

Abstract
The development of poly (adenosine diphosphate [ADP]) ribose polymerase (PARP) inhibitors (PARPi) has progressed greatly over the last few years and has shown encouraging results in the BRCA1/2 mutation–related cancers. This article attempts to summarize the rationale and theory behind PARPi, the clinical trials already reported, as well as ongoing studies designed to determine the role of PARPi in patients with and without germline mutations of BRCA genes. Future plans for PARPi both as monotherapy and in combination with standard cytotoxics, other biological agents, and as radiosensitizers are also covered. The widening scope of PARPi adds another important targeted agent to the growing list of molecular inhibitors; future and ongoing trials will identify the most effective role for PARPi, including for patients other than BRCA germline mutation carriers.
Keywords: PARPi, BRCA genes, germline mutations, cytotoxics, radiosensitizers, BRCA germline mutation carriers
Introduction
The increasing understanding of the functions of the poly (adenosine diphosphate [ADP]) ribose polymerase (PARP) enzymes in DNA repair among other things has led to the investigation of specific inhibitors of PARP in the cancer therapeutics setting.
The PARP proteins are a family of 17 enzymes involved in a wide range of cellular functions including DNA transcription, DNA damage response, genomic stability maintenance, cell cycle regulation, and cell death.1 PARP enzymes are a subgroup of the ADP-ribosyltransferases (ARTs), which transfer ADP-ribose units from nicotinamide adenine dinucleotide (NAD+) molecules to form polymers of ADP ribose (PAR) units. These covalently bind to side chains of specific amino acid residues of acceptor proteins including PARP-1 itself.2 PARP-1 is the most abundant and best characterized protein of this group. In oncology, its integral role in the repair of single-strand DNA breaks (SSBs) via the base excision repair (BER) pathway has been a focus of interest.
PARP-1 recognizes and binds to sites of SSBs (Figure 1B). Not all the PARP enzymes have been fully investigated, but some of them have been shown to catalyze mono-ADP-ribosylation, and thus do not fit the definition of a polymerase.3 Therefore, there have been calls to revise the nomenclature of PARP enzymes to more accurately reflect these differences.2 For the purposes of this review, we will use the term PARP, referring to the PARP-1 enzyme.
Figure 1.
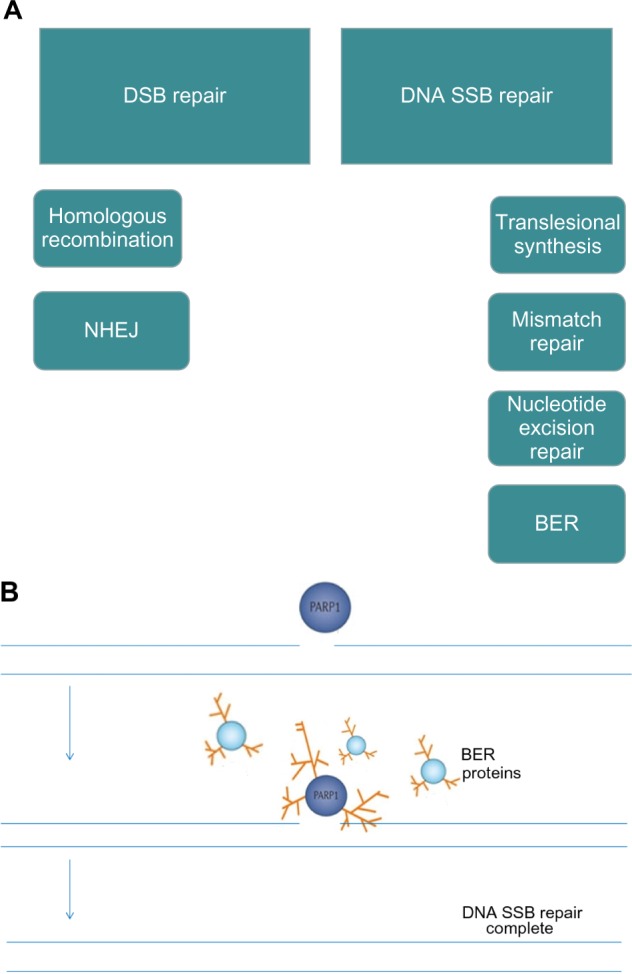
(A) DNA repair pathways; (B) PARP senses DNA SSBs and utilizes NAD+ as a substrate to form PAR, which attach to a range of target proteins including PARP-1 itself and BER proteins. This posttranslational modification is termed PARylation.
Notes: (A) There are six DNA repair pathways, two for repair of DNA DSBs and four for repair of SSBs. PARP is known to be involved in BER and has been shown to be a negative regulator of NHEJ (low-fidelity, error-prone pathway). PARP inhibition therefore leads to disruption of BER and in HR-deficient cells, increased reliance on error-prone NHEJ.53
Abbreviations: BER, base excision repair; DSBs, double-strand DNA breaks; HR, homologous recombination; NAD+, nicotinamide adenine dinucleotide; NHEJ, nonhomologous end joining; PAR, polymers of ADP-ribose; PARP, poly (adenosine diphosphate [ADP]) ribose polymerase; SSBs, single-strand DNA breaks.
In cancer therapeutics, accumulation of SSBs with PARP inhibition leads to the development of double-strand DNA breaks (DSBs), which require competent homologous recombination (HR) repair to allow cell survival.1 PARP has also been shown to be involved in DSB repair pathways.
Much of the development of PARP inhibitors (PARPi) has been focused on targeting cancers associated with a mutation of the breast cancer–related genes, BRCA1 and BRCA2, which are proteins that are integral to the HR repair pathway.4 BRCA mutation carriers have a single functioning BRCA gene (wild type individuals have two copies of the BRCA gene), and when this is lost by mutation, cells are unable to undertake HR;5 this ‘second hit’ makes a cell susceptible to tumor formation. The selective targeting of BRCA-deficient cancer cells by PARPi demonstrates the concept of synthetic lethality, which was first described in 1922 by Bridges,6 whereby a cell harboring one of two gene or protein defects is viable while a cell containing both defects is nonviable (Figure 2).7
Figure 2.

BRCA-deficient cells are highly reliant on BER as well as SSA and the error prone NHEJ pathway for DNA repair, both of which are influenced by PARPi.
Abbreviations: BER, base excision repair; DSB, double-strand DNA breaks; HR, homologous recombination; NHEJ, nonhomologous end joining; SSA, single-strand annealing; SSBs, single-strand DNA breaks; PARPi - poly (adenosine diphosphate [ADP]) ribose polymerase inhibitors.
Epidemiological studies have demonstrated a tight relationship between BRCA1/2 mutations and the development of breast and ovarian cancers,8 as well as other cancers.9 PARPi selectively target BRCA-deficient cancer cells while sparing normal tissues, which retain a normal copy of the BRCA gene and can undergo normal HR repair of persistent SSBs not repaired by PARP.5 Apart from blocking SSB repair, PARPi are also thought to act by ‘trapping’ PARP at sites of DNA damage, resulting in a cytotoxic PARP–DNA complex (Figure 3).10 PARP localization to a point of DNA damage normally stimulates attraction of DNA repair proteins, which in turn prompt PARP dissociation from DNA, allowing repair to take place. The dissociation requires PARP to ‘auto-PARylate’ and is prevented by PARPi. In vitro studies by Murai et al demonstrated that PARP inhibition (by olaparib) and the PARP-1–DNA complexes generated are more cytotoxic than genetic depletion of PARP-1,10 suggesting that there is more than one mechanism of action for PARPi. Interestingly, it has also been shown that the degree of PARP trapping varies between PARPi, niraparib being more potent than olaparib, which is more potent than veliparib. PARP trapping did not correlate with the catalytic inhibitory properties of each drug.10
Figure 3.
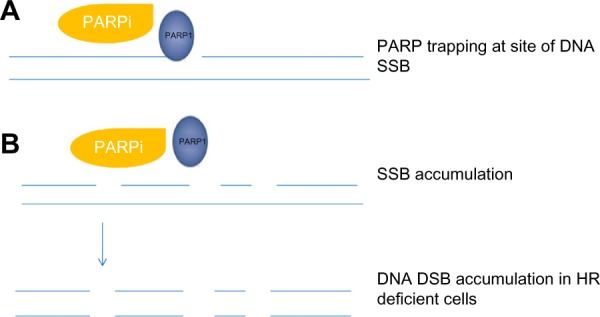
PARP inhibitor – mechanism of action.
Notes: (A) PARP attaches to site(s) of DNA damage and is unable to dissociate due to inhibition of PARP autoPARylation causing PARP trapping and cytotoxic PARP–DNA complexes. The degree of ‘PARP trapping’ appears to vary between the different inhibitors currently under investigation and may explain the differences in toxicity profile observed, with olaparib and niraparib having greater potency compared to veliparib.54 (B) SSBs accumulate, leading to DSBs, which are unrepaired in cells deficient in HR proteins, ultimately leading to cell death.
Abbreviations: DSBs, double-strand DNA breaks; HR, homologous recombination; PARP, poly (adenosine diphosphate [ADP]) ribose polymerase; SSBs, single-strand DNA breaks.
PARPi use in ovarian cancer
The Phase I trial of olaparib (AZD2281)11 was the first clinical demonstration of PARPi activity in the BRCA mutation–associated cancers (Figure 4). In this study, 60 patients with refractory cancers were recruited to evaluate the pharmacokinetics and pharmacodynamics of olaparib. Twenty-two patients (37%) carried a BRCA 1/2 mutation. A clinical benefit rate (CBR) of 63% was observed in this group of patients (12/19). This led to the enrollment of an expanded cohort of 50 patients with BRCA1/2 mutation–associated gynecologic malignancies (ovarian, primary peritoneal, and fallopian tube cancers) where a CBR of 46% was observed. A Response Evaluation Criteria in Solid Tumors (RECIST) radiological response or CA125 response was seen in 40% of patients.12 This early phase study confirmed the BRCA1/2 mutation to be a predictive biomarker of response to PARPi.
Figure 4.

Schema of development of first PARPi, olaparib (AZD2281).
Abbreviations: HR, homologous recombination; PARP, poly (adenosine diphosphate [ADP]) ribose polymerase; PARPi, PARP inhibitor.
In addition, it confirmed a correlation between PARPi response and platinum sensitivity, as platinum-sensitive patients displayed a 69% CBR compared with 46% and 23% in platinum-resistant and refractory patients, respectively. Platinum agents cause intra- and inter-strand DNA crosslinks, which lead to DNA DSBs. In cancer cells with HR deficiency (HRD), such as patients with mutant BRCA, cells become prone to further DNA damage and cell death. There is growing evidence that defects other than that of BRCA1/2 can lead to HRD, which might explain the broader success of platinum agents in cancer,13 the corollary being that cancers sensitive to platinum agents may also be sensitive to PARPi, widening their use beyond the BRCA1/2 mutation carriers (Figure 4). McCabe et al demonstrated via in vitro studies that deficiencies of several proteins in the HR DNA repair pathway, such as the DNA damage sensors ATM (ataxia telangiectasia mutated) and ATR (ataxia telangiectasia and RAD3-related protein), leads to HRD and subsequent PARPi sensitivity.13 This concept, known as ‘BRCAness’ has been used to describe the phenotype arising in sporadic cancers that have intact BRCA1/2 genes but share features with the BRCA1/2 mutation–related tumors, such as profound platinum sensitivity.14
Tumors with BRCA-like features
‘BRCAness’ can be illustrated by the responses in high-grade serous ovarian cancers (HGSOC), where nearly 20% of cases occur in women with germline BRCA1/2 mutations, but a further 35% are thought to have acquired defects of HR15 and display the same sensitivity to platinum chemotherapy, as well as to PARPi. This was highlighted in a Phase II trial of olaparib, which recruited both BRCA1/2 mutated as well as wild type patients with breast cancer and HGSOC. In the cohort of patients who had platinum-sensitive HGSOC without BRCA1/2 mutation, the objective response rate (ORR) was 50% (10/20).16 In addition, among 38 BRCA wild type patients with ovarian cancer, a 26% response rate in CA125 was observed and a combined RECIST or CA125 response rate of 30% was recorded. All these responders had HGSOC. Interestingly, responses were also seen in the platinum-resistant cohort without BRCA 1/2 mutation (17% response rate), suggesting incomplete crossover of platinum sensitivity and PARPi response.16 Similar findings were found by Ledermann et al in a randomized double-blind placebo-controlled Phase II trial of maintenance olaparib in HGSOC, in which patients had responded to two or more lines of platinum chemotherapy.17
Findings such as this have led to Phase III trials of maintenance PARPi treatment after chemotherapy for all ovarian cancer patients; many of the PARPi combination trials are recruiting patients with HGSOC regardless of BRCA status. Currently recruiting is a randomized double-blind placebo-controlled trial of niraparib (MK-4827) as maintenance therapy in platinum-sensitive ovarian cancer patients. This study is recruiting patients who have either a BRCA1/2 mutation or tumors with high-grade serous histology and have had a response to their most recent platinum-containing chemotherapy (NCT01847274). In contrast, the SOLO1 and SOLO2 trials are ongoing Phase III randomized, placebo-controlled trials investigating the use of olaparib maintenance in the first-line (SOLO 1 NCT01844986) and relapsed setting (SOLO 2 NCT01874353) and are only recruiting BRCA mutation carriers with ovarian cancer.
The ARIEL2 and NOVA trials are Phase II trials investigating rucaparib and niraparib, respectively, in platinum-sensitive relapsed ovarian cancer (NCT01891344). One of the aims of ARIEL2 is to identify a molecular signature associated with HRD in HGSOC and fallopian tube cancers, which can then be used to identify patients most likely to respond to rucaparib. A Phase III trial (ARIEL3) is planned once this signature is identified to prospectively assess its use in this patient population.
PARPi use in breast cancer
In breast cancer, a Phase II trial of single-agent olaparib in patients with BRCA1/2 mutation and advanced disease investigated two dose levels (cohort 1: 400 mg twice daily [bd] and cohort 2: 100 mg bd) and showed an ORR of 42% (11/26) and 25% (6/24), respectively. Progression-free survival (PFS) was 5.7 months in cohort 1 and 3.8 months in cohort 2.18
Breast cancer patients with BRCA1 mutations also display marked platinum sensitivity; treatment with neoadjuvant cisplatin in such individuals results in high pathological complete response rates (72%–90%).19,20 The majority of breast cancers in BRCA mutation carriers are found to be negative for the estrogen receptor (ER), progesterone receptor, and human epidermal growth factor receptor (HER2), and are termed triple negative breast cancer (TNBC). Non-BRCA–mutated TNBC appears to harbor a ‘BRCAness’ phenotype due to HRD;21 responses to PARPi in this population are being investigated in a number of Phase I and II trials. However, unlike HGSOC patients with BRCA mutations, TNBC patients are thought to have a poorer prognosis compared to hormone receptor–positive breast cancers. Sporadic TNBC has been shown to have a two-fold lower expression of BRCA1 compared to ER-positive cancers.22 Lower BRCA1 expression is possibly related to a number of factors, such as upregulation of BRCA1 transcriptional repressors such as ID4 and HMG1, and of microRNAs (miR) that negatively regulate BRCA1 expression such as miR-182.23 Many TNBCs share features with the basal-like phenotype classically associated with BRCA1-related cancers, yet only 20% of cases have a germline BRCA1/2 mutation. Although the majority of TNBCs are BRCA-mutation–negative, the shared phenotypic characteristics and molecular aberrations such as those mentioned above lead to HRD, which could sensitize cells to PARP inhibition. The basis of current investigations with respect to TNBC and its response to PARPi revolve around HRD. Potential mechanisms include BRCA1 promoter methylation leading to gene silencing, aberrations of MRE11 protein (involved in DSB detection),24 and ATM, all of which play important roles in HR.
A three-arm, double-blinded, placebo-controlled Phase III trial of veliparib (ABT-888) in TNBC with carboplatin and paclitaxel after neoadjuvant chemotherapy (doxorubicin and cyclophosphamide) in early-stage TNBC is ongoing (NCT02032277).
PARPi in other cancers
Endometrial cancer
The most common genetic aberration seen in up to 80% of endometrioid endometrial cancer (EEC) is loss of the PTEN tumor suppressor gene.25 However, apart from PTEN’s function in the regulation of the PI3 kinase/AKT/mTOR signaling pathway, it is apparent that PTEN competence is also important in maintaining genetic stability.26 Dedes et al showed PTEN-deficient EEC cells lack HR DNA repair,27 which sensitizes the EEC cells to PARPi in a similar fashion to that seen with BRCA-mutated cancers. These in vitro results have also been reflected by in vivo models of PTEN-deficient tumors treated with PARPi where tumor growth was significantly suppressed by PARPi treatment.28 Although this relationship between PTEN loss and sensitivity to PARPi has not been investigated in the context of a clinical trial, a case report of a patient with metastatic endometrial cancer who was treated with olaparib in a Phase I trial showed significant clinical response.29 Subsequent tumor biopsies confirmed no BRCA mutation but did demonstrate PTEN loss.29 Together with preclinical studies, this suggests a potential role for PARPi treatment for PTEN-deficient tumors, including endometrial cancer among others. PTEN loss may perhaps also serve as a predictive biomarker for PARPi sensitivity.
Prostate cancer
A minority of prostate cancers are attributed to BRCA1/2 mutation, and in these cases, PARPi sensitivity may be observed with striking clinical responses and lengthened overall survival (OS).11 Akin to endometrial cancer, 20% of prostate cancers show loss of PTEN expression and similar preclinical studies of PTEN-mutant prostate cancer cell lines have suggested that PTEN loss predicts sensitivity to PARPi with olaparib and veliparib.13
The Phase I trial of niraparib reported in 2013 set out to assess the relationship between PTEN loss in prostate cancer and response.30 Overall, niraparib showed good tolerability, and as seen in ovarian cancer patients, clinical responses were achieved in both BRCA mutation carriers (~50% CBR) as well as sporadic (BRCA wild type) cancers. Surprisingly, molecular defects such as PTEN loss or ETS gene rearrangements, often found in castration-resistant prostate cancer (CRPC), did not correlate with measures of antitumor activity in this Phase I study.30 It is possible that CRPC tumor cells harbor other defects of HR such as ATM, CHEK2, and FANCJ.31,32 The use of PARPi in prostate cancer is thus being further studied in combination trials such as that due to start this year comparing olaparib alone to olaparib and abiraterone in metastatic CRPC regardless of BRCA status (NCT01972217).
Pancreatic cancer
Although hereditary pancreatic cancer is rare, 10% of cases are due to inherited genetic factors,33,34 the most common group being related to BRCA mutations. These account for up to 17% of inherited pancreatic cancer.35 BRCA2 mutation increases the risk of pancreatic cancer development by 3.5-fold.36 As BRCA-associated pancreatic cancer is rare, the data are sparse regarding the use of PARPi, though the responses of patients treated within mixed cohorts in early phase trials of PARPi combinations have been encouraging, with BRCA-mutant patients achieving meaningful partial responses of 1–2 years.37,38
In a Phase II study of olaparib monotherapy for BRCA mutation–associated solid tumors, platinum-based chemotherapy was used first-line in six metastatic pancreatic cancer (MPC) patients; five (83%) achieved a partial response or complete response by RECIST criteria38 suggesting that BRCA-associated MPC may share the platinum sensitivity observed in other BRCA-associated malignancies.
Another trial investigating the use of PARPi in BRCA-associated MPC, with rucaparib monotherapy (NCT02042378), continues to recruit, and a third opening imminently involves maintenance olaparib in patients whose disease has not progressed on platinum-based first-line chemotherapy (NCT02184195).
PARPi in combination therapy
PARPi and ionizing radiation
Like platinum agents, PARPi have been shown to act as radiosensitizers in preclinical studies using both in vitro and in vivo models.39 By combining ionizing radiation with PARPi, the SSB induced by irradiation go unrepaired by PARP-associated BER, leading to cell death and delay in tumor growth. In vitro studies in lung40 and prostate cancers have confirmed this effect, although as described above, in prostate cancer the mechanisms for PARPi sensitization may be as a consequence of PTEN deficiency or upregulation of the fusion gene TMPRSS2-ERG.40–42
Two Phase I trials are underway comparing concomitant PARPi and radiotherapy with radiotherapy alone in esophageal and locally advanced pancreatic cancers (NCT01460888, NCT01908478).
PARPi and chemotherapy
Extensive preclinical studies have shown that PARPi enhance the cytotoxic effects of chemotherapy. The knockout of PARP-1 drastically impairs DNA repair following damage caused by ionizing radiation or cytotoxic treatment.43 Although PARP is expressed in all cells, its expression and activity is increased in actively proliferating cells. Several hematological malignancies as well as solid tumors have been found to have higher levels of PARP expression compared with normal cells.44,45 Therefore, PARPi could selectively target tumor cells. Preclinical studies have shown that PARP inhibition potentiates the cytotoxic effects of agents that cause SSBs in DNA, such as alkylating agents (eg, temozolomide46 or cyclophosphamide) and topoisomerase inhibitors (eg, irinotecan47). However, platinum agents cause DNA crosslinks which are usually repaired either by nucleotide/BER or HR. Therefore, theoretically and in vitro, combined therapy cytotoxics such as these, and PARPi even in normal cells should result in synergism.48 Early combination trials of PARPi and cytotoxics in vivo have been reported49–51 with encouraging results although, as might be expected, responses were more commonly seen in BRCA mutation carriers, suggesting that they have the greatest effect in patients with inherent DNA repair defects.50
Table 1 details trials of PARPi currently underway in combination with chemotherapy. Phase II studies of veliparib with topotecan (in refractory ovarian or primary peritoneal cancer [NCT01012817]) and veliparib with metronomic cyclophosphamide (in refractory gynecological cancers, TNBC, and low-grade non-Hodgkin lymphoma [NCT01306032]) are currently ongoing. A detailed genetic study of DNA repair defects as part of the latter study may identify HRD other than BRCA mutations to predict PARPi sensitivity. Myelosuppression is the main toxicity encountered in these combination studies, and as expected, is more significant when PARPi are combined with topotecan.49,51,52 Similarly, two completed trials, olaparib with paclitaxel in TNBC45 and rucaparib with temozolomide in melanoma,53 have shown trends toward higher response rates and longer PFS than historical controls but both required dose reductions of the cytotoxic agents for myelosuppression.
Table 1.
Currently recruiting trials of PARPi and chemotherapy combinations
| PARPi/chemo combination | Phase of study | Population |
|---|---|---|
| BMN673 + TMZ NCT02116777 | Phase I dose escalation followed by Phase II | Phase II: patients aged >12 months and ≤21 months, relapsed/refractory solid tumors Phase I: >12 months and ≤30 months, Ewing’s sarcoma, PNET, ALL |
| Olaparib + C AUC4 NCT01237067 | Phase I | Recurrent/refractory gynecologic tumors and recurrent/refractory metastatic BC BRCA mutation BC that is locally advanced or metastatic, even if not treated previously |
| AZD2281 + C NCT01445418 | Phase I | BRCA +ve BC and OC Sporadic OC Sporadic TNBC |
| Niraparib + TMZ NCT02044120 | Phase I | Previously treated, incurable Ewing’s sarcoma |
| Veliparib + pegylated liposomal doxorubicin NCT01145430 | Phase I | Recurrent OC, fallopian tube cancer, PPC, metastatic TNBC (BRCA +ve and −ve) |
| Veliparib + floxuridine NCT01749397 | Phase I | Metastatic epithelial ovarian, PPC, or fallopian tube cancer |
| Olaparib + TMZ NCT01390571 | Phase I | Relapsed glioblastoma |
| TMZ +/− veliparib NCT01638546 | Phase II, randomized placebo-controlled | Relapsed or refractory small cell lung cancer |
| E7449 single agent or with TMZ or with C and P NCT01618136 | Phase I dose escalation followed by Phase II | Single agent: advanced solid tumors Combination arms: B cell malignancies (TMZ) or advanced solid tumors (C + P) |
| Veliparib + topotecan NCT01012817 | Phase I dose escalation followed by Phase II | Phase I: advanced solid tumors Phase II: recurrent ovarian or PPC |
| Veliparib + C, P, and bevacizumab NCT00989651 | Phase I | Newly diagnosed stage II, III, or IV OC, fallopian tube cancer, or PPC |
Abbreviations: ALL, acute lymphoblastic leukemia; AUC, area under the curve; BC, breast cancer; C, carboplatin; OC, ovarian cancer; P, paclitaxel; PNET, peripheral primitive neuroectodermal tumor; PPC, primary peritoneal cancer; TMZ, temozolomide; TNBC, triple negative breast cancer.
Current PARPi and chemotherapy combination trials are summarized in Table 1.
PARPi and targeted agents
Combination with antiangiogenics
PARPi in combination with targeted antiangiogenic agents have been studied in the breast and ovarian cancer population. Like PARPi, antiangiogenics such as bevacizumab (anti-vascular endothelial growth factor [VEGF] antibody) and cediranib (a small-molecule inhibitor of VEGF receptor [VEGFR]-1/2/3) have been shown to have single-agent activity in recurrent ovarian cancer, with a 18% response rate to bevacizumab54 and 19% response rate to cediranib.55 Preclinical studies have suggested that PARPi also have an antiangiogenic effect,56 which prompts speculation that synergism may be achieved by combination therapy.
One of the first trials to assess this combination was the Phase I trial of olaparib and cediranib in recurrent ovarian cancer or TNBC.57 In 18 RECIST evaluable ovarian cancer patients, the ORR was 44% and CBR was 61%. These responses were not limited to the BRCA mutation carriers. No responses were seen in seven evaluable breast cancer patients, though two patients had stable disease (both BRCA mutation–positive) for more than 24 weeks. Grade 3 or 4 toxicities were observed in 75% of patients, the most common being hypertension (25%), fatigue (18%), and neutropenia (11%).57
Results from a Phase II trial comparing olaparib alone to olaparib with cediranib in ovarian cancer (NCT01116648) were presented earlier this year (American Society of Clinical Oncology, 2014) and showed a remarkable improvement in ORR (79.6% vs 47.8% and PFS 17.7 months vs 9.0 months, HR: 0.42) for the combination arm versus single-agent olaparib.58 There was a 39%, 23%, and 27% incidence of grade 3 hypertension, diarrhea, and fatigue, respectively, in the combination arm; 77% of patients in the combination arm were dose reduced compared to 24% in the olaparib-alone arm. However, the doses of both agents were relatively high at 30 mg daily cediranib and 200 mg bd olaparib capsules in the combination arm and 400 mg bd olaparib capsules in the single agent arm. OS data are not yet mature, but analysis of the population in relation to BRCA mutation status revealed that the improvement in PFS with combination olaparib/cediranib was much more marked in the BRCA wild type or unknown patients than those with BRCA mutations.58
Combination with PI3K inhibition
Study into the molecular defects of TNBC has identified aberrations involving the PI3 kinase/AKT/mTOR pathway such as PTEN loss or activating mutations of PIK3CA gene encoding PI3K.59 Inhibition of PI3K in TNBC cells causes a downregulation of BRCA1/2 and increased PARP activity, suggesting an increased dependency on BRCA-mediated DNA repair and a potential sensitizing effect to treatment with PARPi when PI3K is inhibited. Additionally, in TNBC xenograft models, dual inhibition of PI3K and PARP significantly suppressed tumor growth by downregulating BRCA1/2.59
On the basis of this work, a Phase I trial of a PI3K inhibitor (buparlisib) combined with olaparib is underway in TNBC and HGSOC patients (NCT01623349). Translational endpoints in this study will be vital to determine predictive biomarkers of response.
Future directions
Clinical trials of PARPi in cancer patients have already shown that these agents are active in patients irrespective of BRCA mutation status. In ovarian cancer, the growing understanding of multiple DNA repair defects other than BRCA mutations makes it likely that these targeted inhibitors can be applied to many BRCA wild type as well as mutation carrier patients. Predictive tools such as molecular profiles to identify responsive patients remain an important translational focus. Preclinical studies have already identified potential markers of PARPi sensitivity other than HR defects but these have yet to be proven clinically useful. Mechanisms of resistance to PARPi are also being unraveled. To date, three have been described: secondary mutations in BRCA1/2 that partially restore the gene reading frame and protein function,60 overexpression of the multidrug efflux transporter protein Pgp,61 and loss of 53BP1 with partial restoration of HR.62 These relationships are not clear, as 36%–45% of BRCA mutation carriers with ovarian cancer who progressed on PARPi therapy went on to respond to further chemotherapy, independent of previous PARPi platinum sensitivity or response to PARPi.63 Understanding these interactions and identifying predictive markers of sensitivity and resistance will inform the treatment strategy for PARPi agents and cytotoxics in the future.
In this era of personalized medicine, combinations of chemotherapy and molecularly targeted agents, such as PARPi, have already become standard treatment in some settings, eg, FOLFOX (fluorouracil and oxaliplatin) and cetuximab in KRAS wild type metastatic colorectal cancer and docetaxel with trastuzumab in HER2-positive breast cancer. Current combination treatment trials will hopefully identify the most effective role for PARPi combinations.
Footnotes
Disclosure
Dr Benafif reports no conflicts of interest in this work. Dr Hall has received honorarium for advisory boards from Roche, Astra Zeneca, Glaxo Smith Kline, Boerhinger Ingelheim, and Merck.
References
- 1.Michels J, Vitale I, Saparbaev M, Castedo M, Kroemer G. Predictive biomarkers for cancer therapy with PARP inhibitors. Oncogene. 2014;33(30):3894–3907. doi: 10.1038/onc.2013.352. [DOI] [PubMed] [Google Scholar]
- 2.Hottiger MO, Hassa PO, Lüscher B, Schüler H, Koch-Nolte F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem Sci. 2010;35(4):208–219. doi: 10.1016/j.tibs.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Vyas S, Matic I, Uchima L, et al. Family-wide analysis of poly(ADP-ribose) polymerase activity. Nat Commun. 2014;5:4426. doi: 10.1038/ncomms5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 5.Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 6.Bridges CB. The origin of variation. Amer Nat. 1922;56(642):51–63. [Google Scholar]
- 7.Shaw HM, Hall M. Emerging treatment options for recurrent ovarian cancer: the potential role of olaparib. Onco Targets Ther. 2013;6:1197–1206. doi: 10.2147/OTT.S30748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72(5):1117–1130. doi: 10.1086/375033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levy-Lahad E, Friedman E. Cancer risks among BRCA1 and BRCA2 mutation carriers. Br J Cancer. 2007;96(1):11–15. doi: 10.1038/sj.bjc.6603535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 12.Fong PC, Yap TA, Boss DS, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28(15):2512–2519. doi: 10.1200/JCO.2009.26.9589. [DOI] [PubMed] [Google Scholar]
- 13.McCabe, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66(16):8109–8115. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 14.Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4(10):814–819. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gelmon KA, Tischkowitz M, Mackay H, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12(9):852–861. doi: 10.1016/S1470-2045(11)70214-5. [DOI] [PubMed] [Google Scholar]
- 17.Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366(15):1382–1392. doi: 10.1056/NEJMoa1105535. [DOI] [PubMed] [Google Scholar]
- 18.Tutt A, Robson M, Garber JE, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376(9737):235–244. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- 19.Byrski T, Huzarski T, Dent R, et al. Response to neoadjuvant therapy with cisplatin in BRCA1-positive breast cancer patients. Breast Cancer Res Treat. 2009;115(2):359–363. doi: 10.1007/s10549-008-0128-9. [DOI] [PubMed] [Google Scholar]
- 20.Byrski T, Gronwald J, Huzarski T, et al. Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J Clin Oncol. 2010;28(3):375–379. doi: 10.1200/JCO.2008.20.7019. [DOI] [PubMed] [Google Scholar]
- 21.Burgess M, Puhalla S. BRCA 1/2-mutation related and sporadic breast and ovarian cancers: more alike than different. Front Oncol. 2014;4:19. doi: 10.3389/fonc.2014.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dedes KJ, Wilkerson PM, Wetterskog D, Weigelt B, Ashworth A, Reis-Filho JS. Synthetic lethality of PARP inhibition in cancers lacking BRCA1 and BRCA2 mutations. Cell Cycle. 2011;10(8):1192–1199. doi: 10.4161/cc.10.8.15273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gonzalez-Angulo AM, Timms KM, Liu S, et al. Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin Cancer Res. 2011;17(5):1082–1089. doi: 10.1158/1078-0432.CCR-10-2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stracker TH, Petrini JH. The MRE11 complex: starting from the ends. Nat Rev Mol Cell Bio. 2011;12(2):90–103. doi: 10.1038/nrm3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mutter GL, Lin MC, Fitzgerald JT, et al. Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. J Nat Cancer Inst. 2000;92(11):924–930. doi: 10.1093/jnci/92.11.924. [DOI] [PubMed] [Google Scholar]
- 26.Gupta A, Yang Q, Pandita RK, et al. Cell cycle checkpoint defects contribute to genomic instability in PTEN deficient cells independent of DNA DSB repair. Cell Cycle. 2009;8(14):2198–2210. doi: 10.4161/cc.8.14.8947. [DOI] [PubMed] [Google Scholar]
- 27.Dedes KJ, Wetterskog D, Mendes-Pereira AM, et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci Transl Med. 2010;2(53):53ra75. doi: 10.1126/scitranslmed.3001538. [DOI] [PubMed] [Google Scholar]
- 28.Mendes-Pereira AM, Martin SA, Brough R, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1(6–7):315–322. doi: 10.1002/emmm.200900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forster MD, Dedes KJ, Sandhu S, et al. Treatment with olaparib in a patient with PTEN-deficient endometrioid endometrial cancer. Nat Rev Clin Oncol. 2011;8(5):302–306. doi: 10.1038/nrclinonc.2011.42. [DOI] [PubMed] [Google Scholar]
- 30.Sandhu SK, Schelman WR, Wilding G, et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013;14(9):882–892. doi: 10.1016/S1470-2045(13)70240-7. [DOI] [PubMed] [Google Scholar]
- 31.Dong X, Wang L, Taniguchi K, et al. Mutations in CHEK2 associated with prostate cancer risk. Am J Hum Genet. 2003;72(2):270–280. doi: 10.1086/346094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beltran H, Yelensky R, Frampton GM, et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol. 2013;63(5):920–926. doi: 10.1016/j.eururo.2012.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lynch HT, Smyrk T, Kern SE, et al. Familial pancreatic cancer: a review. Semin Oncol. 1996;23(2):251–275. [PubMed] [Google Scholar]
- 34.Hruban RH, Petersen GM, Ha PK, Kern SE. Genetics of pancreatic cancer. From genes to families. Surg Oncol Clin N Am. 1998;7(1):1–23. [PubMed] [Google Scholar]
- 35.Murphy KM, Brune KM, Griffin C, et al. Evaluation of candidate genes MAP2K4, MADH4, ACVR1B, and BRCA2 in familial pancreatic cancer, deleterious BRCA2 mutations in 17% Cancer Res. 2002;62(13):3789–3793. [PubMed] [Google Scholar]
- 36.Breast Cancer Linkage Consortium Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst. 1999;91(15):1310–1316. doi: 10.1093/jnci/91.15.1310. [DOI] [PubMed] [Google Scholar]
- 37.Pishvaian MJ, Wang H, Zhuang T, et al. A phase I/II study of ABT-888 in combination with 5-fluorouracil (5-FU) and oxaliplatin (Ox) in patients with metastatic pancreatic cancer (MPC) J Clin Oncol. 2013;31(Suppl 4) abst 147. [Google Scholar]
- 38.Lowery MA, Kelsen DP, Stadler ZK, et al. An emerging entity: pancreatic adenocarcinoma associated with a known BRCA mutation: clinical descriptors, treatment implications, and future directions. Oncologist. 2011;16(10):1397–1402. doi: 10.1634/theoncologist.2011-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Albert JM, Cao C, Kim KW, et al. Inhibition of poly(ADP-ribose) polymerase enhances cell death and improves tumor growth delay in irradiated lung cancer models. Clin Cancer Res. 2007;13(10):3033–3042. doi: 10.1158/1078-0432.CCR-06-2872. [DOI] [PubMed] [Google Scholar]
- 40.Chatterjee P, Choudhary GS, Sharma A, et al. PARP inhibition sensitizes to low dose-rate radiation TMPRSS2-ERG fusion gene-expressing and PTEN-deficient prostate cancer cells. PLoS One. 2013;8(4):e60408. doi: 10.1371/journal.pone.0060408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rubin MA, Maher CA, Chinnaiyan AM. Common gene rearrangements in prostate cancer. J Clin Oncol. 2011;29(27):3659–3668. doi: 10.1200/JCO.2011.35.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barreto-Andrade JC, Efimova E, Mauceri HJ, et al. Response of human prostate cancer cells and tumors to combining PARP inhibition with ionizing radiation. Mol Cancer Ther. 2011;10(7):1185–1193. doi: 10.1158/1535-7163.MCT-11-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tomoda T, Kurashige T, Moriki T, Yamamoto H, Fujimoto S, Taniguchi T. Enhanced expression of poly(ADP-ribose) synthetase gene in malignant lymphoma. Am J Hematol. 1991;37(4):223–227. doi: 10.1002/ajh.2830370402. [DOI] [PubMed] [Google Scholar]
- 44.Shiobara M, Miyazaki M, Ito H, et al. Enhanced polyadenosine diphosphate-ribosylation in cirrhotic liver and carcinoma tissues in patients with hepatocellular carcinoma. J Gastroenterol Hepatol. 2001;16(3):338–344. doi: 10.1046/j.1440-1746.2001.02378.x. [DOI] [PubMed] [Google Scholar]
- 45.Dent AR, Lindeman GJ, Clemons M, et al. Phase I trial of the oral PARP inhibitor olaparib in combination with paclitaxel for first- or second-line treatment of patients with metastatic triple-negative breast cancer. Breast Cancer Res. 2013;15(5):R88. doi: 10.1186/bcr3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Calabrese CR, Almassy R, Barton S, et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst. 2004;96(1):56–67. doi: 10.1093/jnci/djh005. [DOI] [PubMed] [Google Scholar]
- 47.Curtin NJ, Szabo C. Therapeutic applications of PARP inhibitors: anticancer therapy and beyond. Mol Aspects Med. 2013;34(6):1217–1256. doi: 10.1016/j.mam.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Evers B, Drost R, Schut E, et al. Selective inhibition of BRCA2-deficient mammary tumor cell growth by AZD2281 and cisplatin. Clin Cancer Res. 2008;14(12):3916–3925. doi: 10.1158/1078-0432.CCR-07-4953. [DOI] [PubMed] [Google Scholar]
- 49.Kummar S, Chen A, Ji J, et al. Phase I Study of PARP inhibitor ABT-888 in combination with topotecan in adults with refractory solid tumors and lymphomas. Cancer Res. 2011;71(17):5626–5634. doi: 10.1158/0008-5472.CAN-11-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kummar S, Ji J, Morgan R, et al. A phase I study of veliparib in combination with metronomic cyclophosphamide in adults with refractory solid tumors and lymphomas. Clin Cancer Res. 2012;18(6):1726–1734. doi: 10.1158/1078-0432.CCR-11-2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Long HJ, 3rd, Bundy BN, Grendys EC, Jr, et al. Gynecologic Oncology Group Study Randomized phase III trial of cisplatin with or without topotecan in carcinoma of the uterine cervix: a Gynecologic Oncology Group Study. J Clin Oncol. 2005;23(21):4626–4633. doi: 10.1200/JCO.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 52.Samol J, Ranson M, Scott E, et al. Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: a phase I study. Invest New Drugs. 2012;30(4):1493–1500. doi: 10.1007/s10637-011-9682-9. [DOI] [PubMed] [Google Scholar]
- 53.Plummer R, Lorigan P, Steven N, et al. A phase II study of the potent PARP inhibitor, Rucaparib (PF-01367338, AG014699), with temozolomide in patients with metastatic melanoma demonstrating evidence of chemopotentiation. Cancer Chemother Pharmacol. 2013;71(5):1191–1199. doi: 10.1007/s00280-013-2113-1. [DOI] [PubMed] [Google Scholar]
- 54.Burger RA, Sill MW, Monk BJ, Greer BE, Sorosky JI. Phase II trial of bevacizumab in persistent or recurrent epithelial ovarian cancer or primary peritoneal cancer: a Gynecologic Oncology Group Study. J Clin Oncol. 2007;25(33):5165–5171. doi: 10.1200/JCO.2007.11.5345. [DOI] [PubMed] [Google Scholar]
- 55.Matulonis UA, Berlin S, Ivy P, et al. Cediranib, an oral inhibitor of vascular endothelial growth factor receptor kinases, is an active drug in recurrent epithelial ovarian, fallopian tube, and peritoneal cancer. J Clin Oncol. 2009;27(33):5601–5606. doi: 10.1200/JCO.2009.23.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tentori L, Lacal PM, Muzi A, et al. Poly(ADP-ribose) polymerase (PARP) inhibition or PARP-1 gene deletion reduces angiogenesis. Eur J Cancer. 2007;43(14):2124–2133. doi: 10.1016/j.ejca.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 57.Liu JF, Tolaney SM, Birrer M, et al. A Phase 1 trial of the poly(ADP-ribose) polymerase inhibitor olaparib (AZD2281) in combination with the anti-angiogenic cediranib (AZD2171) in recurrent epithelial ovarian or triple-negative breast cancer. Eur J Cancer. 2013;49(14):2972–2978. doi: 10.1016/j.ejca.2013.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu J, Barry WT, Birrer MJ, et al. A randomized phase 2 trial comparing efficacy of the combination of the PARP inhibitor olaparib and the antiangiogenic cediranib against olaparib alone in recurrent platinum-sensitive ovarian cancer; Presented at: ASCO Annual meeting; May 30–June 3, 2014; Chicago, IL. [Google Scholar]
- 59.Ibrahim Y, García-García C, Serra V, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012;2(11):1036–1047. doi: 10.1158/2159-8290.CD-11-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Edwards SL, Brough R, Lord CJ, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451(7182):1111–1115. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 61.Rottenberg S, Jaspers JE, Kersbergen A, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105(44):17079–17084. doi: 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jaspers JE, Kersbergen A, Boon U, et al. Loss of 53BP1 causes PARP inhibitor resistance in BRCA1-mutated mouse mammary tumors. Cancer Discov. 2013;3(1):68–81. doi: 10.1158/2159-8290.CD-12-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee JM, Ledermann JA, Kohn EC. PARP inhibitors for BRCA 1/2 mutation-associated and BRCA-like malignancies. Ann Oncol. 2014;25(1):32–40. doi: 10.1093/annonc/mdt384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bernges F, Zeller WJ. Combination effects of poly(ADP-ribose) polymerase inhibitors and DNA-damaging agents in ovarian tumor cell lines – with special reference to cisplatin. J Cancer Res Clin Oncol. 1996;122(11):665–670. doi: 10.1007/BF01209029. [DOI] [PubMed] [Google Scholar]
- 65.Menear KA, Adcock C, Boulter R, et al. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J Med Chem. 2008;51(20):6581–6591. doi: 10.1021/jm8001263. [DOI] [PubMed] [Google Scholar]