

Abstract
Corticotropin-releasing hormone (CRH) is a central integrator in the brain of endocrine and behavioral stress responses, whereas activation of the endocannabinoid CB1 receptor suppresses these responses. Although these systems regulate overlapping functions, few studies have investigated whether these systems interact. Here we demonstrate a novel mechanism of CRH-induced anxiety that relies on modulation of endocannabinoids. Specifically, we found that CRH, through activation of the CRH receptor type 1 (CRHR1), evokes a rapid induction of the enzyme fatty acid amide hydrolase (FAAH), which causes a reduction in the endocannabinoid anandamide (AEA), within the amygdala. Similarly, the ability of acute stress to modulate amygdala FAAH and AEA in both rats and mice is also mediated through CRHR1 activation. This interaction occurs specifically in amygdala pyramidal neurons and represents a novel mechanism of endocannabinoid–CRH interactions in regulating amygdala output. Functionally, we found that CRH signaling in the amygdala promotes an anxious phenotype that is prevented by FAAH inhibition. Together, this work suggests that rapid reductions in amygdala AEA signaling following stress may prime the amygdala and facilitate the generation of downstream stress-linked behaviors. Given that endocannabinoid signaling is thought to exert “tonic” regulation on stress and anxiety responses, these data suggest that CRH signaling coordinates a disruption of tonic AEA activity to promote a state of anxiety, which in turn may represent an endogenous mechanism by which stress enhances anxiety. These data suggest that FAAH inhibitors may represent a novel class of anxiolytics that specifically target stress-induced anxiety.
Keywords: anxious behavior, basolateral amygdala, endocannabinoid signaling, HPA axis, stress
Introduction
Endocannabinoid (eCB) signaling acts as a gatekeeper over stress and anxiety, fine-tuning neurobehavioral responses in constantly changing environments. Under resting conditions, the eCB, anandamide (AEA), appears to constrain activation of the amygdala, anxiety-like behaviors, and downstream activation of the hypothalamic-pituitary-adrenal (HPA) axis. As such, systemic and intra-amygdalar blockade of the cannabinoid-type 1 (CB1) receptor increases neural activation in the amygdala (Patel et al., 2005; Newsom et al., 2012), anxiety-like behavior (Haller et al., 2004; Dono and Currie, 2012), and HPA axis activity (Ganon-Elazar and Akirav, 2009; Hill et al., 2009; Newsom et al., 2012), indicating that amygdala eCB signaling gates stress- and anxiety-like responses.
In response to stressful stimuli, the amygdala shows dynamic changes in eCB levels, encompassing rapid AEA declines and increases in the activity of the AEA-hydrolyzing enzyme, fatty acid amide hydrolase (FAAH), following acute and chronic stress (Patel et al., 2005; Rademacher et al., 2008; Hill et al., 2009, 2013). Stress exposure is also associated with central increases of the other eCB, 2-arachidonoylglycerol (2-AG), in corticolimbic stress-regulating regions (Evanson et al., 2010; Hill et al., 2011; Wang M et al., 2012), although this occurs on a more delayed temporal scale and appears to be involved in the termination of stress responses (Hill and Tasker, 2012). To date, there is little knowledge regarding the mechanism or functional role of the rapid decline in AEA within the amygdala, although we hypothesized that it may be paramount to the generation of stress responses (Hill et al., 2009). Endocannabinoids are sensitive to glucocorticoid-dependent regulation (Di et al., 2003; Hill et al., 2010; Atsak et al., 2012); however, glucocorticoids increase, rather than decrease, amygdala AEA levels (Hill et al., 2010). This suggests that a distinctly independent stress-linked signaling system, and one that is rapid and upstream of the HPA axis, mediates the stress-induced decline of AEA content occurring in the amygdala.
Corticotropin-releasing hormone (CRH) represents an ideal candidate for linking stress with AEA changes. During stress, CRH receptor type 1 (CRHR1) activity in the limbic forebrain (Smith et al., 1998; Müller et al., 2003; Refojo et al., 2011), and particularly the basolateral amygdala (BLA) (Roozendaal et al., 2002; Gehlert et al., 2005; Jochman et al., 2005; Bruchas et al., 2009; Sztainberg et al., 2010), initiates stress-related and anxiety-like behavioral responses. In downstream stress circuits, such as the anterior pituitary, CRHR1 activation shapes endocrine stress responses by stimulating pituitary release of adrenocorticotropin-releasing hormone to initiate increases in peripheral glucocorticoid production (Chen et al., 1993; Dedic et al., 2012).
Although the eCB system and CRHR1 are both found in stress-regulating structures and exert well-documented opposing roles on stress- and anxiety-like behavior (Timpl et al., 1998; Kathuria et al., 2003), few studies have investigated whether these systems exhibit any functional interactions (Bayatti et al., 2005; Kamprath et al., 2009; Kupferschmidt et al., 2012a, 2012b). Using a range of pharmacological, biochemical, and genetic techniques, we have found that the activity of the postsynaptic AEA-metabolizing intracellular enzyme FAAH is remarkably sensitive to induction by CRHR1 signaling and that this relationship is critical for the regulation of anxiety.
Materials and Methods
Subjects
Male Sprague Dawley rats (65–70 d old; Charles River) were pair-housed with food and water available ad libitum (12 h light/dark cycle; lights on 09:00).
A recently characterized line of conditional knock-out mice was also used, which do not express CRHR1 receptors on dorsal telencephalon glutamatergic neurons (CRHR1-Glu KO). Thorough breeding details have been previously reported (Refojo et al., 2011), but in brief, this breeding strategy uses the neuronal marker Nex (Neurod6/Math2) to drive Cre expression selectively in dorsal telencephalon glutamatergic neurons (Goebbels et al., 2006). CRHR1-Glu KO mice were generated by crossing CRHR1flox/flox mice with Nex-Cre mice to remove CRHR1 receptors from glutamate neurons within a range of brain regions within the dorsal telencephalon, including projection neurons of the cortex, hippocampus, and the BLA (Goebbels et al., 2006; Refojo et al., 2011).
Mice expressing the enzyme β-galactosidase (β-GAL) on CRHR1-positive neurons were also used to visualize and detect CRHR1 expression in the BLA. This line was created using a knock-in approach to introduce a tau-LacZ reporter into the CRHR1 gene locus that allows visualization of CRHR1 expression via the tau-LacZ reporter. The thorough description of the knock-in strategy can be found elsewhere (Kühne et al., 2012).
Male wild-type (WT), CRHR1-Glu KO, and β-GAL/CRHR1 mice were housed 2–4 per cage, were 2–4 months old during testing, and housed under standard laboratory conditions, with food and water available ad libitum (12 h light/dark cycle; lights on 09:00).
All tests began 2 h after lights on and followed protocols approved by the University of Calgary Animal Care Committee, guidelines from the Canadian Council on Animal Care, and the Care and Use of Laboratory Animals from the Government of Bavaria, Germany.
Central surgeries and pharmacological testing
Cannulations.
Rats had 1 week of acclimatization before surgeries, at which time they weighed 270–300 g, then were anesthetized (100 mg/kg ketamine hydrochloride and 7 mg/kg xylazine) and implanted with unilateral cannulae (23-gauge) targeting the lateral ventricle (coordinates: −0.90 mm anteroposterior, 1.4 mm mediolateral, and −2.8 mm dorsoventral from skull). In subsequent experiments, a separate cohort received bilateral BLA cannulations (coordinates: −2.8 mm anteroposterior, ±5.0 mm mediolateral, −6.5 mm dorsoventral). Following cannulations, rats were given 1 week of recovery and exposed to daily mock injections for 3 consecutive days before testing. Intracerebroventricular injections were delivered over 2 min at a flow rate of 1.0 μl/min, whereas intra-BLA injections were delivered 0.2 μl per side, over a period of 32 s. All injectors extended 2 mm from the guide cannula. Injectors were left in place for 90 s following drug administration to ensure the dispersal of the injection.
Drug administration and tissue collection.
Rats were given intracerebroventricular injections of saline or CRH (1 μg, Tocris Bioscience, #1151) and were decapitated 10, 30, or 120 min later. In additional studies, cannulated rats received intracerebroventricular injections of saline, CRHR1 agonist (cortagine; 300 ng; Phoenix Pharmaceuticals, #019-38), or CRHR2 agonist (urocortin 2; 1 μg; Phoenix Pharmaceuticals; #019-34) and then were decapitated 30 min later. Evans blue dye was used (50 μl, 0.02%) to verify ventricular cannula placement, and the amygdala and prefrontal cortex (PFC) were dissected, flash frozen on dry ice, and stored at −80°C for later analysis of eCB levels, FAAH activity, protein levels, and mRNA expression. For dissections, the PFC was dissected from fresh brains following decapitation as a tissue block composed of medial PFC and anterior cingulate cortex, which was anatomically defined as the area dorsal to the anterior olfactory nucleus and medial to the corpus callosum and claustrum formation. The posterior boundary for this tissue block was ∼2.20 mm anterior to bregma. The amygdala was dissected as a tissue block from a coronal slice that spanned from ∼−4.0 mm to −1.6 mm from bregma. Cortical tissue lateral to the external capsule (primarily composed of piriform cortex) was excised, and the amygdala (predominately composed of basolateral nuclei but also containing some tissue from the central and medial nuclei) was dissected as the remaining triangular tissue block lateral to the optic tract. Trunk blood samples were also collected at the time of decapitation for corticosterone (CORT) analysis.
For elevated plus maze (EPM) testing, rats received bilateral intra-BLA infusions of vehicle (PEG/Tween 80/saline, 5:5:90, v/v/v), CRH (10 ng), URB597 (10 ng; Cayman Chemical, #10046), or CRH + URB597 30 min before EPM testing. To survey possible CORT changes, 100 μl tail blood samples were collected 30 min after the onset of EPM exposure. Rats were then anesthetized, and brains were harvested for cryostat slicing and Nissl staining to verify cannula placement.
Blood sampling and CORT measures.
Blood samples were centrifuged at 10,000 rpm for 20 min at 4°C. Plasma was then stored at −20°C until testing with an ELISA CORT kit (Cayman Chemical, #500655). Samples were tested in duplicate and diluted 1:1000 to ensure stress-induced levels fit on the linear portion of the standard curve. The detection limit of the assay was 30 pg/ml at 80% binding.
Stress and behavioral studies
Restraint.
In all restraint studies involving rats, subjects received intraperitoneal injections of vehicle or drug 30 min before a 30 min restraint episode. In these studies, rats received injections of the CRHR1 antagonist antalarmin (20 mg/kg, dissolved in saline; Tocris Bioscience, #2778), the glucocorticoid receptor antagonist RU486 (20 mg/kg, dissolved in a 1:1 v/v mixture of propylene glycol and saline; Sigma, #M8046) or their respective vehicles, before restraint to determine the role of CRH and CORT, respectively, in the effects of stress on eCB levels. For experiments involving mice lacking CRHR1 on dorsal telencephalon glutamatergic neurons, wild-type and CRHR1-Glu KO subjects were restrained in 50 ml Falcon tubes with breathing holes for 30 min. For all restraint experiments, subjects were rapidly decapitated following the end of restraint, or a comparable duration in the home cage. For all groups, brains were removed following rapid decapitation, and brain regions (amygdala, PFC) were dissected out as described above, flash frozen on dry ice, and then stored at −80°C until analysis.
EPM.
The EPM was composed of two open and two closed arms (48 cm long × 15 cm wide × 38 cm height of closed arms) and elevated 48 cm above the floor. All 5 min sessions occurred in a dark room with dim lighting, were video-recorded, commenced with rats being placed in the center facing an open arm, and the maze was cleaned between sessions. Parameters analyzed by a blind observer included total duration spent in open or closed arms (four paws into the arm was considered an entry), the percentage of total time spent in open and closed arms, and the percentage of open arm entries to assess possible treatment effects on anxiety-like behavior. The total number of arm entries made per subject was also measured to survey possible differences in locomotion or activity.
Tissue processing
FAAH activity.
Membrane fractions were prepared from homogenized tissue and samples normalized according to protein content (Hill et al., 2009). Samples were then exposed to 0.2 nm 3H-AEA and varying concentrations of cold AEA (0.05–1.5 μm) to determine the maximal hydrolytic activity (Vmax) that FAAH displayed for AEA (Hill et al., 2009).
Lipid extractions from tissue samples.
Lipid extraction from dissected tissue was performed as described previously (Patel et al., 2005), except that the homogenization volume was spiked with 50 pmol d8-2-AG and 84 pmol d8-AEA per sample. Immediately before liquid chromatography-tandem mass spectrometry analysis, samples were reconstituted in 200 μl of 9:1 methanol/water (v/v), vortexed, and transferred to autosampler vials.
Analysis of endocannabinoid levels.
Analytes were quantified using liquid chromatography-tandem mass spectrometry on a Quantum triple-quadrupole mass spectrometer in positive-ion mode using selected reaction monitoring. Detection of eCBs was performed as previously described (Hermanson et al., 2013).
Western blot analysis of FAAH protein.
Tissue for Western blot analysis was homogenized in RIPA lysis buffer (150 nm NaCl, 50 mm Tris-HCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, Complete protease inhibitors, Roche). Briefly, samples were disrupted using 50 mm stainless steel beads and the TissueLyser LT (QIAGEN), at 50 Hz for 5 min, followed by centrifugation at 16,000 × g at 4°C for 20 min. Protein quantification of supernatant was performed using a BCA kit according to the manufacturer's protocol (ThermoFisher Scientific); 30 μg of protein was denatured using NuPage LDS Sample Buffer and Reducing Agent (Invitrogen) at 95°C for 5 min and then loaded into NuPage 3–8% Tris-acetate Gels (Invitrogen). Gels were run for 1 h at 200 V. Gels were then transferred to nitrocellulose membranes for 1 h at 150 mA. After transferring, membranes were washed in TBST and blocked for 1 h in 5% milk. Primary antibodies (mouse anti-GAPDH, 1:10,000; Abcam, #ab8245; and rabbit anti-FAAH, 1:500; Cayman Chemical, #101600), as described previously (Zabouri et al., 2011), were incubated overnight at 4°C. The following day, membranes were washed and incubated with secondary antibodies (both 1:10,000; donkey anti-mouse IgG IRDye 800 CW, Licor, #926-32212; and donkey anti-rabbit IgG IRDye 680LT, Licor, #926-68023) for 1 h at room temperature, then washed before imaging on a Licor Odyssey CLx (Licor). Images were analyzed for relative differences in optical density using ImageJ software (National Institutes of Health) and the gel analyzer macro to determine area under the curve. The total area under the curve for FAAH was divided by the total area for GAPDH. Values were normalized to the control group for each experiment (i.e., vehicle intracerebroventricular or basal, nonstressed controls).
RT-PCR analysis of FAAH mRNA.
RNA was isolated from tissue using the RNeasy Plus Universal Mini Kit, the TissueLyser LT, and a Qiacube (QIAGEN) according to the manufacturer's protocol. Isolated RNA was converted to cDNA using the qScript cDNA SuperMix (Quanta Biosciences) according to the manufacturer's protocol. Primers for RPLP2 (control gene) and FAAH were designed using Primer Quest Software (IDTDNA) and checked for specificity using the NCBI BLAST tool. Primers for RPLP2 and FAAH are as follows: RPLP2, forward, 5′-CGCTACGTTGCCTCTTATCT-3′, reverse, 5′-GCCCACGCTGTCTAGTATTT-3′; FAAH, forward, 5′-GGAGCTAAGGAGTGAGTATTTC-3′, reverse, 5′-CACCTGTCCTTATCCCATTAC-3′. qPCR was performed using the PerfeCTa SYBR Green FastMix (Quanta Biosciences) according to the manufacturer's protocol using a RotoGeneQ lightcycler (QIAGEN). Cycle thresholds (Ct; the point at which fluorescence crosses threshold) values were assayed for both the control gene and gene of interest for all groups. The ΔCt between the control gene (RPLP2) and gene of interest (FAAH) was calculated for the control groups (intracerebroventricular vehicle or basal, nonstressed control). Finally, ΔΔCt, or the change between the control groups (intracerebroventricular vehicle or basal, nonstressed) and the manipulated groups (intracerebroventricular CRH or stressed, respectively) was calculated. Data were normalized so that the average of the control groups was set to 1.
Immunohistochemistry for CRHR1 and FAAH colabeling.
Given that CRHR1 antibodies are unreliable (Refojo et al., 2011), a line of genetically altered mice was used to survey indirect CRHR1 protein expression. These mice express the enzyme β-GAL within CRHR1-positive cells using a tau-LacZ reporter gene into the CRHR1 gene locus as described above and previously (Kühne et al., 2012).
Brains were harvested as previously described (Kühne et al., 2012), and tissue was cut into 35 μm sections on a cryostat and stored in cryoprotectant (0.1 m potassium phosphate buffered solution [KPBS], 30% ethylene glycol, 30% glycerol). Immunohistochemistry was performed using primary antibodies for FAAH (1:20; Cayman Chemical, #101600), previously validated for specificity in FAAH KO mice (Zabouri et al., 2011), and β-GAL (1:250; Abcam, #AB9361) with an overnight incubation. KPBS rinsed tissue was then exposed to secondary antibodies for 1 h at room temperature (1:1000; Invitrogen), rinsed, and incubated in DAPI solution for 10 min at room temperature (1:1000; Invitrogen), then rinsed and mounted with fluoromount medium (Sigma). Blocking solution contained KPBS, 0.03% Triton-X, 0.1% saponin, 5% normal goat serum, and 1% BSA.
Images were captured on a Leica (DM5500 Q) confocal microscope and analyzed with cellSens software (Olympus). Estimates of coexpression were generated by counting bilaterally across sections the number of total FAAH-positive cells within a standardized analysis box (400 μm2) within the BLA. Then, separately, the total number of β-GAL+ FAAH colabeled cells were counted to determine whether 100%, or only a subset, of FAAH-positive cells were positive for CRHR1 coexpression. Bilateral counts were averaged across sections, and the number of cells positive for coexpression was normalized to total FAAH-positive cell counts. Three mice were used for this analysis, and two or three BLA sections were surveyed for each subject. This analysis was also repeated for the in situ hybridization analysis described below to survey the degree of coexpression among CRHR1 and FAAH mRNA using a standardize box (100 μm2), sampled across 3 subjects, and bilaterally across two or three sections per subject. During the analysis of both protein and mRNA, CRHR1 expression was always found to coexpress FAAH; therefore, additional analyses examining possible subpopulations of FAAH-negative CRHR1 cells were not undertaken.
Fluorescent in situ hybridization.
Detection of mRNA for CRHR1 (gene ID: 58959) and FAAH (gene ID: 29347) was performed using RNAscope probes according to the manufacturer's instructions (ACD). In brief, 30 μm cryostat-prepared sections of unfixed frozen rat tissue were treated as previously described with positive and negative controls (Wang F et al., 2012). Tissue was then examined for coexpression of CRHR1 and FAAH mRNA using the neuronal marker DAPI to visualize individual cells using a Leica confocal microscope.
Statistical analysis
GraphPad Prism 6.02 was used for statistical analyses. Parametric, unpaired t tests were used to analyze the effects of intracerebroventricular vehicle versus CRH on eCB levels, CORT levels, and FAAH activity, the effects of stress and intracerebroventricular CRH administration on FAAH mRNA and protein levels, and the histology analysis. One-way ANOVA was used to analyze the effects of intracerebroventricular vehicle versus CRHR1 and CRHR2 agonism. All other experiments were assessed using a two-way ANOVA. Where appropriate, the Bonferroni method was performed for specific group comparisons. For all data, p < 0.05 indicates statistical significance.
Results
Central CRH signaling rapidly modulates amygdala AEA hydrolysis
Exposure to acute stress has been reliably shown to reduce AEA content within the amygdala of both rats and mice (Patel et al., 2005; Rademacher et al., 2008; Hill et al., 2009). This effect appears to be upstream of the HPA axis (Hill et al., 2009), which led us to investigate the role of CRH in this phenomenon. We additionally surveyed possible changes in the PFC, as both regions are heavily implicated in stress and anxiety regulation and are important sites for the modulatory effects of eCBs on these processes (Rubino et al., 2008; Hill et al., 2009; Hill et al., 2011; Dono and Currie, 2012). Similar to what has been documented following acute stress (Patel et al., 2005; Rademacher et al., 2008; Hill et al., 2009), intracerebroventricular CRH significantly reduced AEA levels in the amygdala 10 min following CRH administration (t(11) = 2.25, p = 0.046; Figure 1A), and significantly increased the hydrolytic activity (Vmax) of FAAH in the amygdala (t(6) = 2.74, p = 0.033; Figure 1B). These effects of intracerebroventricular CRH at 10 min were specific to AEA as there was no effect of CRH on 2-AG levels within the amygdala (t(11) = 0.26, p = 0.80; Figure 1C). At this time point, there was also no effect of CRH administration on circulating plasma CORT (t(19) = 0.74, p = 0.46; Figure 1D).
Figure 1.

Response of amygdala endocannabinoid levels, FAAH activity, and circulating CORT following intracerebroventricular CRH. A–D, Effects of intracerebroventricular vehicle or 1 μg CRH infusions 10 min after administration on (A) AEA, (B) FAAH hydrolytic activity, (C) 2-AG, and (D) circulating plasma CORT levels. E–H, Effects of intracerebroventricular vehicle or 1 μg CRH infusions 30 min after administration on (E) AEA, (F) FAAH hydrolytic activity, (G) 2-AG, and (H) CORT. I–L, Effects of intracerebroventricular vehicle or 1 μg CRH infusions 120 min after administration on (I) AEA, (J) FAAH hydrolytic activity, (K) 2-AG, and (L) CORT. Data are mean + SEM; n = 6–8/group for AEA and 2-AG levels; n = 4/group for FAAH hydrolytic activity; n = 10–12/group for CORT analysis. *p < 0.05.
Similar to what we found at the 10 min time point, intracerebroventricular CRH significantly reduced AEA levels in the amygdala 30 min following administration (t(11) = 2.80, p = 0.017; Figure 1E), significantly increased FAAH activity (t(5) = 2.64, p = 0.046; Figure 1F), and had no effect on 2-AG levels (t(11) = 0.12, p = 0.90; Figure 1G), which is consistent with the effects of acute stress (Patel et al., 2005, 2009; Hill et al., 2009). Also similar to acute stress exposure, intracerebroventricular CRH significantly increased CORT levels at 30 min after injection (t(19) = 3.37, p = 0.003; Figure 1H).
To determine whether our observed eCB changes were temporary or longer lasting, a 120 min time point following intracerebroventricular CRH was assessed. Two hours following CRH administration, there was no longer a significant difference in amygdala AEA levels (t(16) = 1.19, p = 0.24; Figure 1I) or FAAH activity (t(7) = 0.27, p = 0.79; Figure 1J). At this time point, however, CRH administration did elicit a significant increase in 2-AG content within the amygdala (t(16) = 2.21, p = 0.042; Figure 1K), and there was also still a significant increase in circulating CORT (t(13) = 3.51, p = 0.003; Figure 1L).
Unlike the amygdala, 10 min following intracerebroventricular CRH there was no effect on PFC AEA levels (t(11) = 0.49, p = 0.63; Figure 2A) or FAAH activity (t(6) = 0.96, p = 0.37; Figure 2B), but there was a significant increase in 2-AG levels (t(11) = 2.7, p = 0.02; Figure 2C). Similarly, 30 min following CRH administration, there was no effect on PFC AEA levels (t(11) = 1.27, p = 0.23; Figure 2D), PFC FAAH activity (t(6) = 1.65, p = 0.15; Figure 2E), and no effect on PFC 2-AG levels (t(10) = 1.6, p = 0.14; Figure 2F). Analysis of PFC tissue 120 min following CRH-intracerebroventricular administration also revealed no effect of CRH on PFC AEA levels (t(15) = 0.027, p = 0.97; Figure 2G), and PFC FAAH activity (t(6) = 0.76, p = 0.47; Figure 2H), but a significant increase in PFC 2-AG levels (t(16) = 2.22, p = 0.04; Figure 2I). PFC 2-AG levels are positively modulated by CORT (Hill et al., 2011); therefore, a parsimonious assumption would be that our observed PFC 2-AG elevations at the 120 min time point might correspond to CRH-dependent CORT increases. However, given that intracerebroventricular CRH increased 2-AG within the PFC at only 10 min following administration, there also appears to be a more rapid mechanism by which CRH increases 2-AG levels in the PFC. Collectively, these data demonstrate that CRH administration rapidly increases FAAH activity and reduces AEA levels within the amygdala, but not the PFC. Given that the changes in amygdala AEA levels and FAAH activity precede the increase in CORT release driven by CRH, these findings suggest that these effects of CRH on amygdala FAAH/AEA are likely occurring independent of increases in glucocorticoid secretion.
Figure 2.

Response of PFC endocannabinoid levels and FAAH activity following intracerebroventricular CRH. A–C, Effects of intracerebroventricular vehicle or 1 μg CRH infusions 10 min after administration on (A) AEA, (B) FAAH hydrolytic activity, (C) 2-AG. D–F, Effects of intracerebroventricular vehicle or 1 μg CRH infusions 30 min after administration on (D) AEA, (E) FAAH hydrolytic activity, (F) 2-AG. G–I, Effects of intracerebroventricular vehicle or 1 μg CRH infusions 120 min after administration on (G) AEA, (H) FAAH hydrolytic activity, (I) 2-AG. Data are mean + SEM; n = 6–8/group for AEA and 2-AG levels; n = 4/group for FAAH hydrolytic activity. *p < 0.05.
Based on our limited understanding of the factors influencing FAAH activity, we subsequently assessed whether CRH-induced changes in FAAH activity were driven by rapid increases in FAAH mRNA or protein. Examination of FAAH protein, as determined by Western blot analysis, and FAAH mRNA using RT-PCR demonstrated that there were no changes in the amygdala for FAAH protein (t(10) = 0.47, p = 0.63; Figure 3A,E), or mRNA (t(12) = 0.66, p = 0.51; Figure 3B). In the PFC, there was also no effect of CRH on FAAH protein (t(10) = 0.19, p = 0.85; Figure 3C,E) or mRNA (t(10) = 0.8, p = 0.44; Figure 3D).
Figure 3.

FAAH protein and mRNA levels in the amygdala and PFC following intracerebroventricular CRH. A, B, Effects of intracerebroventricular vehicle or 1 μg CRH on amygdala FAAH, (A) protein and (B) mRNA levels, 30 min after intracerebroventricular infusion. C, D, Effects of intracerebroventricular vehicle or 1 μg CRH on PFC FAAH, (C) protein and (D) mRNA levels, 30 min after intracerebroventricular infusion. E, Representative Western blot images of FAAH (63 kDa) and GAPDH (40 kDa) bands in the amygdala and PFC. A, C, For protein analysis, the optical density for FAAH was divided by the optical density for GAPDH and was normalized to the vehicle control group and represented using arbitrary units. B, D, For FAAH mRNA analysis, fold change of FAAH mRNA levels is shown normalized to vehicle controls (relative to RRLP2) and represented as arbitrary units. Data are mean + SEM; n = 5 or 6/group for FAAH protein; n = 6–8/group for FAAH mRNA levels.
Because CRH binds to both CRHR1 and CRHR2, we next tested which receptor might be mediating these amygdala effects. Rats were administered intracerebroventricular injections of either saline, a CRHR1 agonist (cortagine), or CRHR2 agonist (urocortin 2), and decapitated 30 min later to examine FAAH activity and eCB levels. Agonist treatment significantly altered amygdala AEA levels (F(2,18) = 5.85, p = 0.012; Figure 4A), and FAAH activity (F(2,11) = 4.9, p = 0.036; Figure 4B), which post hoc analysis revealed was due to cortagine-induced increases in FAAH activity (p < 0.05), and a concomitant reduction in amygdala AEA levels (p < 0.05; Fig. 4A,B). As expected, and in complement to our previous findings at 10 and 30 min following intracerebroventricular CRH, there was no treatment effect on 2-AG levels in the amygdala 30 min following CRHR1 or CRHR2 agonism (F(2,18) = 1.56, p = 0.24; Figure 4C). CRHR2 activation also had no effect on any of the measures examined (p > 0.05; Fig. 4A–C). These findings suggested that our previous CRH-mediated changes in AEA and FAAH activity were dependent on activation of CRHR1 but not CRHR2. The lack of effect of CRHR2 agonism on FAAH activity or AEA levels in the amygdala is not surprising as CRHR2 is generally thought to be primarily expressed within the medial amygdala (Hauger et al., 2006), a region where FAAH expression is relatively sparse. In contrast, CRHR1 is widely expressed across amygdala subnuclei on both inhibitory and excitatory neurons (Van Pett et al., 2000; Nie et al., 2004; Kühne et al., 2012), with prominent expression on anxiety-regulating pyramidal neurons within the BLA (Müller et al., 2003; Refojo et al., 2011). Further, CRHR1 agonism also significantly increased CORT levels relative to vehicle controls at 30 min (F(2,31) = 10.19, p = 0.0004; data not shown), indicating that our previous CRH-driven increases in CORT were likely mediated by CRHR1 as well.
Figure 4.

Analysis of amygdala and PFC levels of endocannabinoids and FAAH activity after CRH receptor agonism. A–C, Effects of intracerebroventricular vehicle, cortagine (CRHR1 agonist; R1, 300 ng) or urocortin 2 (CRHR2 agonist; R2, 1 μg) on amygdala 30 min after infusion, (A) AEA, (B) FAAH hydrolytic activity, (C) 2-AG. D–F, Effects of intracerebroventricular vehicle, cortagine (CRHR1 agonist; R1, 300 ng) or urocortin 2 (CRHR2 agonist; R2, 1 μg) on PFC 30 min after infusion, (D) AEA, (E) FAAH hydrolytic activity, (F) 2-AG. Data are mean + SEM; n = 6–8/group for AEA and 2-AG levels; n = 4/group for FAAH hydrolytic activity. *p < 0.05.
Similar CRHR1-mediated decreases in AEA were not observed in the PFC (F(2,18) = 0.51, p = 0.61; Figure 4D), but surprisingly CRHR1-dependent increases in FAAH activity were (F(2,10) = 5.73, p = 0.028; post hoc, p < 0.05; Figure 4E) without a corresponding change to its ligand substrate AEA. This suggests that FAAH activity and AEA content within the PFC may be sensitive to CRHR1 stimulation, but the magnitude of this effect is not as reliable or robust as what was seen within the amygdala. Similar to what was seen with CRH administration, activation of CRHR1, but not CRHR2, resulted in an increase in 2-AG levels within the PFC (F(2,19) = 5.18, p = 0.02, post hoc, p < 0.05; Figure 4F) indicating that the ability of CRH to increase prefrontocortical 2-AG levels was also CRHR1, but not CRHR2, mediated.
CRHR1 receptor signaling on glutamatergic neurons within the amygdala mediates stress-induced modulation of FAAH activity and AEA
Having established that CRHR1 activation is sufficient to increase FAAH activity and reduce AEA levels in the amygdala, we next sought to determine whether CRHR1 activation is also necessary for stress to evoke changes in FAAH/AEA signaling. To this extent, we administered a CRHR1 antagonist (intraperitoneal) to rats 30 min before a 30 min restraint stress exposure. For all acute stress studies, our a priori hypothesis was that exposure to stress would reduce AEA levels in untreated animals (consistent with published data in both rats and mice) (Patel et al., 2005; Rademacher et al., 2008; Hill et al., 2009), whereas pharmacological disruption of CRHR1 signaling would prevent this stress-induced decline in AEA levels. Consistent with the previous effects, there was a trend for CRHR1 antagonism to prevent stress-induced reductions in amygdala AEA levels (F(1,23) = 3.16, p = 0.08; Figure 5A). Based on our a priori hypothesis that stress would reduce amygdala AEA levels, a subsequent Bonferroni analysis confirmed this effect in vehicle-treated animals (p < 0.05), but not the CRHR1 antagonist group (p > 0.05). As expected, a significant interaction emerged wherein CRHR1 antagonism prevented stress-induced increases in FAAH activity (F(1,12) = 6.11, p < 0.001; post hoc, p < 0.05; Figure 5B), demonstrating that CRHR1 drives the effects of stress on amygdala eCB signaling. There was also no interaction of stress and antagonism on amygdala 2-AG levels (F(1,23) = 1.56, p = 0.22; Figure 5C). Consistent with previous reports (Deak et al., 1999; Wong et al., 1999; Marinelli et al., 2007) administration of antalarmin had no effect on stress-induced CORT levels (stress vehicle CORT: 268.5 ± 19.1 ng/ml vs stress antalarmin CORT: 269.3 ± 35.0 ng/ml; t(22) = 0.02, p > 0.05). It has been demonstrated that the lack of impact of CRHR1 antagonism on stress-induced HPA axis activation is mediated by additional roles of the vasopressin V1B receptor in maintaining HPA axis drive following stress (Ramos et al., 2006; Bouchez et al., 2012); however, these data do again support the hypothesis that the effects of stress or CRH on amygdala FAAH activity and AEA levels occur independently of CORT.
Figure 5.

Role of CRHR1 receptors in the effects of stress on endocannabinoid levels and FAAH activity in amygdala and PFC. A–F, Effects of intraperitoneal CRHR1 antagonism (R1-ANT, 20 mg/kg) on (A–C) amygdala, (A) AEA, (B) FAAH hydrolytic activity, (C) 2-AG and (D–F) PFC, (D) AEA, (E) FAAH hydrolytic activity, (F) 2-AG, either under basal conditions or immediately following the end of 30 min of restraint stress. G–L, Effects of CRHR1-deletion off of glutamatergic dorsal telencephalon neurons (R1-Glu KO) on (G–I) amygdala, (G) AEA, (H) FAAH hydrolytic activity, (I) 2-AG and (J–L) PFC, (J) AEA, (K) FAAH hydrolytic activity, (L) 2-AG, similarly, under basal conditions or immediately after the end of 30 min of restraint stress. Data are mean + SEM; n = 6–8/group for AEA and 2-AG levels; n = 4/group for FAAH hydrolytic activity. *p < 0.05.
Examination of the PFC found no interaction of stress and CRHR1 antagonism on AEA levels (F(1,22) = 0.0092, p = 0.92; Figure 5D), FAAH activity (F(1,15) = 1.1, p = 0.31; Figure 5E), or 2-AG (F(1,22) = 0.07, p = 0.78; Figure 5F), further underscoring the amygdala as a potentially specialized site for displaying CRH-dependent FAAH changes.
To further determine the neuronal populations involved in this response, we subsequently used a line of genetically modified mice that lack CRHR1 exclusively on dorsal telencephalon glutamatergic neurons (CRHR1-Glu KO). Previous work has demonstrated that limbic CRHR1 exists on glutamatergic neurons exclusively within the cortex, hippocampus, and BLA (Refojo et al., 2011). Similar to the established effects of FAAH inhibition, CRHR1-Glu KO mice are protected against stress-induced anxiety (Refojo et al., 2011). Given that FAAH expression is primarily restricted to glutamatergic pyramidal neurons (Egertová et al., 1998; Tsou et al., 1998), the converging phenotypes of these two manipulations led us to hypothesize that CRHR1-possessing glutamatergic neurons are the site of interaction between these systems. To determine whether CRHR1-glutamatergic neurons contribute to stress-induced changes in AEA tone, we examined the effects of a 30 min restraint stress on AEA levels and FAAH activity in the amygdala of wild-type and CRHR1-Glu KO mice. Similar to our previous antagonism study, again our a priori hypothesis was, staying consistent with the literature, that exposure to stress would reduce AEA levels in wild-type mice (Patel et al., 2005; Rademacher et al., 2008; Hill et al., 2009), whereas genetic disruption of CRHR1 signaling would prevent this stress-induced decline in AEA levels. In complement to our previous rat experiments, and consistent with previous reports in mice (Patel et al., 2005; Rademacher et al., 2008), there was a trend for an interaction between genotype and stress on amygdala AEA levels (F(1,21) = 3.7, p = 0.06; Figure 5G). Based on this existing literature, which has shown that stress reduces amygdala AEA levels, our a priori hypothesis that stress would reduce AEA levels in wild-type, but not CRHR-Glu KO mice, allowed us to perform planned comparisons using Bonferroni analysis between treatment conditions. This analysis determined that stress exposure significantly decreased AEA levels in the amygdala of wild-type (p < 0.05), but not CRHR1-Glu KO mice (p > 0.05). There was also a significant interaction of stress and genotype on amygdala FAAH activity (F(1,12) = 5.06, p = 0.043; Figure 5H), which post hoc analysis revealed was attributed to stress-induced increases in FAAH activity among wild-type (p < 0.05), but not CRHR1-Glu KO mice (p > 0.05). There was no interaction of genotype and stress on amygdala 2-AG levels (F(1,21) = 2.03, p = 0.16; Figure 5I).
Similar to our CRHR1 antagonist findings, there was no interaction of stress and genotype on PFC AEA levels (F(1,21) = 0.60, p = 0.44; Figure 5J), FAAH activity (F(1,12) = 1.99, p = 0.18; Figure 5K), or 2-AG levels (F(1,22) = 2.2, p = 0.15; Figure 5L). Although CORT levels were not examined in these mice within the current study, it has been demonstrated previously that CRHR1-Glu KO mice do not exhibit any differences in stress-induced CORT secretion (Refojo et al., 2011), which is consistent with our lack of effect of antalarmin on stress-induced CORT release in rats.
Similar to what was seen following intracerebroventricular-CRH administration, the effects of stress on FAAH activity do not appear to be due to changes in the transcription or translation of the FAAH enzyme itself as stress had no effect on the level of protein or mRNA transcripts. Following the end of 30 min restraint stress, there was no change in the amygdala for FAAH protein (t(10) = 1.50, p = 0.16; Figure 6A), or mRNA (t(11) = 0.86, p = 0.4; Figure 6B). In the PFC, there was also no effect of stress on FAAH protein (t(10) = 0.74, p = 0.47; Figure 6C), or mRNA (t(12) = 0.58, p = 0.57; Figure 6D).
Figure 6.

FAAH protein and mRNA levels in the amygdala and PFC in response to acute restraint stress. A, B, Effects of 30 min of restraint stress on amygdala FAAH (A) protein and (B) mRNA levels. C, D, Effects of 30 min of restraint stress on PFC FAAH (C) protein and (D) mRNA levels. E, Representative Western blot images of FAAH (63 kDa) and GAPDH (40 kDa) protein bands in the amygdala and PFC. A, C, For protein analysis, the optical density for FAAH was divided by the optical density for GAPDH and was normalized to the basal control group and represented using arbitrary units. B, D, For FAAH mRNA analysis, fold change of FAAH mRNA levels is shown normalized to basal controls (relative to RRLP2) and represented as arbitrary units. Data are mean + SEM; n = 5 or 6/group for FAAH protein; n = 6–8/group for FAAH mRNA levels.
Given that the data we have collected so far strongly indicate that the effects of stress on amygdala AEA levels are driven by direct activation of CRHR1, and not due to a consequential increase in CORT, we next sought to examine directly whether CORT had any effect on stress-induced declines in amygdala AEA levels. To examine this process further, the effects of stress-induced eCB changes in the amygdala were investigated in the presence and absence of a glucocorticoid antagonist RU486 (20 mg/kg). As stress is known to produce local increases in CRH release within the amygdala (Merlo Pich et al., 1995; Roozendaal et al., 2002) and increase the release of CORT, this approach offers the ability to dissociate the effects of endogenous amygdala CRH versus the effects of stress-induced CORT on changes in eCB levels. There was a main effect of stress on amygdala AEA levels (F(1,22) = 10.01, p = 0.004; Table 1), which is consistent with previous studies that have demonstrated stress-induced AEA decreases in the amygdala (Patel et al., 2005; Rademacher et al., 2008; Hill et al., 2009), but no effect of RU486 (F(1,22) = 0.12, p = 0.72) and no interaction (F(1,22) = 1.43, p = 0.24). There was also no interaction of stress and RU486 treatment on 2-AG levels in the amygdala (F(1,22) = 0.57, p = 0.46; Table 1). Together, these data confirm that stress-induced decreases in amygdala AEA content are not modulated by glucocorticoid signaling but are reversed by CRHR1 antagonism, further supporting the importance of physiologically released CRH in this process.
Table 1.
Glucocorticoids do not mediate acute stress-induced reductions in AEA content within the amygdalaa
| Treatment | AEA (pmol/g) | 2-AG (nmol/g) |
|---|---|---|
| Vehicle–basal | 14.96 ± 0.39 | 10.63 ± 0.50 |
| RU486–basal | 14.58 ± 0.51 | 11.07 ± 0.72 |
| Vehicle–stress* | 13.00 ± 0.39 | 10.02 ± 0.61 |
| RU486–stress* | 13.70 ± 0.47 | 9.34 ± 1.05 |
aData are mean ± SEM: amygdala levels of AEA (n = 6 or 7/group) and 2-AG (n = 6 or 7/group) under basal conditions and following the end of 30 min restraint stress in subjects that were administered intraperitoneal injections of vehicle or the glucocorticoid antagonist RU486 (20 mg/kg).
*p < 0.05, main effect of stress.
Anatomical colocalization of CRHR1 and FAAH within the BLA
FAAH expression is predominately limited to glutamatergic principal neurons (Egertová et al., 1998; Tsou et al., 1998), and it has been determined that, within the BLA, CRHR1 is also primarily localized to glutamatergic neurons (Refojo et al., 2011). Given that our findings indicated that deletion of CRHR1 from this neuronal population abrogated the effects of stress on amygdala FAAH activity and AEA levels, we sought to determine whether there could be coexpression of these proteins within glutamatergic principal neurons of the BLA, providing an anatomical basis for this interaction. To test this, we used a recently developed reporter mouse line that expresses the enzyme β-GAL on CRHR1-positive cells, as there are currently no reliable antibodies for the CRHR1 receptor (Refojo et al., 2011; Kühne et al., 2012). To further establish what percentage of FAAH-positive cells in the BLA are susceptible to CRHR1 modulation, and hence coexpress CRHR1, immunohistochemical analysis of the BLA found that 100% of FAAH-immunoreactive cells coexpressed β-GAL (CRHR1) labeling, with no detectable significant difference between FAAH-positive cell counts or counts of colabeled cells (t(14) = 0.53, p = 0.61; Figure 7B–E). To further validate this coexpression, we subsequently performed in situ hybridization for FAAH and CRHR1 mRNA in the BLA (Fig. 7F–I), and similar to our protein analysis, 100% of FAAH-positive cells were also found to reliably coexpress CRHR1 gene transcripts in the BLA (t(14) = 0.97, p = 0.34), with no significant difference in expression between cells positive for FAAH mRNA, or cells positive for both FAAH/CRHR1 mRNA. Given the consistent overlap between expression of CRHR1 and FAAH within the same cells in the BLA, these data create a compelling argument for an anatomical basis for the functional interaction between CRHR1 and FAAH within glutamatergic neurons of the BLA.
Figure 7.

Coexpression of CRHR1 and FAAH protein and mRNA in the BLA. A, Illustration demonstrating the approximate area (blue box) of the amygdala where microscope images of protein and mRNA were taken. B–E, Representative images of immunohistochemical expression of (B) the neuronal marker DAPI, (C) FAAH, and (D) β-GAL, which is expressed only in CRHR1-expressing cells (Kühne et al., 2012). E, Merged image, demonstrating that FAAH and β-GAL (indirect CRHR1 label) are coexpressed (white arrows) in cells of the BLA. Scale bar, 40 μm. F–I, Representative in situ hybridization images of (F) DAPI, (G) FAAH, and (H) CRHR1 in the BLA. I, Merged image demonstrating that FAAH and CRHR1 transcripts are coexpressed in the BLA (white arrows). Scale bar, 24 μm.
Inhibition of FAAH activity in the amygdala blocks CRH-mediated anxiety
Inhibition of FAAH and deletion of CRHR1 from glutamatergic neurons both attenuate stress-induced anxiety without altering basal anxiety (Haller et al., 2009; Refojo et al., 2011; Hill et al., 2013), suggesting a potentially overlapping, if not interrelated, process involving these factors. The BLA, and particularly the activation of principal neurons in the BLA, is also widely implicated in regulating anxiety-like behavior (Sajdyk et al., 1999; Rainnie et al., 2004; Tye et al., 2011). Thus, the final arm of this study examined the possibility of a CRH–eCB interaction within the BLA as a mediating influence on anxiety-like behavior. To test this possibility, we examined the effects of local intra-BLA administration of CRH and FAAH inhibition (URB597), alone or in combination, on anxiety-like responses and HPA axis activation. Examination of animals where bilateral cannulae were within the boundaries of the BLA (see Fig. 8) revealed an interaction between CRH and FAAH inhibition on the total open arm time (F(1,44) = 4.83, p = 0.03; Figure 8A) and the percentage of open arm time during EPM testing (F(1,44) = 4.93, p = 0.03; Figure 8B). Specifically, intra-BLA CRH decreased both the total time and percentage of time spent in the EPM open arms relative to vehicle-treated animals (p < 0.05), and these effects were attenuated by coadministration of FAAH inhibitor. Similarly, an interaction was also found for the total time spent in closed arms of the EPM (F(1,44) = 6.25, p = 0.02; Figure 8C) and the percentage of time spent in closed arms (F(1,44) = 6.16, p = 0.02; Figure 8D) where CRH increased both the duration and percentage of time spent in EPM closed arms (p < 0.05), and these effects were attenuated by FAAH inhibition. No interaction of CRH and FAAH inhibition was found for the number of total entries made during EPM testing (F(1,44) = 0.27, p = 0.61, Figure 8E). However, there was a trend for an interaction of CRH and FAAH inhibition on the percentage of open arm entries made (F(1,44) = 2.65, p = 0.11; Figure 8F). With respect to the HPA axis, there was a significant interaction between CRH and FAAH inhibition on CORT levels (F(1,44) = 4.35, p = 0.04; Figure 8G), such that intra-BLA CRH increased circulating CORT concentrations relative to vehicle-treated animals following exposure to the EPM (p < 0.05), and this effect was attenuated by coadministration of FAAH inhibitor. The specificity of these effects to the BLA is highlighted by the fact that there was no effect of CRH on anxiety-like behavior in the EPM test or on CORT levels for off target cannulae (data not shown). Together, these findings show that the anxiogenic behavioral effects of CRH in the BLA involve the recruitment of FAAH activity, and consistent with previous work, demonstrate that FAAH inhibition selectively attenuates stress-induced anxiety but has no effect on basal anxiety (Haller et al., 2009; Hill et al., 2013). The model for this interaction can be visualized in Figure 9, where we hypothesize that the ability of AEA signaling to gate activation of the pyramidal cells within the BLA during stress is due to a preferential regulation of glutamatergic transmission. As such, we predict that stress evokes the release of CRH, which activates CRHR1 and triggers FAAH activity to deplete the signaling pool of AEA. The loss of this AEA signal results in a disinhibition of glutamatergic inputs to the BLA, which facilitates activation of anxiety-inducing BLA pyramidal neurons (Tye et al., 2011). This model is based upon several lines of converging evidence that clearly demonstrate that the ability of eCB signaling to dampen anxiety involves a suppression of glutamatergic, and not GABAergic, neurotransmission (Rossi et al., 2010; Rey et al., 2012; Ruehle et al., 2013).
Figure 8.
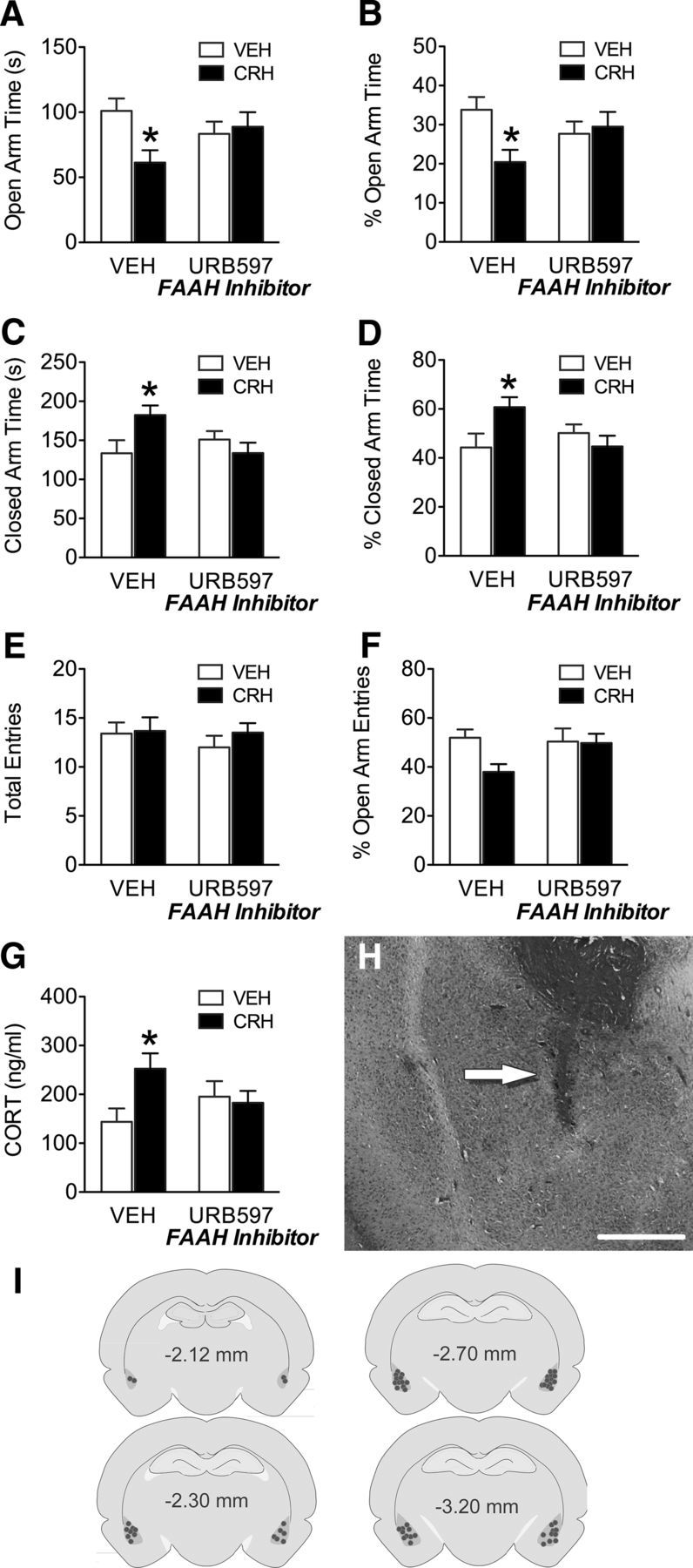
CRH administration and FAAH inhibition on anxiety-like behavior. A–G, Effects of vehicle, CRH (10 ng), FAAH inhibition (URB597; 10 ng), or CRH + URB597, 30 min before testing on the EPM on (A) open arm time, (B) percentage open arm time, (C) closed arm time, (D) percentage closed arm time, (E) total entries, (F) percentage open arm entries, and (G) circulating plasma CORT levels 30 min following EPM testing. H, Representative photomicrograph showing injector placement (arrow). Scale bar, 350 μm. I, Illustration of coronal sections showing representative cannula placements. Data are mean + SEM; n = 10–14/group. *p < 0.05.
Figure 9.

Model of CRH and FAAH interactions within the amygdala following exposure to stress. Under resting basal conditions (left), AEA activation of CB1 receptors provides an inhibitory tone on presynaptic glutamate release within the BLA, constraining excess glutamate release and neuronal activation within the BLA. Following exposure to stress (right panel), release of CRH, and the subsequent activation of postsynaptic CRHR1, rapidly increases the enzymatic activity of FAAH, resulting in a depletion of the signaling pool of AEA within the BLA. The loss of this inhibitory AEA tone may in turn result in a facilitation of glutamate release and subsequent activation of excitatory pyramidal projection neurons of the BLA targeting downstream regions regulating stress-related endocrine and behavioral outputs.
Discussion
These data reveal a novel relationship between the stress-regulating CRH and eCB systems within the amygdala, which has an essential contribution to the initiation of stress and anxiety-like behavior. This model builds upon existing literature that has established CRHR1 signaling in the BLA as a catalyst promoting stress and anxiety, by demonstrating that CRHR1, but not CRHR2, activation rapidly suppresses AEA levels in the amygdala, which in turn, contributes to the generation of an anxious state. Given that AEA signaling in the BLA has been linked to modulating both intrinsic excitability of this nucleus (Shin et al., 2010; Gunduz-Cinar et al., 2013) and the regulation of HPA axis activity and anxiety (Hill et al., 2009, 2013; John and Currie, 2012; Gunduz-Cinar et al., 2013; Bedse et al., 2014), these data support a putative model whereby a CRH-mediated disruption of AEA signaling represents an early ‘initiation’ response to stress. This raises the interesting possibility that a disruption of amygdalar AEA signaling may be an endogenous mechanism to prime the amygdala in response to stress and facilitate the development of an anxious state. This is consistent with the fact that pharmacological blockade of eCB signaling produces a stress-like state, whereas FAAH inhibition exerts minimal effects under basal conditions but restricts anxiety following exposure to stress or aversive environments (Kathuria et al., 2003; Haller et al., 2009; Hill et al., 2013; Bluett et al., 2014). As well, these findings support the emerging view that FAAH inhibition helps maintain “emotional homeostasis” and resiliency to stress-induced behaviors in adverse contexts (Haller et al., 2013). Consistent with this model, it is interesting to note that there is a remarkable parallel between the effects of local activation of CRHR1 within the BLA and a disruption of eCB signaling, such as increased grooming, vigilance, anorexia, anxiety, impaired fear extinction, and hyperactivation of the HPA axis (Rubino et al., 2000; Marsicano et al., 2002; Jochman et al., 2005; Ganon-Elazar and Akirav, 2009; Hill et al., 2009; Dono and Currie, 2012; Abiri et al., 2014). As these effects are all prototypical manifestations of a stress response, these data support a putative model whereby CRHR1-FAAH/AEA crosstalk within the BLA is an integral step in the manifestation of a stress response.
The data generated herein create a compelling argument for the importance of CRH signaling in mediating the effects of stress on amygdala AEA and FAAH in a manner that is independent of glucocorticoids. Previous studies have demonstrated biochemical and behavioral effects of intracerebroventricular CRH as rapid as 5–10 min (Spina et al., 2002; Refojo et al., 2005); and accordingly, our initial experiments examining the effects of intracerebroventricular CRH on amygdala eCB levels did suggest that the effects of CRH on FAAH and AEA in the amygdala occur within 10 min from CRH administration, a time at which CORT levels were not significantly elevated. Moreover, genetic and pharmacological disruption of CRHR1 signaling reversed the effects of stress on AEA and FAAH within the amygdala but did not prevent stress-induced HPA axis activation. Although surprising, this is consistent with previous reports (Deak et al., 1999; Wong et al., 1999; Marinelli et al., 2007) and supports the argument that limbic CRH signaling can coordinate anxiety-like behavior in a manner distinct from its regulation of the HPA axis (Refojo et al., 2011). This hypothesis also receives support from the fact that pharmacological blockade of glucocorticoid receptors did not alter stress-induced suppression of AEA levels within the amygdala. Collectively, these data clearly demonstrate that stress-induced suppression of AEA signaling in the amygdala is driven by the release of CRH and not a subsequent increase in glucocorticoids.
This is not to suggest that glucocorticoids are not important for the regulation of AEA within the amygdala. We have previously reported that administration of CORT in the absence of stress actually increases, not decreases, amygdala AEA levels (Hill et al., 2010). Given that the reduction in amygdala AEA levels had normalized at 120 min following CRH administration, a time at which CORT levels remained elevated, the possibility exists that the subsequent elevation in CORT following the release of CRH actually functions as part of a feedback loop that acts to elevate and normalize AEA levels within the amygdala, thus resulting in the termination of stress-induced anxiety. This hypothesis is consistent with existing evidence that has shown that stress-induced CORT increases normalize amygdala functionality following stress (Henckens et al., 2010, 2012). This is also consistent with the ability of CORT to reduce stress-induced increases in amygdala excitability through an eCB-dependent mechanism (Karst et al., 2010). As such, the possibility exists that CORT-mediated AEA elevations in the amygdala may be involved in a negative feedback loop terminating the neurobehavioral effects of stress.
The mechanisms by which CRHR1 and FAAH are functionally connected remain elusive. Our current data indicate that the increase in FAAH activity produced by administration of CRH or exposure to stress is not accompanied by increases in either FAAH mRNA or protein levels, suggesting that this effect is not mediated by a rapid effect on mRNA transcription or protein translation. Given the rapid onset of the effects, it seems that the most parsimonious explanation is that CRHR1 activation recruits an intracellular signaling cascade that exerts post-translational modification of the FAAH enzyme, resulting in rapid and dynamic changes in enzyme activity. Unfortunately, to date, there are no published accounts of any post-translational modifications, such as phosphorylation possibly influencing FAAH activity. Of interest, however, is that the intracellular signaling cascades driven by activation of CRHR1 differ between the amygdala and PFC (Refojo et al., 2005), suggesting that there may be biased agonism of the CRHR1 depending on the cell type it resides in (glutamatergic vs GABAergic). This divergence in cellular responses is consistent with the region-specific coupling of CRHR1 and FAAH we have documented here and thus may help to shed light on the signaling mechanisms linking these two pathways.
Although these data strongly implicate a direct functional interaction between CRHR1 and FAAH within glutamatergic neurons of the BLA, the methods used do not rule out alternative possibilities. For example, it is possible that the effects of CRH and stress could be mediated through an indirect noradrenergic mechanism because both of these manipulations reliably activate the locus ceruleus, which in turn will release norepinephrine into the amygdala. Although investigation of this possibility is beyond the scope of the current paper, future work will examine the role of norepinephrine in these processes. Additionally, the fact that local CRH and FAAH manipulations within the BLA proper exerted clear effects on anxiety does highlight the importance of this structure in this relationship but does not rule out the possibility that similar interactions are occurring in other structures as many of the documented effects of stress on AEA levels occur throughout many other limbic regions (Patel et al., 2004, 2005; Dubreucq et al., 2012; McLaughlin et al., 2012; Wang M et al., 2012; Marco et al., 2013; Bluett et al., 2014).
In addition to the effects of CRH on AEA content within the amygdala, the current data indicate that CRH also resulted in a delayed elevation in 2-AG levels within both the PFC and amygdala (seen 120 min following CRH administration in both structures), which is consistent with a delayed increase in 2-AG driven through a CORT-mediated process, as has been documented within the PFC (Hill et al., 2011). However, unlike the amygdala, the effect of CRH on 2-AG within the PFC occurs on a very rapid temporal scale as we were able to detect elevations in tissue levels of 2-AG within the PFC 10 min following CRH administration. Because these effects precede glucocorticoid secretion, they are likely driven by the direct actions of CRH acting on CRHR1 to mobilize 2-AG, and not a consequential increase due to CORT secretion. Future work is required to fully understand the mechanisms and functional relevance of CRH-mediated elevations in 2-AG content within the PFC.
In a broader context, these data are also consistent with the findings in humans that genetic polymorphisms in the FAAH gene, which reduce protein levels of FAAH and are accompanied by constitutive elevations in AEA (Chiang et al., 2004; Sipe et al., 2010), result in reduced activation and accelerated habituation of the amygdala in response to stress, as well as lower levels of trait anxiety (Hariri et al., 2009; Gunduz-Cinar et al., 2013; Dincheva et al., 2015). This would suggest that, consistent with the present rodent data, in humans reduced FAAH activity limits activation of the amygdala by stress, likely by tempering the capacity of CRH to prime and engage this system to evoke anxiety. This model, in turn, may have significant implications for understanding the neurobiology of anxiety disorders, as well as drug development for novel anxiolytics. As well, this study importantly highlights that other conditions associated with elevated CRH and negative affect, such as drug withdrawal (Contarino and Papaleo, 2005; Heilig and Koob, 2007), depression, and panic disorder (Sajdyk et al., 1999), may represent clinical conditions that could benefit significantly from pharmacological interventions that inhibit FAAH activity.
Footnotes
This work was supported by the Canadian Institutes of Health Research and the Natural Sciences and Engineering Research Council of Canada to M.N.H. and National Institutes of Health Grants MH090412 and R21 MH103515 to S.P., J.M.G. was supported by a Postdoctoral Fellowship from Alberta Innovates Health Solutions. D.J.H. was supported by National Institutes of Health predoctoral fellowship DA031572. T.T.Y.L. was the recipient of a doctoral fellowship from Canadian Institutes of Health Research. H.A.V. was the recipient of a summer research support award from the Canadian Consortium for the Investigation of Cannabinoids. K.I.H. was the recipient of a studentship from the Branch Out Neurological Foundation. We thank Caitlin Riebe, Judith Reichel, and Heather Leduc-Pessah for technical assistance; Reg Sidhu for microscopy help; Drs. Quentin Pittman, Tuan Trang, and Cam Teskey for sharing surgical space and/or laboratory equipment; Dr. Carol Schuurmans for input with the β-GAL staining; Dr. Jaideep Bains for thoughtful suggestions on the manuscript; and the Vanderbilt University Mass Spectrometry Research Center.
M.N.H. is a scientific consultant to Pfizer. The remaining authors declare no competing financial interests. All funding agencies had no influence on the design, execution, or publishing of this work.
References
- Abiri D, Douglas CE, Calakos KC, Barbayannis G, Roberts A, Bauer EP. Fear extinction learning can be impaired or enhanced by moduation of the CRF system in the basolateral nucleus of the amygdala. Behav Brain Res. 2014;271:234–239. doi: 10.1016/j.bbr.2014.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atsak P, Hauer D, Campolongo P, Schelling G, McGaugh JL, Roozendaal B. Glucocorticoids interact with the hippocampal endocannabinoid system in impairing retrieval of contextual fear memory. Proc Natl Acad Sci U S A. 2012;109:3504–3509. doi: 10.1073/pnas.1200742109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayatti N, Hermann H, Lutz B, Behl C. Corticotropin-releasing hormone-mediated induction of intracellular signaling pathways and brain-derived neurotrophic factor expression is inhibited by the activation of the endocannabinoid system. Endocrinology. 2005;146:1205–1213. doi: 10.1210/en.2004-1154. [DOI] [PubMed] [Google Scholar]
- Bedse G, Colangeli R, Lavecchia AM, Romano A, Altieri F, Cifani C, Cassano T, Gaetani S. Role of the basolateral amygdala in mediating the effects of the fatty acid amide hydrolase inhibitor URB597 on HPA axis response to stress. Eur Neuropsychopharmacol. 2014;24:1511–1523. doi: 10.1016/j.euroneuro.2014.07.005. [DOI] [PubMed] [Google Scholar]
- Bluett RJ, Gamble-George JC, Hermanson DJ, Hartley ND, Marnett LJ, Patel S. Central anandamide deficiency predicts stress-induced anxiety: behavioral reversal through endocannabinoid augmentation. Transl Psychiatry. 2014;4:e408. doi: 10.1038/tp.2014.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchez G, Millan MJ, Rivet JM, Billiras R, Boulanger R, Gobert A. Quantification of extracellular levels of corticosterone in the basolateral amygdaloid complex of freely-moving rats: a dialysis study of circadian variation and stress-induced modulation. Brain Res. 2012;1452:47–60. doi: 10.1016/j.brainres.2012.01.010. [DOI] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Lemos JC, Chavkin C. CRF1-R activation of the dynorphin/kappa opioid system in the mouse basolateral amygdala mediates anxiety-like behavior. PLoS One. 2009;4:e8528. doi: 10.1371/journal.pone.0008528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Lewis KA, Perrin MH, Vale WW. Expression cloning of a human corticotropin-releasing-factor receptor. Proc Natl Acad Sci U S A. 1993;90:8967–8971. doi: 10.1073/pnas.90.19.8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang KP, Gerber AL, Sipe JC, Cravatt BF. Reduced cellular expression and activity of the P129T mutant of human fatty acid amide hydrolase: evidence for a link between defects in the endocannabinoid system and problem drug use. Hum Mol Genet. 2004;13:2113–2119. doi: 10.1093/hmg/ddh216. [DOI] [PubMed] [Google Scholar]
- Contarino A, Papaleo F. The corticotropin-releasing factor receptor-1 pathway mediates the negative affective states of opiate withdrawal. Proc Natl Acad Sci U S A. 2005;102:18649–18654. doi: 10.1073/pnas.0506999102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deak T, Nguyen KT, Ehrlich AL, Watkins LR, Spencer RL, Maier SF, Licinio J, Wong ML, Chrousos GP, Webster E, Gold PW. The impact of the nonpeptide corticotropin-releasing hormone antagonist antalarmin on behavioral and endocrine responses to stress. Endocrinology. 1999;140:79–86. doi: 10.1210/endo.140.1.6415. [DOI] [PubMed] [Google Scholar]
- Dedic N, Touma C, Romanowski CP, Schieven M, Kühne C, Ableitner M, Lu A, Holsboer F, Wurst W, Kimura M, Deussing JM. Assessing behavioural effects of chronic HPA axis activation using conditional CRH-overexpressing mice. Cell Mol Neurobiol. 2012;32:815–828. doi: 10.1007/s10571-011-9784-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di S, Malcher-Lopes R, Halmos KC, Tasker JG. Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci. 2003;23:4850–4857. doi: 10.1523/JNEUROSCI.23-12-04850.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dincheva I, Drysdale AT, Hartley CA, Johnson DC, Jing D, King EC, Ra S, Gray JM, Yang R, DeGruccio AM, Huang C, Cravatt BF, Glatt CE, Hill MN, Casey BJ, Lee FS. FAAH genetic variation enhances fronto-amygdala function in mouse and human. Nat Commun. 2015 doi: 10.1038/ncomms7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dono LM, Currie PJ. The cannabinoid receptor CB1 inverse agonist AM251 potentiates the anxiogenic activity of urocortin I in the basolateral amygdala. Neuropharmacology. 2012;62:192–199. doi: 10.1016/j.neuropharm.2011.06.019. [DOI] [PubMed] [Google Scholar]
- Dubreucq S, Matias I, Cardinal P, Häring M, Lutz B, Marsicano G, Chaouloff F. Genetic dissection of the role of cannabinoid type-1 receptors in the emotional consequences of repeated social stress in mice. Neuropsychopharmacology. 2012;37:1885–1900. doi: 10.1038/npp.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egertová M, Giang DK, Cravatt BF, Elphick MR. A new perspective on cannabinoid signalling: complementary localization of fatty acid amide hydrolase and the CB1 receptor in rat brain. Proc Biol Sci. 1998;265:2081–2085. doi: 10.1098/rspb.1998.0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evanson NK, Tasker JG, Hill MN, Hillard CJ, Herman JP. Fast feedback inhibition of the HPA axis by glucocorticoids is mediated by endocannabinoid signaling. Endocrinology. 2010;151:4811–4819. doi: 10.1210/en.2010-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganon-Elazar E, Akirav I. Cannabinoid receptor activation in the basolateral amygdala blocks the effects of stress on the conditioning and extinction of inhibitory avoidance. J Neurosci. 2009;29:11078–11088. doi: 10.1523/JNEUROSCI.1223-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehlert DR, Shekhar A, Morin SM, Hipskind PA, Zink C, Gackenheimer SL, Shaw J, Fitz SD, Sajdyk TJ. Stress and central urocortin increase anxiety-like behavior in the social interaction test via the CRF1 receptor. Eur J Pharmacol. 2005;509:145–153. doi: 10.1016/j.ejphar.2004.12.030. [DOI] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, Nave KA. Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis. 2006;44:611–621. doi: 10.1002/dvg.20256. [DOI] [PubMed] [Google Scholar]
- Gunduz-Cinar O, Macpherson KP, Cinar R, Gamble-George J, Sugden K, Williams B, Godlewski G, Ramikie TS, Gorka AX, Alapafuja SO, Nikas SP, Makriyannis A, Poulton R, Patel S, Hariri AR, Caspi A, Moffitt TE, Kunos G, Holmes A. Convergent translational evidence of a role for anandamide in amygdala-mediated fear extinction, threat processing and stress-reactivity. Mol Psychiatry. 2013;7:813–823. doi: 10.1038/mp.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller J, Varga B, Ledent C, Barna I, Freund TF. Context-dependent effects of CB1 cannabinoid gene disruption on anxiety-like and social behaviour in mice. Eur J Neurosci. 2004;19:1906–1912. doi: 10.1111/j.1460-9568.2004.03293.x. [DOI] [PubMed] [Google Scholar]
- Haller J, Barna I, Barsvari B, Gyimesi Pelczer K, Yasar S, Panlilio LV, Goldberg S. Interactions between environmental aversiveness and the anxiolytic effects of enhanced cannabinoid signaling by FAAH inhibition in rats. Psychopharmacology (Berl) 2009;204:607–616. doi: 10.1007/s00213-009-1494-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller J, Goldberg SR, Pelczer KG, Aliczki M, Panlilio LV. The effects of anandamide signaling enhanced by the FAAH inhibitor URB597 on coping styles in rats. Psychopharmacology (Berl) 2013;230:353–362. doi: 10.1007/s00213-013-3161-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariri AR, Gorka A, Hyde LW, Kimak M, Halder I, Ducci F, Ferrell RE, Goldman D, Manuck SB. Divergent effects of genetic variation in endocannabinoid signaling on human threat- and reward-related brain function. Biol Psychiatry. 2009;66:9–16. doi: 10.1016/j.biopsych.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauger RL, Risbrough V, Brauns O, Dautzenberg FM. Corticotropin releasing factor (CRF) receptor signaling in the central nervous system: new molecular targets. CNS Neurol Disord Drug Targets. 2006;5:453–479. doi: 10.2174/187152706777950684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig M, Koob GF. A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci. 2007;30:399–406. doi: 10.1016/j.tins.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henckens MJ, van Wingen GA, Joëls M, Fernández G. Time-dependent effects of corticosteroids on human amygdala processing. J Neurosci. 2010;30:12725–12732. doi: 10.1523/JNEUROSCI.3112-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henckens MJ, van Wingen GA, Joëls M, Fernández G. Corticosteroid induced decoupling of the amygdala in men. Cereb Cortex. 2012;22:2336–2345. doi: 10.1093/cercor/bhr313. [DOI] [PubMed] [Google Scholar]
- Hermanson DJ, Hartley ND, Gamble-George J, Brown N, Shonesy BC, Kingsley PJ, Colbran RJ, Reese J, Marnett LJ, Patel S. Substrate-selective COX-2 inhibition decreases anxiety via endocannabinoid activation. Nat Neurosci. 2013;16:1291–1298. doi: 10.1038/nn.3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, Tasker JG. Endocannabinoid signaling, glucocorticoid-mediated negative feedback, and regulation of the hypothalamic-pituitary-adrenal axis. Neuroscience. 2012;204:5–16. doi: 10.1016/j.neuroscience.2011.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, McLaughlin RJ, Morrish AC, Viau V, Floresco SB, Hillard CJ, Gorzalka BB. Suppression of amygdalar endocannabinoid signaling by stress contributes to activation of the hypothalamic-pituitary-adrenal axis. Neuropsychopharmacology. 2009;34:2733–2745. doi: 10.1038/npp.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, Karatsoreos IN, Hillard CJ, McEwen BS. Rapid elevations in limbic endocannabinoid content by glucocorticoid hormones in vivo. Psychoneuroendocrinology. 2010;35:1333–1338. doi: 10.1016/j.psyneuen.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, McLaughlin RJ, Pan B, Fitzgerald ML, Roberts CJ, Lee TT, Karatsoreos IN, Mackie K, Viau V, Pickel VM, McEwen BS, Liu QS, Gorzalka BB, Hillard CJ. Recruitment of prefrontal cortex endocannabinoid signaling by glucocorticoids contributes to termination of the stress response. J Neurosci. 2011;31:10506–10515. doi: 10.1523/JNEUROSCI.0496-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, Kumar SA, Filipski SB, Iverson M, Stuhr KL, Keith JM, Cravatt BF, Hillard CJ, Chattarji S, McEwen BS. Disruption of fatty acid amide hydrolase activity prevents the effects of chronic stress on anxiety and amygdalar microstructure. Mol Psychiatry. 2013;18:1125–1135. doi: 10.1038/mp.2012.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jochman KA, Newman SM, Kalin NH, Bakshi VP. Corticotropin-releasing factor-1 receptors in the basolateral amygdala mediate stress-induced anorexia. Behav Neurosci. 2005;119:1448–1458. doi: 10.1037/0735-7044.119.6.1448. [DOI] [PubMed] [Google Scholar]
- John CS, Currie PJ. N-Arachidonoyl-serotonin in the basolateral amygdala increases anxiolytic behavior in the elevated plus maze. Behav Brain Res. 2012;233:382–388. doi: 10.1016/j.bbr.2012.05.025. [DOI] [PubMed] [Google Scholar]
- Kamprath K, Plendl W, Marsicano G, Deussing JM, Wurst W, Lutz B, Wotjak CT. Endocannabinoids mediate acute fear adaptation via glutamatergic neurons independently of corticotropin-releasing hormone signaling. Genes Brain Behav. 2009;8:203–211. doi: 10.1111/j.1601-183X.2008.00463.x. [DOI] [PubMed] [Google Scholar]
- Karst H, Berger S, Erdmann G, Schütz G, Joëls M. Metaplasticity of amygdalar responses to the stress hormone corticosterone. Proc Natl Acad Sci U S A. 2010;107:14449–14454. doi: 10.1073/pnas.0914381107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valiño F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Kühne C, Puk O, Graw J, Hrabì de Angelis M, Schütz G, Wurst W, Deussing JM. Visualizing corticotropin-releasing hormone receptor type 1 expression and neuronal connectivities in the mouse using a novel multifunctional allele. J Comp Neurol. 2012;520:3150–3180. doi: 10.1002/cne.23082. [DOI] [PubMed] [Google Scholar]
- Kupferschmidt DA, Klas PG, Erb S. Cannabinoid CB1 receptors mediate the effects of corticotropin-releasing factor on the reinstatement of cocaine seeking and expression of cocaine-induced behavioural sensitization. Br J Pharmacol. 2012a;167:196–206. doi: 10.1111/j.1476-5381.2012.01983.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupferschmidt DA, Newman AE, Boonstra R, Erb S. Antagonism of cannabinoid 1 receptors reverses the anxiety-like behavior induced by central injections of corticotropin-releasing factor and cocaine withdrawal. Neuroscience. 2012b;204:125–133. doi: 10.1016/j.neuroscience.2011.07.022. [DOI] [PubMed] [Google Scholar]
- Marco EM, Scattoni ML, Rapino C, Ceci C, Chaves N, Macrì S, Maccarrone M, Laviola G. Emotional, endocrine and brain anandamide response to social challenge in infant male rats. Psychoneuroendocrinology. 2013;38:2152–2162. doi: 10.1016/j.psyneuen.2013.04.004. [DOI] [PubMed] [Google Scholar]
- Marinelli PW, Funk D, Juzytsch W, Harding S, Rice KC, Shaham Y, Lê AD. The CRF1 receptor antagonist antalarmin attenuates yohimbine-induced increases in operant alcohol self-administration and reinstatement of alcohol seeking in rats. Psychopharmacology (Berl) 2007;195:345–355. doi: 10.1007/s00213-007-0905-x. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgänsberger W, Di Marzo V, Lutz B. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- McLaughlin RJ, Hill MN, Bambico FR, Stuhr KL, Gobbi G, Hillard CJ, Gorzalka BB. Prefrontal cortical anandamide signaling coordinates coping responses to stress through a serotonergic pathway. Eur Neuropsychopharmacol. 2012;22:664–671. doi: 10.1016/j.euroneuro.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlo Pich E, Lorang M, Yeganeh M, Rodriguez de Fonseca F, Raber J, Koob GF, Weiss F. Increase of extracellular corticotropin-releasing factor-like immunoreactivity levels in the amygdala of awake rats during restraint stress and ethanol withdrawal as measured by microdialysis. J Neurosci. 1995;15:5439–5447. doi: 10.1523/JNEUROSCI.15-08-05439.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller MB, Zimmermann S, Sillaber I, Hagemeyer TP, Deussing JM, Timpl P, Kormann MS, Droste SK, Kühn R, Reul JM, Holsboer F, Wurst W. Limbic corticotropin-releasing hormone receptor 1 mediates anxiety-related behavior and hormonal adaptation to stress. Nat Neurosci. 2003;6:1100–1107. doi: 10.1038/nn1123. [DOI] [PubMed] [Google Scholar]
- Newsom RJ, Osterlund C, Masini CV, Day HE, Spencer RL, Campeau S. Cannabinoid receptor type 1 antagonism significantly modulates basal and loud noise induced neural and hypothalamic-pituitary-adrenal axis responses in male Sprague-Dawley rats. Neuroscience. 2012;204:64–73. doi: 10.1016/j.neuroscience.2011.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z, Schweitzer P, Roberts AJ, Madamba SG, Moore SD, Siggins GR. Ethanol augments GABAergic transmission in the central amygdala via CRF1 receptors. Science. 2004;303:1512–1514. doi: 10.1126/science.1092550. [DOI] [PubMed] [Google Scholar]
- Patel S, Roelke CT, Rademacher DJ, Cullinan WE, Hillard CJ. Endocannabinoid signaling negatively modulates stress-induced activation of the hypothalamic-pituitary-adrenal axis. Endocrinology. 2004;145:5431–5438. doi: 10.1210/en.2004-0638. [DOI] [PubMed] [Google Scholar]
- Patel S, Roelke CT, Rademacher DJ, Hillard CJ. Inhibition of restraint stress-induced neural and behavioural activation by endogenous cannabinoid signalling. Eur J Neurosci. 2005;21:1057–1069. doi: 10.1111/j.1460-9568.2005.03916.x. [DOI] [PubMed] [Google Scholar]
- Patel S, Kingsley PJ, Mackie K, Marnett LJ, Danny G. Repeated homotypic stress elevates 2-arachidonoylglycerol levels and enhances short-term endocannabinoid signaling at inhibitory synapses in basolateral amygdala. Neuropharmacology. 2009;34:2699–2709. doi: 10.1038/npp.2009.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademacher DJ, Meier SE, Shi L, Ho WS, Jarrahian A, Hillard CJ. Effects of acute and repeated restraint stress on endocannabinoid content in the amygdala, ventral striatum, and medial prefrontal cortex in mice. Neuropharmacology. 2008;54:108–116. doi: 10.1016/j.neuropharm.2007.06.012. [DOI] [PubMed] [Google Scholar]
- Rainnie DG, Bergeron R, Sajdyk TJ, Patil M, Gehlert DR, Shekhar A. Corticotrophin releasing factor-induced synaptic plasticity in the amygdala translates stress into emotional disorders. J Neurosci. 2004;24:3471–3479. doi: 10.1523/JNEUROSCI.5740-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos AT, Troncone LR, Tufik S. Suppression of adrenocorticotrophic hormone secretion by simultaneous antagonism of vasopressin 1b and CRH-1 receptors on three different stress models. Neuroendocrinology. 2006;84:309–316. doi: 10.1159/000097587. [DOI] [PubMed] [Google Scholar]
- Refojo D, Echenique C, Müller MB, Reul JM, Deussing JM, Wurst W, Sillaber I, Paez-Pereda M, Holsboer F, Arzt E. Corticotropin-releasing hormone activates ERK1/2 MAPK in specific brain areas. Proc Natl Acad Sci U S A. 2005;102:6183–6188. doi: 10.1073/pnas.0502070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Refojo D, Schweizer M, Kuehne C, Ehrenberg S, Thoeringer C, Vogl AM, Dedic N, Schumacher M, von Wolff G, Avrabos C, Touma C, Engblom D, Schütz G, Nave KA, Eder M, Wotjak CT, Sillaber I, Holsboer F, Wurst W, Deussing JM. Glutamatergic and dopaminergic neurons mediate anxiogenic and anxiolytic effects of CRHR1. Science. 2011;333:1903–1907. doi: 10.1126/science.1202107. [DOI] [PubMed] [Google Scholar]
- Rey AA, Purrio M, Viveros MP, Lutz B. Biphasic effects of cannabinoids in anxiety responses: CB1 and GABA(B) receptors in the balance of GABAergic and glutamatergic neurotransmission. Neuropsychopharmacology. 2012;37:2624–2634. doi: 10.1038/npp.2012.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roozendaal B, Brunson KL, Holloway BL, McGaugh JL, Baram TZ. Involvement of stress-released corticotropin-releasing hormone in the basolateral amygdala in regulating memory consolidation. Proc Natl Acad Sci U S A. 2002;99:13908–13913. doi: 10.1073/pnas.212504599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi S, De Chiara V, Musella A, Sacchetti L, Cantarella C, Castelli M, Cavasinni F, Motta C, Studer V, Bernardi G, Cravatt BF, Maccarrone M, Usiello A, Centonze D. Preservation of striatal cannabinoid CB1 receptor function correlates with the antianxiety effects of fatty acid amide hydrolase inhibition. Mol Pharmacol. 2010;78:260–268. doi: 10.1124/mol.110.064196. [DOI] [PubMed] [Google Scholar]
- Rubino T, Viganò D, Zagato E, Sala M, Parolaro D. In vivo characterization of the specific cannabinoid receptor antagonist, SR141716A: behavioral and cellular responses after acute and chronic treatments. Synapse. 2000;35:8–14. doi: 10.1002/(SICI)1098-2396(200001)35:1<8::AID-SYN2>3.0.CO%3B2-Y. [DOI] [PubMed] [Google Scholar]
- Rubino T, Realini N, Castiglioni C, Guidali C, Viganó D, Marras E, Petrosino S, Perletti G, Maccarrone M, Di Marzo V, Parolaro D. Role in anxiety behavior of the endocannabinoid system in the prefrontal cortex. Cereb Cortex. 2008;18:1292–1301. doi: 10.1093/cercor/bhm161. [DOI] [PubMed] [Google Scholar]
- Ruehle S, Remmers F, Romo-Parra H, Massa F, Wickert M, Wörtge S, Häring M, Kaiser N, Marsicano G, Pape HC, Lutz B. Cannabinoid CB1 receptor in dorsal telencephalic glutamatergic neurons: distinctive sufficiency for hippocampus-dependent and amygdala-dependent synaptic and behavioral functions. J Neurosci. 2013;33:10264–10277. doi: 10.1523/JNEUROSCI.4171-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajdyk TJ, Schober DA, Gehlert DR, Shekhar A. Role of corticotropin-releasing factor and urocortin within the basolateral amygdala of rats in anxiety and panic responses. Behav Brain Res. 1999;100:207–215. doi: 10.1016/S0166-4328(98)00132-6. [DOI] [PubMed] [Google Scholar]
- Shin RM, Tully K, Li Y, Cho JH, Higuchi M, Suhara T, Bolshakov VY. Hierarchical order of coexisting pre- and postsynaptic forms of long-term potentiation at synapses in amygdala. Proc Natl Acad Sci U S A. 2010;107:19073–19078. doi: 10.1073/pnas.1009803107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipe JC, Scott TM, Murray S, Harismendy O, Simon GM, Cravatt BF, Waalen J. Biomarkers of endocannabinoid system activation in severe obesity. PLoS One. 2010;5:e8792. doi: 10.1371/journal.pone.0008792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GW, Aubry JM, Dellu F, Contarino A, Bilezikjian LM, Gold LH, Chen R, Marchuk Y, Hauser C, Bentley CA, Sawchenko PE, Koob GF, Vale W, Lee KF. Corticotropin releasing factor receptor 1-deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron. 1998;20:1093–1102. doi: 10.1016/S0896-6273(00)80491-2. [DOI] [PubMed] [Google Scholar]
- Spina MG, Merlo-Pich E, Akwa Y, Balducci C, Basso AM, Zorrilla EP, Britton KT, Rivier J, Vale WW, Koob GF. Time-dependent induction of anxiogenic-like effects after central infusion of urocortin or corticotropin-releasing factor in the rat. Psychopharmacology (Berl) 2002;160:113–121. doi: 10.1007/s00213-001-0940-y. [DOI] [PubMed] [Google Scholar]
- Sztainberg Y, Kuperman Y, Tsoory M, Lebow M, Chen A. The anxiolytic effect of environmental enrichment is mediated via amygdalar CRF receptor type 1. Mol Psychiatry. 2010;15:905–917. doi: 10.1038/mp.2009.151. [DOI] [PubMed] [Google Scholar]
- Timpl P, Spanagel R, Sillaber I, Kresse A, Reul JM, Stalla GK, Blanquet V, Steckler T, Holsboer F, Wurst W. Impaired stress response and reduced anxiety in mice lacking a functional corticotropin-releasing hormone receptor 1. Nat Genet. 1998;19:162–166. doi: 10.1038/520. [DOI] [PubMed] [Google Scholar]
- Tsou K, Nogueron MI, Muthian S, Sañudo-Pena MC, Hillard CJ, Deutsch DG, Walker JM. Fatty acid amide hydrolase is located preferentially in large neurons in the rat central nervous system as revealed by immunohistochemistry. Neurosci Lett. 1998;254:137–140. doi: 10.1016/S0304-3940(98)00700-9. [DOI] [PubMed] [Google Scholar]
- Tye KM, Prakash R, Kim SY, Fenno LE, Grosenick L, Zarabi H, Thompson KR, Gradinaru V, Ramakrishnan C, Deisseroth K. Amygdala circuitry mediating reversible and bidirectional control of anxiety. Nature. 2011;471:358–362. doi: 10.1038/nature09820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Pett K, Viau V, Bittencourt JC, Chan RK, Li HY, Arias C, Prins GS, Perrin M, Vale W, Sawchenko PE. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol. 2000;428:191–212. doi: 10.1002/1096-9861(20001211)428:2<191::AID-CNE1>3.0.CO%3B2-U. [DOI] [PubMed] [Google Scholar]
- Wang F, Flanagan J, Su N, Wang LC, Bui S, Nielson A, Wu X, Vo HT, Ma XJ, Luo Y. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J Mol Diagn. 2012;14:22–29. doi: 10.1016/j.jmoldx.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Hill MN, Zhang L, Gorzalka BB, Hillard CJ, Alger BE. Acute restraint stress enhances hippocampal endocannabinoid function via glucocorticoid receptor activation. J Psychopharmacol. 2012;26:56–70. doi: 10.1177/0269881111409606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M, Webster E, Spokes H, Phu P, Ehrhart-Borstein M, Bornstein S, Park C, Rice KC, Chrousos GP, Julio L, Gold PW. Chronic administration of the non-peptide CRH type 1 receptor antagonist antalarmin does not blunt hypothalamic-pituitary-adrenal axis responses to acute immobilization stress. Life Sci. 1999;65:53–58. doi: 10.1016/S0024-3205(99)00268-4. [DOI] [PubMed] [Google Scholar]
- Zabouri N, Ptito M, Casanova C, Bouchard JF. Fatty acid amide hydrolase expression during retinal postnatal development in rats. Neuroscience. 2011;195:145–165. doi: 10.1016/j.neuroscience.2011.08.008. [DOI] [PubMed] [Google Scholar]