is important to consider the competing O2 concentration when evaluating CO actions. For example, Zhang et al45 have shown that CO concentrations of only 15 ppm have a major anti-apoptotic effect on cultured cells that have undergone an anoxia–reoxygenation protocol, consistent with the prediction that CO–COX inhibition will be maximized in anoxic conditions.
Specific CO sensors or receptors
Initially, a second messenger role for CO was based on the concept that this gas interacted with specific receptors and thus might have a variety of specific actions (in contrast to the non-specific activity that seemingly would result from COX inhibition). However, there are surprisingly little data with regard to the identity or structure of such CO receptors. Initial enthusiasm for a messenger role for CO was based on the demonstration that exposure to this gas activated soluble guanylate cyclase (sGC), a heme-containing protein, resulting in production of cyclic guanosine monophosphate (analogous to the action of NO).46,47 This concept was based on the demonstration of the activation of whole cell K+ currents in corneal epithelial cells and jejunal smooth muscle, when equilibrated with a very high PCO of 7.6 mmHg.47,48 Subsequent observations showing that CO produced only a maximum 4.4-fold activation of purified sGC compared to the 130-fold activation with NO48 and that the sGC–CO affinity was low (with a Km of about 10 μM49) raised serious doubts about sGC as a mediator of CO action.50 This sGC example is illustrative of a major limitation of many proposed specific CO second messenger actions. The observed responses to CO could simply be a non-specific reaction to the tissue asphyxiation that results from the massively superphysiological CO concentrations employed (eg, 7.6 mmHg in the sGC example, which is 4,000× the normal plasma CO).
The classical second messenger reacts with a receptor that provides specificity to the signaling. Given the extensive literature ascribing a messenger function for CO, there is a surprising paucity of data identifying the CO-specific receptors. The best understood CO-mediated regulatory protein is the bacterial transcription factor CO oxidation activator (CooA), whose structure and function have been characterized in great detail.9,51 This protein is a CO-sensitive transcription activator in the bacterium Rhodospirillum rubrum, which can live on CO in anaerobic conditions. The activation of CooA is specific for CO and does not occur with O2, indicating that heme binding sites can be specific for CO sensing. The only reported homologous mammalian protein is involved in the neuronal circadian rhythm control system,52 and there is suggestive evidence that CO may have a role in the mammalian control of diurnal rhythm.53–55 The CooA affinity is relatively low, with a P50 of 2.2 μM (or 2 mmHg),56 about 1,000× higher than the normal blood CO (Table 1), and, as discussed in detail in the “Quantitative modeling of the steady state and time-dependent tissue CO in the presence of the blood sink” section, it is unlikely that normal tissue concentrations can reach this level.
The best characterized mammalian high-affinity CO receptors are the large conductance Ca++- and voltage-activated K+ channels (BKCa, also referred to as Slo1 or MaxiK), which are expressed in almost every cell type and are involved in vasodilation, oxygen sensing, neuronal excitability, transmitter release, and multiple other functions.57 The strongest evidence that BKCa channels are CO receptors is that they can be activated by 1 μM CO in excised cell membrane patches that are free of COX, and presumably, other regulatory heme proteins.58,59 The main experimental focus has been on the involvement of this regulatory system in the control of smooth muscle vascular resistance, reviewed in great detail by Leffler et al.11 The most sensitive tissue described to date is the piglet pial vessels, with applied concentrations as low as 0.1 μM (0.09 mmHg) producing a transient dilatation.60 Arterioles in adult pigs and in other mammals of all ages are appreciably less sensitive to CO.61 For example, the rat tail artery model requires CO concentrations of about 10 μM to produce a relaxation that is about 25% of the maximum CO-induced relaxation.62 The constitutive form of HO (HO-2) is expressed in high concentrations in central nervous system arterial endothelial cells and in their closely associated astrocytes,62 and the inducible form (HO-1) is highly expressed in peripheral vascular smooth muscle.63 The structural mechanism of the CO activation of BKCa is not well understood, but seems to involve an interaction between BKCa and cellular heme.58,64 A similar interaction of a calcium-activated potassium channel, heme, and CO has been described in a plant (pollen tube) system,65 suggesting that this CO regulatory mechanism may have an ancient lineage. Based on the above, it appears that tissue HO must raise the local tissue CO to at least 0.1 μM to activate arteriolar BKCa. The physiology required for the generation of such a tissue concentration in the face of diffusion to blood is addressed quantitatively in the “Quantitative modeling of the steady state and time-dependent tissue CO in the presence of the blood sink” section.
Oxygen is a substrate for the HO reaction (Equation 1), and it has been suggested that HO is the carotid body O2 receptor, with high O2 increasing HO activity that then increases the rate of CO production, activating a BKCa channel.66 One difficultly with this concept is that HO has a very high O2 affinity, with a P50 of 0.013 to 0.03 μM,67 corresponding to a of 0.0094 to 0.022 mmHg (using an O2 solubility of 1.38 μmol/L/mmHg68), far below the O2 sensitivity range of the carotid body. A further complication is that HO knockout mice apparently have normal carotid body oxygen sensing.69
There is also suggestive evidence that the epithelial Na+ channel (ENaC) is a high-affinity CO receptor. The ENaC activity of excised membrane patches from mouse kidney cortical collecting duct cells is stimulated by the addition of soluble heme at a concentration similar to that in the intact cell, with a Kd of 23 nM, and this heme activation is increased by the addition of CO at concentrations as low as 0.1 μM.70 In addition, O2 is required to maintain the stimulatory effect of heme, suggesting that the CO production by membrane-associated HO is required for the heme–ENaC interaction and that this system might be another O2 sensor.70 A potentially important clinical application of CO is based on the observation that inhalation of CO at concentrations of only 100 ppm (0.076 mmHg; 0.084 μM) provides protection against hypoxic lung injury.71 It has been proposed that ENaC is the receptor responsible for this protection,72 and Althaus et al72 demonstrated that ventilation of isolated rabbit lungs with 250 ppm CO (0.19 μM) decreased alveolar sodium absorption and fluid clearance. Although they also showed that CO inhibited the apical amiloride-sensitive current (presumably ENaC), this might be an indirect result of inhibiting COX, since it required very high concentrations (100 μM) of a CO-releasing molecule, CORM-3.73 Extrapolation from such studies to a messenger role for endogenously produced CO requires demonstration that locally produced CO raises the tissue CO concentration to levels comparable to those achieved with exogenous administration of CO.
Although CO has been proposed to be a regulator of many other ion channel types, including voltage-activated K+, L-type Ca++ channels, ligand-gated P2X receptors, and TREK1 P domain K+ channels, the evidence that these channels are specific CO receptors is much weaker.73 One limitation of many of these studies is that the exact CO concentration used has not been well-characterized, because the CO was administered as various forms of CO-releasing molecules (CORMs).73 These molecules release CO, either spontaneously or when exposed to light,74 in cell culture media or organ perfusion systems. The resulting CO concentration depends on the balance between the rate of CORM CO release and the rate of loss of CO to the environment, factors that are poorly characterized. Depending on the CORM molecule used and the environmental conditions (eg, pH), these agents release CO with half-times varying from a few minutes to hours.75 A conventional application is to use a CORM bathing concentration of 30 μM, which could produce a CO concentration ranging from 30 μM, if completely and rapidly released, down to much lower concentrations if released slowly. However, the final CO concentration is unlikely to be less than about 5 μM, a relatively high concentration that markedly inhibits COX. Consistent with this evidence, there is suggestive evidence that the CO activation of the L-type Ca++ channel76 and the delayed rectifier Kv2.1 potassium channel77 are secondary to COX inhibition and the corresponding ROS production.
The binding of CO to several clotting factors (fibrinogen, plasmin, and α2-antiplasmin) results in increased rate of clot formation and stronger clots.78 It is thought that the CO interacts with these proteins via an associated heme.79,80 Since the CO concentrations required in in vitro assays are very large (eg, 50 μM CORM80), it is unlikely that this CO binding is physiologically significant.
HO activity, heme substrate concentration, and the endogenous rate of CO tissue production
As discussed in the “Specific CO sensors or receptors” section, the lowest PCO concentration shown to produce an observable specific second messenger effect in the most sensitive tissues (pig pial vessels) is about 0.09 mmHg (0.1 μM). Thus, the tissue CO concentration must be raised at least 50× (and in most systems 500×) higher than the usual blood concentration of 0.0019 μM (Table 1) to produce a physiological response. In other words, CO production by tissue HO must be sufficiently rapid to at least temporarily maintain a concentration of 0.1 μM in the presence of diffusion to the local blood sink. This section and Table 2 summarize the very limited available experimental data on the rates of the endogenous tissue CO production rates.
Table 2.
Hemoxygenase activity and endogenous CO production rates
| Tissue | Rate nmol/g cell/h | Method | Comments |
|---|---|---|---|
| Average human CO output | |||
| Total daily output81 | 0.27 | ||
| Maximum available CO signaling | 0.02 | Assume 7.5% of total | = MaxCO |
| Rat heme oxygenase activity110 | Hematin substrate | Optimal conditions | |
| Spleen | 580** | ||
| Brain | 531** | ||
| Heart | 96** | ||
| Aortic smooth muscle cells111 | 8* | CO production | Maximally stimulated |
| Olfactory receptor neurons49 | 26* | 14CO from 14C glycine | Maximally stimulated |
| Olfactory receptor neurons49 | 10* | Bilirubin production | Maximally stimulated |
| Astrocyte112 | 10* | CO production | Maximally stimulated |
| Piglet cerebral vessel113 | 140* | CO production | Maximally stimulated |
| Cerebellar slices114 | 3,400 | CO capture by Hb | Basal |
Whole animal measurements of CO production provide insight into the maximal possible rate of second messenger CO production. The first entry in Table 2 is the total rate of human CO production of about 0.27 nmol/g/h.81 Of this, about 75% is accounted for by red cell turnover, and another 10%–15% is produced by the liver (eg, cytochrome P450 turnover).82 To this should be added the basal turnover of the rest of the body’s heme. For example, a 70 kg human has about 30 kg83 of muscle containing about 4.4 g Mb/kg muscle,84 or a total of 7.8 mmole of Mb. If this muscle Mb turns over just once/year it would release about 0.012 nmol/g/h, or another 5% of the total CO turnover. In addition, another 5%–10% of the total production could result from the turnover of muscle cytochrome. Thus, the routine “housekeeping” turnover of heme can readily account for the entire CO output of healthy subjects, and CO produced for signaling can only be a trivial fraction of the normal total production. For the purposes of discussion in this paper, we will assume that a maximum 7.5% of the total daily CO production, or about 0.02 nmol/g/h, could possibly be associated with specific CO second messenger signaling. This value, which is possibly an overestimate, will be referred to as the MaxCO rate (Table 2).
Endogenous tissue CO production is severely substrate limited. Tissue HO activity assayed in the presence of excess substrate (hematin) releases CO at a rate thousands of times greater than MaxCO (Table 2). Even a tissue with a relatively low HO activity, such as the heart, in the presence of unlimited substrate, produces CO at a rate that is 4,800× MaxCO. These tissue HO assays measure the total HO activity (HO-1 plus HO-2). Tissue HO activity is variable because HO-1 induction results in marked changes in the total activity, with 5-fold increases measured in whole liver85 and 10-fold increases seen in cell culture.86
In theory, if the physiological endogenous tissue heme substrate concentration was known, HO enzyme activities could be used to predict endogenous CO production. However, since there is very little knowledge of what constitutes the heme source for signaling reactions, much less the effective concentration of this substrate, it is not possible to predict CO production from HO activity, and such information must be derived from direct measurements of tissue CO release.
There have been only a few direct experimental measurements of endogenous CO tissue production in the absence of additional heme substrate (summarized in Table 2). Since CO production is low under physiological conditions, virtually all non-heme-enriched studies have employed non-physiological conditions (eg, enrichment with glutamate) to enhance CO release. It can be seen from Table 2 that the reported local tissue production rates are from 400 to 170,000× greater than MaxCO. The non-physiological nature of some of these values is illustrated by the production rate reported for cerebellar slices (3,400 nmol/g cell/h). Just 6 g of this cerebellar tissue would generate CO at the same rate as does the total human body. Obviously, these measurements cannot represent the average CO production of these tissues and must correspond to some sort of stimulated condition, presumably initiated by the experimental tissue conditions.
The next section, the “Quantitative modeling of the steady state and time-dependent tissue CO in the presence of the blood sink” section, discusses the tissue PCO produced by the endogenous tissue CO production. As a benchmark, the reported rate (M) of CO produced by piglet cerebral vessels (140 nmol/g cell/h; Table 2) will be assumed to be the best estimate of the maximum tissue M value. Since extrapolation of this rate to all cells in the body yields a value 7,000× greater than MaxCO, this putative messenger CO production rate must occur either in very limited tissue regions (eg, pial vessels) and/or for very short time periods. For example, just 10 g of tissue producing CO at this rate equals the maximum total human CO production rate available for signaling.
A neglected but important question is the nature of the heme substrate that supports this messenger CO production, a question that remains unresolved.87 Although a “free heme pool” has been proposed, there is no evidence to support this proposal.87 Liver and erythroid cells are known to have rapid rates of heme enzyme and Hb synthesis, respectively, and many of the details of the control of heme biosynthesis in these tissues are well understood. Although other tissues are thought not to possess extensive biochemical pathways dedicated to heme synthesis,87 this contention is not supported by measurements of messenger ribonucleic acid (mRNA) levels of 5-aminolevulinic acid synthase-1, which is thought to be the rate-limiting step in heme synthesis, with the brain, kidney, and heart having 5-aminolevulinic acid synthase-1 levels at about 40% of the liver.88 There seems to be no question that about 75% of the total human CO production (and correspondingly, heme production and breakdown) is due to red blood cell turnover, with most of the remaining CO production due to turnover of heme in the liver.89 The rates of endogenous CO production listed in Table 2 for maximally stimulated tissues, if correct, correspond to huge rates of heme turnover. For example, of the total human daily CO production of 19 μmole/70 kg/h,81 about 15% takes place in the liver,90 corresponding to a rate of liver CO production (or heme synthesis and breakdown) of about 1.9 nmol/g/h. This rate is only 1/10th to 1/100th of the rates listed in Table 2 for arteriolar or nerve cells. Although these rates might be present for only relatively short time periods, they would still require large amount of heme synthesis. For example, the major heme protein in smooth muscle is COX, with a maximal concentration of 30 nmol/g91 (smooth muscle has very low Mb concentrations92). The reported pial arterial CO production of 140 nmol/g/h (Table 2) would require turnover of the entire heme COX content of the arteriole wall (two hemes per COX) every 25 minutes. Clearly, much more study of the details of the heme substrate utilized by HO is required for an understanding of the biochemistry of the CO signaling mechanism.
Quantitative modeling of the steady state and time-dependent tissue CO in the presence of the blood sink
As discussed, the local CO concentration required for a biological effect, eg, in piglet pial arterioles, is at least 50× greater than the blood concentration. Although it is conventionally assumed that such high tissue concentrations can be achieved locally via HO-catalyzed release of CO,10 the quantitative requirements of this assumption have not been previously evaluated. In this section, a physical model of a tissue region bathed by the blood Hb sink will be presented. Two different geometric arrangements will be considered. The first, representative of an arteriole or artery, consists of a cylindrical shell (the arteriolar wall) with a blood-perfused lumen. The second is representative of a solid tissue (eg, brain) penetrated by an array of capillaries. Although both of these models are idealized, they provide first-order estimates of the predicted local tissue CO concentration as a function of the rate of production of CO and the geometric diffusion distances between tissue and blood.
Figure 3 shows a diagram of the arteriolar model. It is assumed that CO is produced at a uniform rate M throughout the wall of the pial arteriole or the rat tail artery. The PCO in the vascular lumen is fixed at PB, the normal blood value. It is assumed that the outer wall of the vessel is impermeable to CO. This assumption actually leads to an overestimate of the true concentration in the wall, because there will be some diffusion through the outer wall toward more distant circulating blood. For pial vessels, there may be an additional CO production from the astrocytes that form a sheath around the arteriole.93 Since this sheath is only about 1 μm thick,94 it will be assumed that it can be simply added to the vessel wall thickness.
Figure 3.
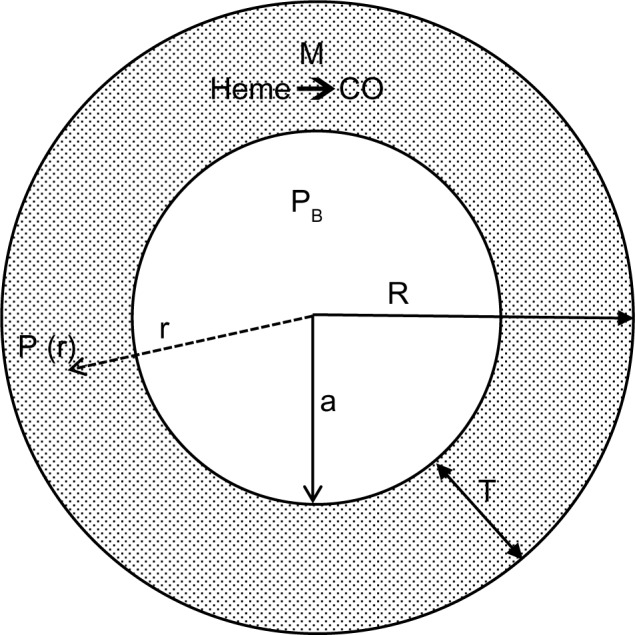
Diagram of the model used to model the pial arteriole or the rat tail artery. It is assumed that there is uniform rate of CO production (M) in the arterial wall (thickness = T). The outer wall of the artery is assumed to be impermeable to CO, and the diffusion of CO to the blood sink (PCO = PB) sets up a steady state PCO gradient (= P(r)) in the vessel wall.
Abbreviation: PCO, CO partial pressure; P(r), CO partial pressure at radial position r in arteriole wall; PB, CO partial pressure of blood; a, radius of arteriole lumen; R, outer wall arteriole radius; T, arteriole wall thickness.
In the steady state, the PCO in the vessel wall (P(r)) as a function of the radial distance (r) from the center of the vessel is described by the Krogh–Erlang equation (with a rate M of CO production in place of the classically employed rate of oxygen consumption):95
| (14) |
where PB is the blood PCO, a is the radius of the blood lumen, and R is the outer radius of the arteriole (plus astrocyte for pial vessels), which is equal to (a + T), where T is the wall thickness, and K is the Krogh diffusivity = αD, where D is the tissue CO diffusion coefficient and α is the CO solubility (K =1.77×10−5 nmol/cm/mmHg/s; Table 1). The maximum tissue CO concentration (Pmax) is at the outer vessel wall (r = R):
| (15) |
The pial arteriole radius (R) is about 30 μm,60,67 and because the ratio of (arterial wall thickness)/R is about 0.2,96 T (including the 1 μm thick astrocyte) is about 7 μm, and a =23 μm. It will be assumed that PB is equal to the normal human blood value of 0.0019 mmHg (Table 1). Note that although most of the tissue CO is bound by tissue heme and is not free, this binding does not enter the steady state solution (Equation 14), because only the diffusion of the free CO, characterized by the PCO, needs to be considered.
Figure 4 shows a plot of Equation 14 of the pial arteriole tissue PCO as a function of the radial distance from the inner wall of the vessel. (All calculations described here were performed using the Maple (Maplesoft®) plotting and equation solvers). The solid lines correspond to different values of M, the rate of tissue CO production. It can be seen that for M =280 nmol/g tissue/h (the black line; twice the reported maximal experimental value for pial vessels), the maximum tissue PCO is only 0.0031 mmHg (63% greater than the blood), 30-fold less than the minimum PCO required for an observable biological effect (0.1 mmHg; Table 1). To raise tissue PCO to the biologically effective value of 0.1 mmHg requires an M of 28,000 nanomole/g tissue/h (green line, Figure 4), 200-fold greater than the maximal observed rate of pial arteriole tissue CO production. To put this requisite CO production rate (28,000 nmol/g/h) into physiological perspective, just 1.5 g of tissue producing CO at this rate for 1 hour would yield 7.5% of the total daily body CO production, which is the maximal estimate of the CO available for signaling in the entire human body. The above analysis indicates that the maximally observed rates of tissue CO production cannot raise the pial arteriolar PCO to the 0.1 mmHg value required for dilation, raising the question of what the function of this well-characterized CO response mechanism is. One possibility is that this CO response mechanism is only important during pathological conditions of low arterial perfusion, conditions when the blood would no longer function as an infinite sink. This is discussed in more detail in the concluding discussion (the “Discussion: does HO function via its release of CO?” section).
Figure 4.
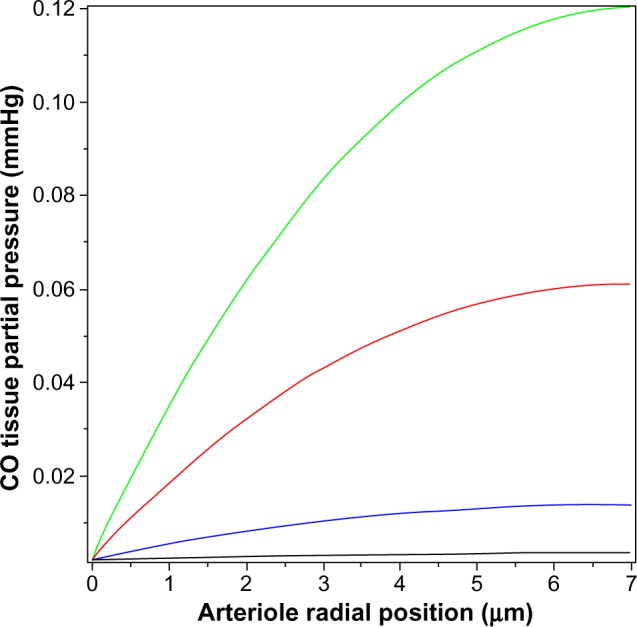
Steady state PCO in wall of pial arteriole as a function of the radial distance from the blood surface for varying rates of tissue CO production (M) (black line =280, blue line =2,800, red line =14,000, and green line =28,000 nmol/g cell/h).
Abbreviation: PCO, CO partial pressure.
The value of the tissue Pmax (Equation 15) for a given rate of CO production increases as the thickness of the tissue-producing region and the distance the CO must diffuse to reach the blood sink increases. The relative thinness of the pial arteriole (plus astrocyte) wall (≈7 μm) is the major factor constraining the ability of endogenous production to raise the tissue PCO. The rat tail artery, another CO responsive experimental model,62 has a much thicker wall of about 117 μm, with a luminal radius of about 93 μm,97 and would be expected to have a much higher tissue PCO for a given endogenous production rate. (The walls of vessels of this size are apparently avascular98,99). However, the lowest stimulatory CO concentration observed for these vessels is about 10 mmHg.62 Figure 5 shows the corresponding plot of tissue PCO for a rat tail artery with these dimensions. It can be seen that the tissue PCO is much greater than that of the pial arteriole (Figure 4) for the same values of M. The lowest production rate plotted in Figure 5 is 280 nmol/g/h (35× the 8 nmol/g/h production rate observed for cultured aortic muscle tissue), which yields a Pmax of less than 0.4 mmHg. As shown in Figure 5, the CO production rate would have to be on the order of 14,000 nmol/g/h to develop a CO concentration in excess of the 10 mmHg required for significant rat tail artery relaxation. Thus, even for this much thicker wall artery, the measured local rate of CO production is not sufficient to produce the 10 mmHg PCO values that are required for activation of the effector mechanism.
Figure 5.
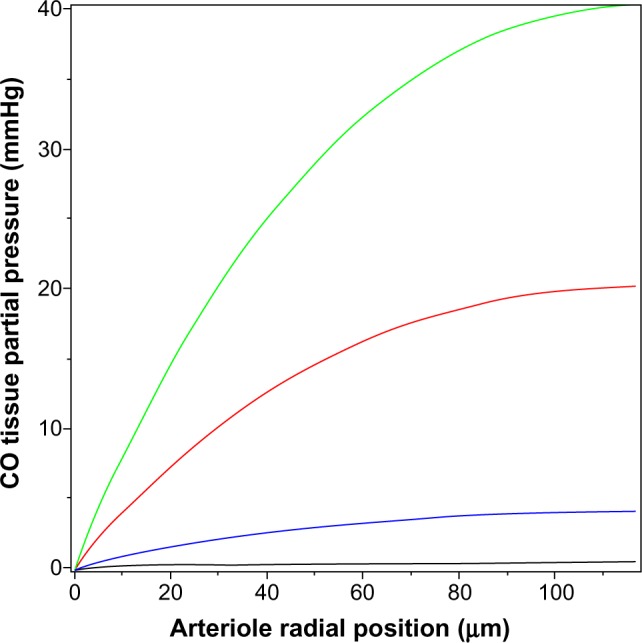
Steady state PCO in wall of rat tail artery as a function of the radial distance from the blood surface for varying rates of tissue CO production (M) (black line =280, blue line =2,800, red line =14,000, and green line =28,000 nmol/g cell/h).
Abbreviation: PCO, CO partial pressure.
The above two cases of CO responsiveness in vessel walls are special cases that lend themselves to relatively straightforward modeling. In contrast, most of the putative signaling actions of CO are for solid tissues, such as the brain,8 which have much more complicated blood perfusion geometry. As a first approximation, the tissue CO in the brain tissue can again be modeled using the Krogh cylinder model,95 based on the tissue cross-section shown in Figure 6. It is assumed that the capillaries have a symmetric hexagonal arrangement. Since the cylindrical surface represented by the dashed line in Figure 6 is equidistant between neighboring capillaries, there will be no diffusion of CO across this surface, and it can be assumed to be an impermeable barrier. Thus, the cross-hatched cylindrical region becomes mathematically identical to the arteriole wall (Figure 3), with a tissue radius R equal to one-half the intercapillary distance, and a radius (a) equal to the capillary radius. In the following calculation, it will be assumed that R =50 μm and a =3 μm, and that the capillary blood acts as a sink, with a PCO equal to that of the blood (PB). This value for R is about twice the anatomical value and assumes that only every other capillary is perfused at rest. Figure 7 shows the corresponding plot of the Krogh cylinder tissue PCO as a function of the radial distance from the capillary wall for tissue production rates (M) of 22, 220, and 2,200 nmol/g/h. It can be seen that in order to reach a CO Pmax of 1 mmHg (eg, the concentration required for BKCa channel activation) an M of about 2,200 nmol/g/h is required. This is 16× the maximal experimental pial artery M, and 220× the maximal M for astrocytes (Table 2), and 1 g of tissue producing CO at this rate would account for the total maximal rate of CO production potentially available for signaling purposes. Thus, it seems unlikely that endogenous tissue CO production could raise tissue PCO to 1 mmHg in the presence of a normal blood perfusion.
Figure 6.

Diagram of the Krogh cylinder model. Because of the hexagonally symmetrical arrangement of the capillaries, there should be no diffusive CO flux across the cylindrical surface, indicated by the dashed lines, and thus, this surface can be assumed to be an impermeable barrier. The cross-hatched cylindrical region then becomes an isolated tissue region, and the diffusion equation for this region becomes mathematically identical to the arterial tissue region described in Figure 3.
Abbreviation: M, tissue CO production rate.
Figure 7.
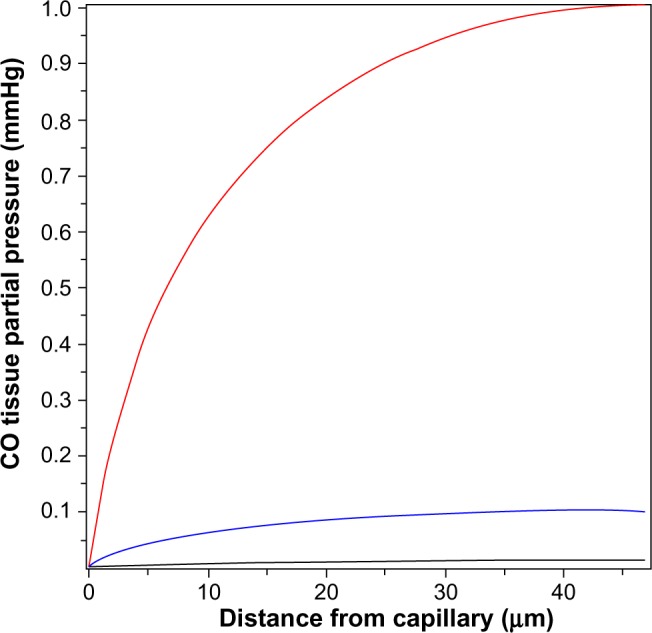
Steady state PCO in wall of Krogh cylinder solid tissue model as a function of the radial distance from the capillary surface for endogenous CO tissue production rates of 22 (black line), 220 (blue line), and 2,200 (red line) nmol/g cell/h.
Abbreviation: PCO, CO partial pressure.
In contrast, to raise tissue CO Pmax (Equation 15) to the 10 ppm (0.0076 mmHg) required to produce a significant ROS signal (as described in the “COX binding and ROS formation” section) only requires an M of 13 nmol/g/h, roughly equal to the rate of CO tissue production of olfactory neurons or astrocytes (Table 2). This suggests that the control of ROS production is a realistic candidate for a CO signaling mechanism (as described in more detail in the “Discussion: does HO function via its release of CO?” section).
The tissue CO production required to raise the tissue PCO to 1 mmHg also can be estimated in a totally independent fashion using the following calculation. Figure 8 shows a schematic diagram of the blood–brain tissue exchange for both O2 and CO. The driving force for O2 tissue uptake is the difference between the average in the capillary blood (about 45 mmHg) and the in the brain tissue (about 5 mmHg), and the driving force for CO leaving the tissue is the difference between the average in the tissue (1 mmHg) and the in the blood (0.0019 mmHg). Since the complex tissue geometry and diffusion distances are identical for O2 and CO, the CO flux (JCO) should be related to the O2 flux by the relation:
| (16) |
where KCO and are the CO and O2 tissue diffusivities, and the ratio ( /KCO) is about 1.32.68,100 A typical value of human brain oxygen consumption is about 120 μm/g/h.101 Thus, the CO flux JCO from the brain to the blood, ie, the rate of tissue CO production for a of 1 mmHg and , is about 2,270 nmol/g/h – surprisingly close to the value of 2,200 nmol/g/h estimated using the brain Krogh cylinder model, as described earlier in this section.
Figure 8.
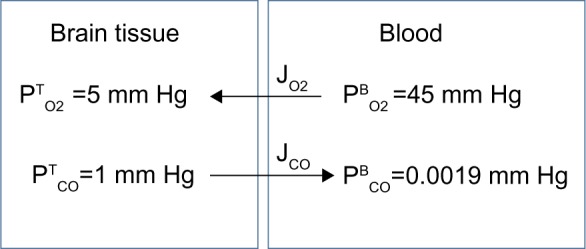
Schematic diagram showing the diffusional exchange of O2 and CO between brain tissue and capillary blood where and JCO are the diffusional O2 and CO fluxes, respectively. Because the geometry is identical for O2 and CO, the fluxes should be related by the relative O2 and CO tissue diffusivities.
Another question that can be examined quantitatively is the time constant for changes in the tissue PCO. If, for example, CO was a regulator of arteriolar dilation, then changes in PCO presumably should occur in times of minutes or less. To answer this question, one must solve the general time-dependent equation in cylindrical coordinates:
| (17) |
where α is the CO solubility (Table 1), K is the tissue CO diffusivity, and M is the rate of tissue CO production. This equation depends on tissue heme CO binding and buffering, and the critical factor that accounts for this buffering in Equation 17 is f, which is defined as the fraction of the tissue CO that is free (ie, unbound). Assuming that the binding results from a single saturable binding site with concentration A and association constant KA, f is described by:
| (18) |
This model (Equations 17 and 18) will be applied in this discussion to determine the time course of the rise in tissue CO in the rat tail artery model described previously in this section. It will be assumed that at time (t) =0, the CO production is turned on at a rate (M) of 2,800 nmol/g cell/h, which, at the steady state, produces the tissue profile described by the blue line in Figure 5. Because smooth muscle has very low Mb concentration,92 it is assumed that the main CO-binding protein is COX. As discussed in the “Hb and Mb CO binding and pharmacokinetics” section, O2 and CO are competing for the same binding site in COX, described by Equation 12, so that the apparent KA for CO will depend on the . In the following calculation, it will be assumed that the tissue is fixed at 3 mmHg and that the apparent CO Kd is 1 μM, corresponding to a KA of 1 μM−1. The tissue COX concentration (A) will be set at 30 μM, the value reported for heart muscle.91 Equations 17 and 18 were numerically integrated to generate the curves shown in Figure 9, which plot the tissue PCO as a function of distance from the blood for times of 10 seconds (black curve), 50 seconds (red curve), 100 seconds (green curve), and infinity (blue curve) after turning on the CO tissue production. It can be seen that for the large rate of production (M =2,800 nmol/g cell/h) required to raise the steady state tissue PCO to 4 mmHg, the buffering effect is relatively small, and the steady state (t = infinity) is reached in about 2 minutes. Thus, for example, CO might be able to regulate arteriolar blood flow on a minute-to-minute time scale if this extremely high rate of tissue CO production could be reached.
Figure 9.
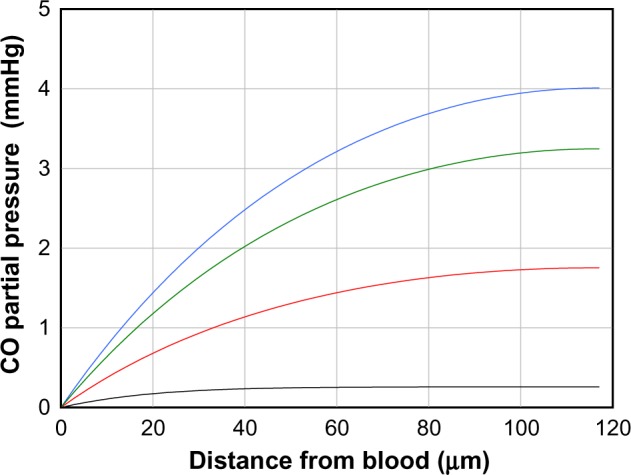
Artery wall CO partial pressure (PCO) as a function of distance from the blood surface for times of 10 seconds (black curve), 50 seconds (red curve), 100 seconds (green curve), and an infinite time (blue curve), after suddenly turning on tissue CO production at a rate of 2,800 nM/g cell/h. The artery has a thickness of 117 μm.
Discussion: does HO function via its release of CO?
It is surprising that the above question should be raised, given the voluminous literature devoted to HO measurements and CO signaling. However, as discussed in this review, the available data do not provide clear-cut evidence that CO serves as the intracellular effector of HO activity in normally functioning tissue. The modulation of HO-1 activity that has been observed in nearly every tissue studied strongly suggests that this enzyme must have an important regulatory role in cell physiology. As emphasized in the “Introduction” section, HO has three other functions besides CO production: 1) catabolism of heme, 2) recycling of Fe2+, and 3) production of the anti-oxidant bilirubin. The factors that induce HO-1 are primarily events that tend to produce apoptosis or cell death, such as oxidative stress, heat shock, ischemia, radiation, hypoxia, or hyperoxia, all of which would be expected to increase heme turnover, with a consequent need for increased HO. In the great majority of studies,4 the question of the effector associated with this HO induction is simply ignored. Rather, HO modulation is regarded as the central event, and CO is assumed to be the mediator of HO activity. Rarely discussed are important quantitative physiological questions concerning this pathway, such as the tissue PCO required for the biological effect, the rate of tissue CO production, the identity of the CO receptor, and the influence of diffusion from tissue to blood on intracellular CO. These questions are the focus of this review.
The belief in CO as the mediator of HO activity is primarily based on the observations that CO is produced by tissue and that CO exposure alters tissue function in both in vitro and in vivo experiments. As discussed in the “HO activity, heme substrate concentration, and the endogenous rate of CO tissue production” section, CO released during the routine turnover of Hb, hepatic heme, or other heme proteins (eg, muscle Mb) can readily account for the total daily CO production (about 0.5 mmol) of normal subjects. Thus, there is no need to teleologically hypothesize a useful function for CO, eg, intracellular signaling, simply based on CO production in the body. While it is not possible to exclude the possibility that a small fraction of CO is produced as a messenger, this production must be very small, ranging from 0%–7.5% of total production, or a maximal rate of 0.02 nmol/g/h (previously defined as MaxCO). To put this value of MaxCO in perspective, a tissue production rate of 100× MaxCO will only raise tissue PCO to 0.0028 mmHg (using the brain Krogh cylinder model, discussed in the “Quantitative modeling of the steady state and time-dependent tissue CO in the presence of the blood sink” section), less than twice the plasma PCO and far below the level required for any known CO signaling mechanism.
The reported rates of “physiological” endogenous CO production by a variety of tissue preparations are surprisingly large, 400 to 170,000× greater than MaxCO (Table 2). These clearly cannot represent the daily average rate of CO production of these tissues. For example, if the relative low reported rate of 10 nmol/g/h for astrocytes and olfactory neurons were representative of brain tissue, then the human brain (weighing 1.5 kg) would produce 0.35 mmol/day, 70% of the total daily human CO output, which is far in excess (10×) the maximal total CO possibly available for signaling in humans. One possible explanation for these high CO production rates measured in vitro is that the supposedly physiological experimental tissue conditions actually induce some cell damage, increasing the heme substrate concentration for HO.
Even though these CO production rates cannot represent average daily rates, production in highly localized tissue regions for short time periods could create very high local CO production rates that could trigger second messenger effects. The crucial experimental evidence to support a messenger function for CO would be the demonstration that intracellular CO production is capable of raising the cellular CO concentration to a level that is known to alter cell function. Not commonly considered is the extent to which rapid diffusion of CO from cells to blood limits such increases in intracellular CO concentration. As discussed in the “Specific CO sensors or receptors” section, the most sensitive specific CO receptor-mediated function is the dilatation of the piglet pial artery, which responds to an experimental PCO of as low as 0.1 mmHg (presumably via activation of the BKCa channel receptor). However, as shown in the “Quantitative modeling of the steady state and time-dependent tissue CO in the presence of the blood sink” section, because of the short diffusion distances between the arteriole wall and luminal blood, even the relatively high experimental in vitro pial arteriolar production rate of 140 nmol/g/h can only raise the tissue CO concentration to a maximum PCO of 0.0025 mmHg. This is only 30% greater than the blood PCO (0.0019 mmHg), and 40-fold less than the 0.1 mmHg, required for the minimal arteriolar dilation observed with exogenous CO administration. In order to raise the tissue piglet artery PCO to 0.1 mmHg, given the rapid diffusion of CO into blood, requires rates of tissue CO production that are 100× the experimentally observed maximum tissue production rate (Figure 4).
If uptake by blood renders endogenous CO release grossly insufficient to serve as an effector in the most CO-responsive tissue, it seems necessary to reconsider the concept that CO acts as a tissue messenger, as nitric oxide does, in normally perfused tissue. The possibility remains that a CO regulatory mechanism might become important under conditions in which the blood sink is disabled, ie, conditions with significantly decreased blood flow. For example, piglets’ brain CO production (as estimated from changes in cerebral spinal fluid CO) was acutely increased by a factor of 2.3 in response to hypotension,102 an increase that would not be expected to cause a rise in cellular CO sufficient to induce vascular dilatation if tissue perfusion remained normal. However, the combination of this relatively modest increase in CO production with poor perfusion could appreciably increase intracellular CO. Although entirely speculative, this CO might serve as an autoregulatory factor, acting only when blood flow has dropped to low enough levels that the perfusing blood ceases to act as a CO sink.
The failure to clearly identify specific CO receptor mechanism(s) has re-directed attention to the possibility that CO acts relatively non-specifically via its binding to and inhibition of COX. As discussed in the “COX binding and ROS formation” section, concentrations of CO of as low as 10 ppm (0.0076 mmHg) have been shown to increase ROS production secondary to COX inhibition. Thus, ROS production is by far the most sensitive potential CO signaling mechanism described to date, and this mechanism has been invoked as the explanation for the multiple physiological alterations observed with in vivo, low-dose CO exposure. For example, inhaled PCO of 25–250 ppm for 1 hour/day, 3 days/week for 10 weeks produces statistically significant decreases in vascular inflammation in a sickle cell mouse model.103 A similar regimen of inhaled 250 ppm CO for 1 hour/day has been shown to have an amazing variety of other effects such as reducing the rate of growth of a prostatic tumor cell line in nude mice,104 reducing pulmonary hypertension in rats,105 improving myeloid cell engraftment,106 and enhancing liver regeneration.107 This evidence suggests that this short exposure to CO and subsequent ROS production turns on a variety of downstream processes that can have profound physiological effects. As discussed in the “COX binding and ROS formation” section, because CO and O2 are competing for the same COX binding site, the rate of CO-induced COX ROS production should be critically dependent on the effective pO2. This might provide some specificity to low-dose whole animal CO administration – directing it to anoxic tissue regions such as tumors (eg, action of inhaled CO on prostatic tumors104) or regions where red cell sickling occurs (eg, action of inhaled CO on vascular inflammation in a sickle cell mouse model103).
These actions of a relatively low dose of CO exposure on COX ROS production are dramatic and raise the possibility that they reflect normal physiological control systems involving the endogenous release of CO via HO enzyme activity. However, it remains to be shown that endogenous CO production can raise tissue PCO to the required levels. Raising tissue PCO to 10 ppm (0.0076 mmHg), which is only 4× the normal blood PCO and is the lowest concentration for which a biological effect has been reported,42 requires a tissue CO production rate of about 13 nmol/g/h (using the brain Krogh cylinder model, discussed in the “Quantitative modeling of the steady state and time-dependent tissue CO in the presence of the blood sink” section). This endogenous rate of CO production seems feasible, because it is only 1.3× the CO production rate that has been reported for maximally stimulated astrocytes and olfactory receptor neurons (Table 2). However, the total body CO balance analysis (as discussed in the “HO activity, heme substrate concentration, and the endogenous rate of CO tissue production” section) places strong constraints on the normal spatial and temporal distribution of these enhanced rates. This rate of 13 nmol/g/h is 650× the maximum possible average signaling rate of CO production for the entire body (MaxCO). Only 93 g of tissue producing CO at this rate would account for our maximal estimate of total human CO production that might serve a signaling function. However, an important proviso of this CO balance analysis is that it is based on the average normal rate of total body CO production. The pathological conditions associated with HO-1 induction, such as oxidative stress, heat shock, ischemia, radiation, hypoxia, or hyperoxia would be expected to produce increased cell death, with a corresponding increased heme turnover and CO production. Just a 20% increase in total body CO produced via such a mechanism would correspond to a 370% increase in the CO production that might serve a signaling function. Support for this hypothesis would be provided by measurements showing increased total body CO production under stress conditions.
As discussed above, endogenous CO production could raise tissue CO to the levels that are produced by exogenous administration of 10 ppm CO, but it seems much less likely that endogenous CO production could mimic the tissue concentrations resulting from the 250 ppm inhaled CO that has become a staple of experimental whole animal studies. In mice, at the end of 1 hour dosing with 250 ppm inhaled CO, the plasma (and tissue) PCO should have reached a value of about 200 mmHg (as described in the “Hb and Mb CO binding and pharmacokinetics” section), which requires a 350 nmol/g/h rate of CO production, significantly larger than most of the reported tissue rates (Table 2). Just 100 g of tissue producing CO at this rate will produce 35 μmole/h of CO, twice the normal total human CO production rate, tripling the normal human CO excretion rate. Such levels of CO excretion have only been reported in patients with hemolysis.
In summary, we utilized the uniquely non-metabolizable nature of CO to quantitatively model the endogenous CO production required to achieve various intracellular CO concentrations, given the rapid diffusion of CO from cell to blood. This modeling suggests that under normally perfused conditions, intracellular CO production is seemingly insufficient to induce the specific second messenger functions that have been attributed to CO in healthy tissue. However, it is also clear that exogenous CO administration can induce multiple physiological effects. The effector in these studies is thought to be ROS, released secondary to CO-induced inhibition of COX. Whether such effects can be considered a “signaling” system, as opposed to a non-specific response to cell injury, is uncertain. Our calculations suggest that endogenous CO production is insufficient to induce the cellular CO concentrations achieved in the usual 250 ppm CO inhalation study. However, a few studies have shown that ROS production and alteration of tissue function can be produced by exposure to as low as 10 ppm CO. Since this low-dose CO exposure produces cellular CO concentrations that might be achieved via endogenous production, such ROS release appears to provide the most plausible mechanism by which CO could provide an intracellular messenger function.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Bernard C. Lecons sur les effects des substances toxiques et medicamenteuses. Paris: Bailliere; 1857. French. [PubMed] [Google Scholar]
- 2.Sjostrand T. The formation of carbon monoxide by the decomposition of haemoglobin in vivo. Acta Physiol Scand. 1952;26(4):338–344. doi: 10.1111/j.1748-1716.1952.tb00915.x. [DOI] [PubMed] [Google Scholar]
- 3.Ghosh D, LeVault KR, Brewer GJ. Dual-energy precursor and nuclear erythroid-related factor 2 activator treatment additively improve redox glutathione levels and neuron survival in aging and Alzheimer mouse neurons upstream of reactive oxygen species. Neurobiol Aging. 2014;35(1):179–190. doi: 10.1016/j.neurobiolaging.2013.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–554. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 5.Wu L, Wang R. Carbon monoxide: endogenous production, physiological functions, and pharmacological applications. Pharmacol Rev. 2005;57(4):585–630. doi: 10.1124/pr.57.4.3. [DOI] [PubMed] [Google Scholar]
- 6.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86(2):583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 7.Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annual review of pharmacology and toxicology. 2010;50:323–354. doi: 10.1146/annurev.pharmtox.010909.105600. [DOI] [PubMed] [Google Scholar]
- 8.Queiroga CS, Vercelli A, Vieira HL. Carbon monoxide and the CNS: challenges and achievements. Br J Pharmacol. 2014 Apr 24; doi: 10.1111/bph.12729. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gullotta F, di Masi A, Coletta M, Ascenzi P. CO metabolism, sensing, and signaling. Biofactors. 2012;38(1):1–13. doi: 10.1002/biof.192. [DOI] [PubMed] [Google Scholar]
- 10.Heinemann SH, Hoshi T, Westerhausen M, Schiller A. Carbon monoxide – physiology, detection and controlled release. Chem Commun (Camb) 2014;50(28):3644–3660. doi: 10.1039/c3cc49196j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leffler CW, Parfenova H, Jaggar JH. Carbon monoxide as an endogenous vascular modulator. Am J Physiol Heart Circ Physiol. 2011;301(1):H1–H11. doi: 10.1152/ajpheart.00230.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maines MD, Gibbs PE. 30 some years of heme oxygenase: from a “molecular wrecking ball” to a “mesmerizing” trigger of cellular events. Biochem Biophys Res Commun. 2005;338(1):568–577. doi: 10.1016/j.bbrc.2005.08.121. [DOI] [PubMed] [Google Scholar]
- 13.Hanafy KA, Oh J, Otterbein LE. Carbon monoxide and the brain: time to rethink the dogma. Curr Pharm Des. 2013;19(15):2771–2775. doi: 10.2174/1381612811319150013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agostoni A, Stabilini R, Viggiano G, Luzzana M, Samaja M. Influence of capillary and tissue PO2 on carbon monoxide binding to myoglobin: a theoretical evaluation. Microvasc Res. 1980;20(1):81–87. doi: 10.1016/0026-2862(80)90021-7. [DOI] [PubMed] [Google Scholar]
- 15.Roughton FJW, DeLand EC, Kernohan JC, Serveringhaus JW. Some recent studies of the oxyhaemoglobin dissociation curve of human blood under physiological conditions and the fitting of the Adair equation to the standard curve; Paper presented at: Proceeding of the Alfred Benzon Symposium IV; Copenhagen, Denmark. 1972. [Google Scholar]
- 16.Roughton FJ. The equilibrium of carbon monoxide with human hemoglobin in whole blood. Ann N Y Acad Sci. 1970;174(1):177–188. doi: 10.1111/j.1749-6632.1970.tb49784.x. [DOI] [PubMed] [Google Scholar]
- 17.Furne JK, Springfield JR, Ho SB, Levitt MD. Simplification of the end-alveolar carbon monoxide technique to assess erythrocyte survival. J Lab Clin Med. 2003;142(1):52–57. doi: 10.1016/S0022-2143(03)00086-6. [DOI] [PubMed] [Google Scholar]
- 18.Coburn RF, Forster RE, Kane PB. Considerations of the physiological variables that determine the blood carboxyhemoglobin concentration in man. J Clin Invest. 1965;44(11):1899–1910. doi: 10.1172/JCI105296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peterson JE, Stewart RD. Absorption and elimination of carbon monoxide by inactive young men. Arch Environ Health. 1970;21(2):165–171. doi: 10.1080/00039896.1970.10667215. [DOI] [PubMed] [Google Scholar]
- 20.Drabkin DL. The distribution of the chromoprotein, hemoglobin, myoglobin, and cytochrome c, in the tissues of different species and the relationship of the total content of each chromoprotein to body mass. J Biol Chem. 1950;1950(182):317–334. [Google Scholar]
- 21.Bruce EN, Bruce MC. A multicompartment model of carboxyhemoglobin and carboxymyoglobin responses to inhalation of carbon monoxide. J Appl Physiol (1985) 2003;95(3):1235–1247. doi: 10.1152/japplphysiol.00217.2003. [DOI] [PubMed] [Google Scholar]
- 22.Riches AC, Sharp JG, Thomas DB, Smith SV. Blood volume determination in the mouse. J Physiol. 1973;228(2):279–284. doi: 10.1113/jphysiol.1973.sp010086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guyton AC. Measurement of the respiratory volumes of laboratory animals. Am J Physiol. 1947;150(1):70–77. doi: 10.1152/ajplegacy.1947.150.1.70. [DOI] [PubMed] [Google Scholar]
- 24.Verma K, Penney DG, Helfman CC, Sutariya BB. Carboxyhemoglobin in the rat: improvements in the spectrophotometric measurement and comparison to other studies. J Appl Toxicol. 1989;9(5):323–330. doi: 10.1002/jat.2550090508. [DOI] [PubMed] [Google Scholar]
- 25.Reedy CJ, Elvekrog MM, Gibney BR. Development of a heme protein structure-electrochemical function database. Nucleic Acids Res. 2008;36(Database issue):D307–D313. doi: 10.1093/nar/gkm814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Piantadosi CA. Biological chemistry of carbon monoxide. Antioxidant Redox Signal. 2002;4(2):259–270. doi: 10.1089/152308602753666316. [DOI] [PubMed] [Google Scholar]
- 27.Alonso JR, Cardellach F, López S, Casademont J, Miró O. Carbon monoxide specifically inhibits cytochrome c oxidase of human mitochondrial respiratory chain. Pharmacol Toxicol. 2003;93(3):142–146. doi: 10.1034/j.1600-0773.2003.930306.x. [DOI] [PubMed] [Google Scholar]
- 28.Zuckerbraun BS, Chin BY, Bilban M, et al. Carbon monoxide signals via inhibition of cytochrome c oxidase and generation of mitochondrial reactive oxygen species. FASEB J. 2007;21(4):1099–1106. doi: 10.1096/fj.06-6644com. [DOI] [PubMed] [Google Scholar]
- 29.Petersen LC. The effect of inhibitors on the oxygen kinetics of cytochrome c oxidase. Biochim Biophys Acta. 1977;460(2):299–307. doi: 10.1016/0005-2728(77)90216-x. [DOI] [PubMed] [Google Scholar]
- 30.Hansen FB, Nicholls P. Control of respiration in proteoliposomes containing cytochrome aa3. II. Inhibition by carbon monoxide and azide. Biochim Biophys Acta. 1978;502(3):400–408. doi: 10.1016/0005-2728(78)90073-7. [DOI] [PubMed] [Google Scholar]
- 31.Yoshikawa S, Choc MG, O’Toole MC, Caughey WS. An infrared study of CO binding to heart cytochrome c oxidase and hemoglobin A. Implications re O2 reactions. J Biol Chem. 1977;252(15):5498–5508. [PubMed] [Google Scholar]
- 32.Wohlrab H, Ogunmola GB. Carbon monoxide binding studies of cytochrome a3 hemes in intact rat liver mitochondria. Biochemistry. 1971;10(7):1103–1106. doi: 10.1021/bi00783a001. [DOI] [PubMed] [Google Scholar]
- 33.Gnaiger E, Lassnig B, Kuznetsov A, Rieger G, Margreiter R. Mitochondrial oxygen affinity, respiratory flux control and excess capacity of cytochrome c oxidase. J Exp Biol. 1998;201(Pt 8):1129–1139. doi: 10.1242/jeb.201.8.1129. [DOI] [PubMed] [Google Scholar]
- 34.Wilson DF, Rumsey WL, Green TJ, Vanderkooi JM. The oxygen dependence of mitochondrial oxidative phosphorylation measured by a new optical method for measuring oxygen concentration. J Biol Chem. 1988;263(6):2712–2718. [PubMed] [Google Scholar]
- 35.Coburn RF. Mechanisms of carbon monoxide toxicity. Prev Med. 1979;8(3):310–322. doi: 10.1016/0091-7435(79)90008-2. [DOI] [PubMed] [Google Scholar]
- 36.Glabe A, Chung Y, Xu D, Jue T. Carbon monoxide inhibition of regulatory pathways in myocardium. Am J Physiol. 1998;274(6 Pt 2):H2143–H2151. doi: 10.1152/ajpheart.1998.274.6.H2143. [DOI] [PubMed] [Google Scholar]
- 37.Chance B, Erecinska M, Wagner M. Mitochondrial responses to carbon monoxide toxicity. Ann N Y Acad Sci. 1970;174(1):193–204. doi: 10.1111/j.1749-6632.1970.tb49786.x. [DOI] [PubMed] [Google Scholar]
- 38.Cooper CE, Brown GC. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. J Bioenerg Biomembr. 2008;40(5):533–539. doi: 10.1007/s10863-008-9166-6. [DOI] [PubMed] [Google Scholar]
- 39.McLennan HR, Degli Esposti M. The contribution of mitochondrial respiratory complexes to the production of reactive oxygen species. J Bioenerg Biomembr. 2000;32(2):153–162. doi: 10.1023/a:1005507913372. [DOI] [PubMed] [Google Scholar]
- 40.Mittler R, Vanderauwera S, Suzuki N, et al. ROS signaling: the new wave? Trends Plant Sci. 2011;16(6):300–309. doi: 10.1016/j.tplants.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 41.Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Molecular cell. 2012;48(2):158–167. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thom SR, Fisher D, Xu YA, Notarfrancesco K, Ischiropoulos H. Adaptive responses and apoptosis in endothelial cells exposed to carbon monoxide. Proc Natl Acad Sci U S A. 2000;97(3):1305–1310. doi: 10.1073/pnas.97.3.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Middleton ET, Morice AH. Breath carbon monoxide as an indication of smoking habit. Chest. 2000;117(3):758–763. doi: 10.1378/chest.117.3.758. [DOI] [PubMed] [Google Scholar]
- 44.Peng CA, Palsson BO. Determination of specific oxygen uptake rates in human hematopoietic cultures and implications for bioreactor design. Ann Biomed Eng. 1996;24(3):373–381. doi: 10.1007/BF02660886. [DOI] [PubMed] [Google Scholar]
- 45.Zhang X, Shan P, Otterbein LE, et al. Carbon monoxide inhibition of apoptosis during ischemia-reperfusion lung injury is dependent on the p38 mitogen-activated protein kinase pathway and involves caspase 3. J Biol Chem. 2003;278(2):1248–1258. doi: 10.1074/jbc.M208419200. [DOI] [PubMed] [Google Scholar]
- 46.Ryter SW, Morse D, Choi AM. Carbon monoxide: to boldly go where NO has gone before. Sci STKE. 2004;2004(230):RE6. doi: 10.1126/stke.2302004re6. [DOI] [PubMed] [Google Scholar]
- 47.Verma A, Hirsch DJ, Glatt CE, Ronnett GV, Snyder SH. Carbon monoxide: a putative neural messenger. Science. 1993;259(5093):381–384. doi: 10.1126/science.7678352. [DOI] [PubMed] [Google Scholar]
- 48.Stone JR, Marletta MA. Soluble guanylate cyclase from bovine lung: activation with nitric oxide and carbon monoxide and spectral characterization of the ferrous and ferric states. Biochemistry. 1994;33(18):5636–5640. doi: 10.1021/bi00184a036. [DOI] [PubMed] [Google Scholar]
- 49.Ingi T, Chiang G, Ronnett GV. The regulation of heme turnover and carbon monoxide biosynthesis in cultured primary rat olfactory receptor neurons. J Neurosci. 1996;16(18):5621–5628. doi: 10.1523/JNEUROSCI.16-18-05621.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cary SP, Marletta MA. The case of CO signaling: why the jury is still out. J Clin Invest. 2001;107(9):1071–1073. doi: 10.1172/JCI12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aono S. Biochemical and biophysical properties of the CO-sensing transcriptional activator CooA. Acc Chem Res. 2003;36(11):825–831. doi: 10.1021/ar020097p. [DOI] [PubMed] [Google Scholar]
- 52.Uchida T, Sato E, Sato A, Sagami I, Shimizu T, Kitagawa T. CO-dependent activity-controlling mechanism of heme-containing CO-sensor protein, neuronal PAS domain protein 2. J Biol Chem. 2005;280(22):21358–21368. doi: 10.1074/jbc.M412350200. [DOI] [PubMed] [Google Scholar]
- 53.Gilun P, Stefanczyk-Krzymowska S, Romerowicz-Misielak M, Tabecka-Lonczynska A, Przekop F, Koziorowski M. Carbon monoxide-mediated humoral pathway for the transmission of light signal to the hypothalamus. J Physiol Pharmacol. 2013;64(6):761–772. [PubMed] [Google Scholar]
- 54.Dioum EM, Rutter J, Tuckerman JR, Gonzalez G, Gilles-Gonzalez MA, McKnight SL. NPAS2: a gas-responsive transcription factor. Science. 2002;298(5602):2385–2387. doi: 10.1126/science.1078456. [DOI] [PubMed] [Google Scholar]
- 55.Artinian LR, Ding JM, Gillette MU. Carbon monoxide and nitric oxide: interacting messengers in muscarinic signaling to the brain’s circadian clock. Exp Neurol. 2001;171(2):293–300. doi: 10.1006/exnr.2001.7781. [DOI] [PubMed] [Google Scholar]
- 56.Puranik M, Nielsen SB, Youn H, et al. Dynamics of carbon monoxide binding to CooA. J Biol Chem. 2004;279(20):21096–21108. doi: 10.1074/jbc.M400613200. [DOI] [PubMed] [Google Scholar]
- 57.Vetri F, Choudhury MSR, Pelligrino DA, Sundivakkam P. BKCa channels as physiological regulators: a focused review. J Receptor Ligand Channel Res. 2014;7:3–13. [Google Scholar]
- 58.Hou S, Xu R, Heinemann SH, Hoshi T. The RCK1 high-affinity Ca2+ sensor confers carbon monoxide sensitivity to Slo1 BK channels. Proc Natl Acad Sci U S A. 2008;105(10):4039–4043. doi: 10.1073/pnas.0800304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xi Q, Tcheranova D, Parfenova H, Horowitz B, Leffler CW, Jaggar JH. Carbon monoxide activates KCa channels in newborn arteriole smooth muscle cells by increasing apparent Ca2+ sensitivity of alpha-subunits. Am J Physiol Heart Circ Physiol. 2004;286(2):H610–H618. doi: 10.1152/ajpheart.00782.2003. [DOI] [PubMed] [Google Scholar]
- 60.Knecht KR, Milam S, Wilkinson DA, Fedinec AL, Leffler CW. Time-dependent action of carbon monoxide on the newborn cerebrovascular circulation. Am J Physiol Heart Circ Physiol. 2010;299(1):H70–H75. doi: 10.1152/ajpheart.00258.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Holt DC, Fedinec AL, Vaughn AN, Leffler CW. Age and species dependence of pial arteriolar responses to topical carbon monoxide in vivo. Exp Biol Med (Maywood) 2007;232(11):1465–1469. doi: 10.3181/0705-BC-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang R, Wang Z, Wu L. Carbon monoxide-induced vasorelaxation and the underlying mechanisms. Br J Pharmacol. 1997;121(5):927–934. doi: 10.1038/sj.bjp.0701222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morita T, Perrella MA, Lee ME, Kourembanas S. Smooth muscle cell-derived carbon monoxide is a regulator of vascular cGMP. Proc Natl Acad Sci U S A. 1995;92(5):1475–1479. doi: 10.1073/pnas.92.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yi L, Morgan JT, Ragsdale SW. Identification of a thiol/disulfide redox switch in the human BK channel that controls its affinity for heme and CO. J Biol Chem. 2010;285(26):20117–20127. doi: 10.1074/jbc.M110.116483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu JY, Qu HY, Shang ZL, et al. Reciprocal regulation of Ca(2)+-activated outward K+ channels of Pyrus pyrifolia pollen by heme and carbon monoxide. New Phytol. 2011;189(4):1060–1068. doi: 10.1111/j.1469-8137.2010.03564.x. [DOI] [PubMed] [Google Scholar]
- 66.Williams SE, Wootton P, Mason HS, et al. Hemoxygenase-2 is an oxygen sensor for a calcium-sensitive potassium channel. Science. 2004;306(5704):2093–2097. doi: 10.1126/science.1105010. [DOI] [PubMed] [Google Scholar]
- 67.Migita CT, Matera KM, Ikeda-Saito M, et al. The oxygen and carbon monoxide reactions of heme oxygenase. J Biol Chem. 1998;273(2):945–949. doi: 10.1074/jbc.273.2.945. [DOI] [PubMed] [Google Scholar]
- 68.Altman PL, Dittmer DS. Respiration and circulation. Bethesda, MD: Federation of American Societies for Experimental Etiology; 1971. p. 16. http://www2.epa.gov/sites/production/files/2014-03/documents/leidyref.pdf. [Google Scholar]
- 69.Ortega-Sáenz P, Pascual A, Piruat JI, López-Barneo J. Mechanisms of acute oxygen sensing by the carotid body: lessons from genetically modified animals. Respir Physiol Neurobiol. 2007;157(1):140–147. doi: 10.1016/j.resp.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 70.Wang S, Publicover S, Gu Y. An oxygen-sensitive mechanism in regulation of epithelial sodium channel. Proc Natl Acad Sci U S A. 2009;106(8):2957–2962. doi: 10.1073/pnas.0809100106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Otterbein LE, Mantell LL, Choi AM. Carbon monoxide provides protection against hyperoxic lung injury. Am J Physiol. 1999;276(4 Pt 1):L688–L694. doi: 10.1152/ajplung.1999.276.4.L688. [DOI] [PubMed] [Google Scholar]
- 72.Althaus M, Fronius M, Buchackert Y, et al. Carbon monoxide rapidly impairs alveolar fluid clearance by inhibiting epithelial sodium channels. Am J Respir Cell Mol Biol. 2009;41(6):639–650. doi: 10.1165/rcmb.2008-0458OC. [DOI] [PubMed] [Google Scholar]
- 73.Wilkinson WJ, Kemp PJ. Carbon monoxide: an emerging regulator of ion channels. J Physiol. 2011;589(Pt 13):3055–3062. doi: 10.1113/jphysiol.2011.206706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Motterlini R. Carbon monoxide-releasing molecules (CO-RMs): vasodilatory, anti-ischaemic and anti-inflammatory activities. Biochem Soc Trans. 2007;35(Pt 5):1142–1146. doi: 10.1042/BST0351142. [DOI] [PubMed] [Google Scholar]
- 75.Motterlini R, Otterbein LE. The therapeutic potential of carbon monoxide. Nat Rev Drug Discov. 2010;9(9):728–743. doi: 10.1038/nrd3228. [DOI] [PubMed] [Google Scholar]
- 76.Scragg JL, Dallas ML, Wilkinson JA, Varadi G, Peers C. Carbon monoxide inhibits L-type Ca2+ channels via redox modulation of key cysteine residues by mitochondrial reactive oxygen species. J Biol Chem. 2008;283(36):24412–24419. doi: 10.1074/jbc.M803037200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dallas ML, Boyle JP, Milligan CJ, et al. Carbon monoxide protects against oxidant-induced apoptosis via inhibition of Kv2.1. FASEB J. 2011;25(5):1519–1530. doi: 10.1096/fj.10-173450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nielsen VG, Pearson EC, Smith MC. Increased carbon monoxide production by hemeoxygenase-1 caused by device-mediated hemolysis: thrombotic phantom menace? Artif Organs. 2013;37(11):1008–1014. doi: 10.1111/aor.12122. [DOI] [PubMed] [Google Scholar]
- 79.Arkebauer MR, Kanaparthy SS, Malayaman SN, Vosseller K, Nielsen VG. Carbon monoxide and nitric oxide modulate α-antiplasmin and plasmin activity: role of heme. Blood Coagul Fibrinolysis. 2011;22(8):712–719. doi: 10.1097/MBC.0b013e32834c73f9. [DOI] [PubMed] [Google Scholar]
- 80.Nielsen VG, Cohen JB, Malayaman SN, Nowak M, Vosseller K. Fibrinogen is a heme-associated, carbon monoxide sensing molecule: a preliminary report. Blood Coagul Fibrinolysis. 2011;22(5):443–447. doi: 10.1097/MBC.0b013e328345c069. [DOI] [PubMed] [Google Scholar]
- 81.Berk PD, Howe RB, Bloomer JR, Berlin NI. Studies of bilirubin kinetics in normal adults. J Clin Invest. 1969;48(11):2176–2190. doi: 10.1172/JCI106184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Blanckaert N, Fevery J. Physiology and pathophysiology of bilirubin metabolism. In: Zakim D, Boyer TD, editors. Hepatology: A textbook of Liver Disease. 2nd ed. Vol. 1. Philadelphia: WB Saunders; 1990. pp. 254–302. [Google Scholar]
- 83.Dickerson JW, Widdowson EM. Chemical changes in skeletal muscle during development. Biochem J. 1960;74:247–257. doi: 10.1042/bj0740247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kagen LJ, Christian CL. Immunologic measurements of myoglobin in human adult and fetal skeletal muscle. Am J Physiol. 1966;211(3):656–660. doi: 10.1152/ajplegacy.1966.211.3.656. [DOI] [PubMed] [Google Scholar]
- 85.Vreman HJ, Stevenson DK. Heme oxygenase activity as measured by carbon monoxide production. Anal Biochem. 1988;168(1):31–38. doi: 10.1016/0003-2697(88)90006-1. [DOI] [PubMed] [Google Scholar]
- 86.Ishikawa K, Navab M, Leitinger N, Fogelman AM, Lusis AJ. Induction of heme oxygenase-1 inhibits the monocyte transmigration induced by mildly oxidized LDL. J Clin Invest. 1997;100(5):1209–1216. doi: 10.1172/JCI119634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ponka P. Cell biology of heme. Am J Med Sci. 1999;318(4):241–256. doi: 10.1097/00000441-199910000-00004. [DOI] [PubMed] [Google Scholar]
- 88.Zheng J, Tian Q, Hou W, Watts JA, Schrum LW, Bonkovsky HL. Tissue-specific expression of ALA synthase-1 and heme oxygenase-1 and their expression in livers of rats chronically exposed to ethanol. FEBS Lett. 2008;582(13):1829–1834. doi: 10.1016/j.febslet.2008.04.047. [DOI] [PubMed] [Google Scholar]
- 89.Berk PD, Bloomer JR, Howe RB, Blaschke TF, Berlin NI. Bilirubin production as a measure of red cell life span. J Lab Clin Med. 1972;79(3):364–378. [PubMed] [Google Scholar]
- 90.Ajioka RS, Phillips JD, Kushner JP. Biosynthesis of heme in mammals. Biochim Biophys Acta. 2006;1763(7):723–736. doi: 10.1016/j.bbamcr.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 91.Balaban RS, Mootha VK, Arai A. Spectroscopic determination of cytochrome c oxidase content in tissues containing myoglobin or hemoglobin. Anal Biochem. 1996;237(2):274–278. doi: 10.1006/abio.1996.0239. [DOI] [PubMed] [Google Scholar]
- 92.Qiu Y, Sutton L, Riggs AF. Identification of myoglobin in human smooth muscle. J Biol Chem. 1998;273(36):23426–23432. doi: 10.1074/jbc.273.36.23426. [DOI] [PubMed] [Google Scholar]
- 93.Leffler CW, Parfenova H, Fedinec AL, Basuroy S, Tcheranova D. Contributions of astrocytes and CO to pial arteriolar dilation to glutamate in newborn pigs. Am J Physiol Heart Circ Physiol. 2006;291(6):H2897–H2904. doi: 10.1152/ajpheart.00722.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zahs KR, Wu T. Confocal microscopic study of glial-vascular relationships in the retinas of pigmented rats. J Comp Neurol. 2001;429(2):253–269. doi: 10.1002/1096-9861(20000108)429:2<253::aid-cne6>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 95.Krogh A. The number and distribution of capillaries in muscles with calculations of the oxygen pressure head necessary for supplying the tissue. J Physiol. 1919;52(6):409–415. doi: 10.1113/jphysiol.1919.sp001839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hart MN, Heistad DD, Brody MJ. Effect of chronic hypertension and sympathetic denervation on wall/lumen ratio of cerebral vessels. Hypertension. 1980;2(4):419–423. doi: 10.1161/01.hyp.2.4.419. [DOI] [PubMed] [Google Scholar]
- 97.Staszyk C, Bohnet W, Gasse H, Hackbarth H. Blood vessels of the rat tail: a histological re-examination with respect to blood vessel puncture methods. Lab Anim. 2003;37(2):121–125. doi: 10.1258/00236770360563750. [DOI] [PubMed] [Google Scholar]
- 98.Heistad DD, Marcus ML. Role of vasa vasorum in nourishment of the aorta. Blood Vessels. 1979;16(5):225–238. doi: 10.1159/000158209. [DOI] [PubMed] [Google Scholar]
- 99.Wolinsky H, Glagov S. Nature of species differences in the medial distribution of aortic vasa vasorum in mammals. Circ Res. 1967;20(4):409–421. doi: 10.1161/01.res.20.4.409. [DOI] [PubMed] [Google Scholar]
- 100.Cussler EL. Diffusion – Mass Transfer in Fluid Systems. 2nd ed. Cambridge: University of Cambridge; 1997. [Google Scholar]
- 101.Lenzi GL, Frackowiak RS, Jones T, et al. CMRO2 and CBF by the oxygen-15 inhalation technique. Results in normal volunteers and cerebrovascular patients. Eur Neurol. 1981;20(3):285–290. doi: 10.1159/000115248. [DOI] [PubMed] [Google Scholar]
- 102.Kanu A, Whitfield J, Leffler CW. Carbon monoxide contributes to hypotension-induced cerebrovascular vasodilation in piglets. Am J Physiol Heart Circ Physiol. 2006;291(5):H2409–H2414. doi: 10.1152/ajpheart.01368.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Beckman JD, Belcher JD, Vineyard JV, et al. Inhaled carbon monoxide reduces leukocytosis in a murine model of sickle cell disease. Am J Physiol Heart Circ Physiol. 2009;297(4):H1243–H1253. doi: 10.1152/ajpheart.00327.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wegiel B, Gallo D, Csizmadia E, et al. Carbon monoxide expedites metabolic exhaustion to inhibit tumor growth. Cancer Res. 2013;73(23):7009–7021. doi: 10.1158/0008-5472.CAN-13-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zuckerbraun BS, Chin BY, Wegiel B, et al. Carbon monoxide reverses established pulmonary hypertension. J Exp Med. 2006;203(9):2109–2119. doi: 10.1084/jem.20052267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wegiel B, Hedblom A, Li M, et al. Heme oxygenase-1 derived carbon monoxide permits maturation of myeloid cells. Cell Death Dis. 2014;5:e1139. doi: 10.1038/cddis.2014.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kuramitsu K, Gallo D, Yoon M, et al. Carbon monoxide enhances early liver regeneration in mice after hepatectomy. Hepatology. 2011;53(6):2016–2026. doi: 10.1002/hep.24317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wise D, Houghton G. Diffusion coefficients of neon, krypton, xenon, carbon monoxide and nitric oxide in water at 10–60 C. Chem Eng Sci. 1968;23(10):1211–1216. [Google Scholar]
- 109.Kawashiro T, Nüsse W, Scheid P. Determination of diffusivity of oxygen and carbon dioxide in respiring tissue: results in rat skeletal muscle. Pflugers Arch. 1975;359(3):231–251. doi: 10.1007/BF00587382. [DOI] [PubMed] [Google Scholar]
- 110.Maines MD. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988;2(10):2557–2568. [PubMed] [Google Scholar]
- 111.Morimoto Y, Durante W, Lancaster DG, Klattenhoff J, Tittel FK. Real-time measurements of endogenous CO production from vascular cells using an ultrasensitive laser sensor. Am J Physiol Heart Circ Physiol. 2001;280(1):H483–H488. doi: 10.1152/ajpheart.2001.280.1.H483. [DOI] [PubMed] [Google Scholar]
- 112.Mentré P, Escaig F. Preservation of the diffusible cations for secondary ion mass spectrometry. II. Artefacts in material embedded in araldite or melamine. Biol Cell. 1992;74(1):119–126. doi: 10.1016/0248-4900(92)90017-u. [DOI] [PubMed] [Google Scholar]
- 113.Leffler CW, Balabanova L, Fedinec AL, Parfenova H. Nitric oxide increases carbon monoxide production by piglet cerebral microvessels. Am J Physiol Heart Circ Physiol. 2005;289(4):H1442–H1447. doi: 10.1152/ajpheart.00464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nathanson JA, Scavone C, Scanlon C, McKee M. The cellular Na+ pump as a site of action for carbon monoxide and glutamate: a mechanism for long-term modulation of cellular activity. Neuron. 1995;14(4):781–794. doi: 10.1016/0896-6273(95)90222-8. [DOI] [PubMed] [Google Scholar]
- 115.Sohlenius-Sternbeck AK. Determination of the hepatocellularity number for human, dog, rabbit, rat and mouse livers from protein concentration measurements. Toxicol In Vitro. 2006;20(8):1582–1586. doi: 10.1016/j.tiv.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 116.Iwatsubo T, Suzuki H, Sugiyama Y. Prediction of species differences (rats, dogs, humans) in the in vivo metabolic clearance of YM796 by the liver from in vitro data. J Pharmacol Exp Ther. 1997;283(2):462–469. [PubMed] [Google Scholar]