

Abstract
Mitochondria fulfill a number of biological functions which inherently depend on ATP and O2−•/H2O2 production. Both ATP and O2−•/H2O2 are generated by electron transfer reactions. ATP is the product of oxidative phosphorylation whereas O2−• is generated by singlet electron reduction of di-oxygen (O2). O2−• is then rapidly dismutated by superoxide dismutase (SOD) producing H2O2. O2−•/H2O2 were once viewed as unfortunately by-products of aerobic respiration. This characterization is fitting considering over production of O2−•/H2O2 by mitochondria is associated with range of pathological conditions and aging. However, O2−•/H2O2 are only dangerous in large quantities. If produced in a controlled fashion and maintained at a low concentration, cells can benefit greatly from the redox properties of O2−•/H2O2. Indeed, low rates of O2−•/H2O2 production are required for intrinsic mitochondrial signaling (e.g. modulation of mitochondrial processes) and communication with the rest of the cell. O2−•/H2O2 levels are kept in check by anti-oxidant defense systems that sequester O2−•/H2O2 with extreme efficiency. Given the importance of O2−•/H2O2 in cellular function, it is imperative to consider how mitochondria produce O2−•/H2O2 and how O2−•/H2O2 genesis is regulated in conjunction with fluctuations in nutritional and redox states. Here, I discuss the fundamentals of electron transfer reactions in mitochondria and emerging knowledge on the 11 potential sources of mitochondrial O2−•/H2O2 in tandem with their significance in contributing to overall O2−•/H2O2 emission in health and disease. The potential for classifying these different sites in isopotential groups, which is essentially defined by the redox properties of electron donator involved in O2−•/H2O2 production, as originally suggested by Brand and colleagues is also surveyed in detail. In addition, redox signaling mechanisms that control O2−•/H2O2 genesis from these sites are discussed. Finally, the current methodologies utilized for measuring O2−•/H2O2 in isolated mitochondria, cell culture and in vivo are reviewed.
Keywords: Mitochondria, Reactive oxygen species, Bioenergetics, Redox signaling
Graphical abstract
Highlights
-
•
Use of electrons for the production of ATP or O2−•/H2O2 by mitochondria and the factors that influence the production of either molecule.
-
•
Control of O2−•/H2O2 levels by antioxidant systems.
-
•
Up to date assessment of novel technologies being utilized to accurately measure O2−•/H2O2 production in mitochondria ex vivo and in vivo.
Basic principles in oxidative metabolism and aerobic respiration
Di-oxygen (O2) initially appeared in significant amounts on Earth around 2.2 billion years ago due to the action of photosynthesizing cyanobacteria [1]. At first, most of the O2 reacted with solubilized iron (Fe) forming insoluble oxide minerals [1]. After this initial event O2 started to build up in substantial amounts in the surrounding environment and atmosphere. The sharp increase in O2 concentration in the atmosphere and formation of various mineral oxides is referred to as the Great Oxygenation Event (GOE) [2]. It is also referred to as the Great Oxygen Catastrophe since the rise in O2 led to the first mass extinction on Earth. Indeed, O2 is highly toxic towards anaerobic organisms. The damaging effects of O2 is associated with its free radical properties which deplete essential thiols and dismantle Fe–S clusters required for metabolism and the biosynthesis of macromolecular structures in anaerobic organisms [3]. The definition of “free radical” refers to “any species capable of independent existence that contains one or more unpaired electrons”, where an unpaired electron occupies an atomic orbital by itself [1]. Considering that ground state O2 has two unpaired electrons in its outer most anti-bonding orbital it can thus be classified as a free radical species [4]. The two lone electrons in the outer most orbital of O2 also have the same spin quantum number which imposes a spin restriction on electron acceptance [5]. Thus, O2 can only accept one electron at a time when it is being reduced to H2O which can generate several free radical intermediates namely, superoxide (O2−•), hydrogen peroxide (H2O2), and hydroxyl radical (OH•) [6]. This also makes O2 dangerous since its univalent reduction leads to the genesis of highly reactive intermediates (Fig. 1).
Fig. 1.
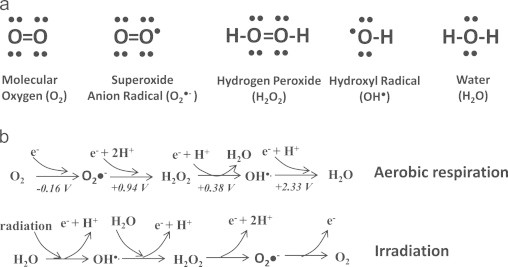
Reduction of O2 to H2O and its free radical intermediates (A) Lewis structures for molecular oxygen (O2) and its singlet electron derivatives superoxide (O2•−), hydrogen peroxide (H2O2), and hydroxyl radical (OH•). (B) The step wise reduction of O2 to H2O during aerobic respiration. The standard redox potential for reduction of each intermediate is also shown (reproduced and modified from [23]). Irradiation-mediated cleavage of H2O which produces OH• accounts for the damaging effects of radiation therapy.
Adaptation to an oxidizing environment provided a major selective advantage for organisms that could couple enzyme activities to O2 utilization [3,7]. In aerobic cells, O2 is utilized by many enzyme systems but is primarily the driving force behind aerobic ATP production. The use of O2 in nutrient metabolism maximized energy conservation among aerobic eukaryotes prompting an increase in biological complexity culminating the evolution of humanity [8,9]. In aerobic eukaryotes, production of ATP by oxidative phosphorylation occurs in mitochondria, double membrane organelles with prokaryotic origins that house the necessary enzymatic machinery required for O2-dependent production of ATP from carbon oxidation [10,11]. In this complicated process nutrients in the form of carbohydrates, fatty acids, or amino acids are converted into common intermediates such as acetyl-CoA, oxaloacetate, and 2-oxoglutarate which enter the Krebs cycle and undergo further oxidation (Fig. 2) [6,12]. Acetyl-CoA and oxaloacetate, which are generated by the metabolism of either monosaccharides or fatty acids, are condensed by citrate synthase yielding citric acid which is then systematically oxidized by the concerted action of 7 other Krebs cycle enzymes (Fig. 2). Amino acids can also feed in to the Krebs cycle at various levels. Although there are 20 different amino acids that can be degraded to form either ketogenic or gluconeogenic Krebs cycle intermediates, the most common intermediate formed is 2-oxoglutarate. Indeed, 2-oxoglutarate either serves as an ammonia acceptor forming glutamate during amino acid catabolism or is formed following degradation of glutamate or use of glutamate for amino acid biosynthesis. Fatty acids which are the product of triglyceride hydrolysis also feed into the Krebs cycle at the level of acetyl-CoA. The complex process of extracting electrons from fat molecules for ATP production is called β-oxidation. Entry of fatty acyl-CoA into the matrix is prohibited unless the fatty acid is coupled via an ester linkage to carnitine which facilitates mitochondrial uptake of acyl molecules, a reaction catalyzed by carnitine palmitoyl transferase 1 (Cpt1). Upon entry into the matrix, carnitine is immediately exchanged with CoASH by Cpt2 and acyl-CoA enters into β-oxidation. Note that 1 FADH2 and 1 NADH along with an acetyl-CoA are yielded from the oxidation of two carbons on the fatty acyl chain (Fig. 2). Acetyl-CoA then enters the Krebs cycle where it is oxidized further. Thus, in contrast to glucose, fatty acid oxidation yields far more ATP, e.g. palmitate which is 16 carbons long produces 129 ATP vs the 36 garnered from glucose metabolism. It is not surprising then that the human heart, which turns over 30 kg of ATP daily due to contraction relaxation coupling, produces most of its ATP by fatty acid oxidation [13].
Fig. 2.
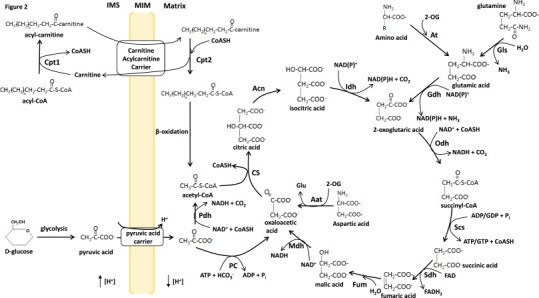
The oxidative Krebs cycle. The Krebs cycle is the most central metabolic pathway for all life on Earth since it yields the necessary carbon intermediates required for either ATP formation or genesis of biomolecules like amino acids, lipids, and nucleotides. Different sources of carbon including glucose (and other monosaccharides), fatty acids, and amino acids are converted into common intermediates acetyl-CoA, oxaloacetate, and 2-oxoglutarate, Krebs cycle intermediates that are then systematically stripped of electrons (oxidized) to yield electron carriers NADH and succinate which are then oxidized by the respiratory chain for ATP production. Note that enzymes Idh and Gdh can utilize either NAD+ or NADP+ as cofactors which is isozyme dependent. Irreversible steps of the Krebs cycle involve enzymes that couple decarboxylation to electron movement. For clarity key metabolic cascades involved in the conversion of glucose and fatty acids into key intermediates, pyruvate and acetyl-CoA, have been omitted. Pdh; pyruvate dehydrogenase, PC; pyruvate carboxylase, CS; citrate synthase, Acn; aconitase, Idh; isocitrate dehydrogenase, Odh; 2-oxoglutarate dehydrogenase, Scs; succinyl-CoA synthetase, Sdh; succinate dehydrogenase, Fum; fumarase, Mdh; malate dehydrogenase, Gcl; glutaminase, Gdh; glutamate dehydrogenase, At; aminotransferase, Aat; aspartate aminotransferase, Cpt; carnitine palmitoyltransferase, 2-OG; 2-oxoglutarate, MIM; mitochondrial inner membrane, IMS; intermembrane space.
Removal of electrons during oxidation is coupled to the reduction of NAD forming NADH which is then oxidized by Complex I. Succinate is also oxidized by Krebs cycle enzyme Complex II (succinate dehydrogenase; Sdh) producing fumarate and reducing FAD to FADH2 [14]. Electrons from Complex I and II are then passed through a series or prosthetic groups positioned according to increasing affinity for electrons to ubiquinone (Q) producing ubiquinol (QH2) which is then oxidized by Complex III (Fig. 3) [15]. Electrons can also be fed into the Q pool by several other enzymes associated with the mitochondrial inner membrane (MIM) including sn-glycerol-3-phosphate dehydrogenase (G3PDH), proline dehydrogenase, dihydroorotate dehydrogenase, sulfide:quinone oxidoreductase (SQR), electron transfer flavoprotein oxidoreductase (ETFQO) [16–21]. The electrons are then passed to Complex IV and utilized to reduce O2 forming H2O. Importantly electron transfer from NADH (E°′=−340 mV) or FADH2 (E°′=+31 mV) through the Q pool (E°′=+45 mV) to O2 (E°′=+840 mV) is an energetically favorable process and is thus coupled to the pumping of protons across the mitochondrial inner membrane (MIM) into the intermembrane space (IMS) (Fig. 3). This creates a transmembrane electrochemical gradient of protons (Δμm) which is composed of both an electrical (ΔΨm) and chemical (ΔpH) potential. Notably ΔΨm accounts for ~90% of the energy associated with the transmembrane electrochemical gradient of protons. The Gibbs free energy stored across the MIM in the form of Δμm is then used to drive ATP synthesis by Complex V (Fig. 3) [22]. Complex V is a multisubunit transmembrane protein complex that couples the energy liberated from electron transport to the terminal electron acceptor O2 to the conversion of ADP and Pi into ATP, a process referred to as oxidative phosphorylation. Basically Complex V is composed of two portions, the F0 portion which is buried in the MIM and the F1 part which makes contact with the matrix [23]. F0 is composed of subunits A, B, and C and subunits alpha (α), beta (β), gamma (γ), delta (δ), and epsilon (ε) comprise F1. The process of oxidative phosphorylation involves the diffusion of protons from the IMS into the matrix, which is facilitated by the F0 portion of Complex V. As protons are translocated through F0 the stalk rotates inducing alternating conformational changes in α and β subunits of F1 resulting in the binding of ADP and Pi and the release of ATP [23,24]. ATP is then translocated into the cytosolic environment in exchange for ADP, which is catalyzed by ATP:ADP antiporter complex, to do work in the cell (Fig. 3). It is important to realize that ATP is a major driving force for all life on Earth which is the reason it is often referred to as the universal energy currency. Indeed, even in mammalian cells, mitochondria are strategically positioned to ensure that energy demands for even the most energy intensive processes like heart contraction and relaxation can be met.
Fig. 3.
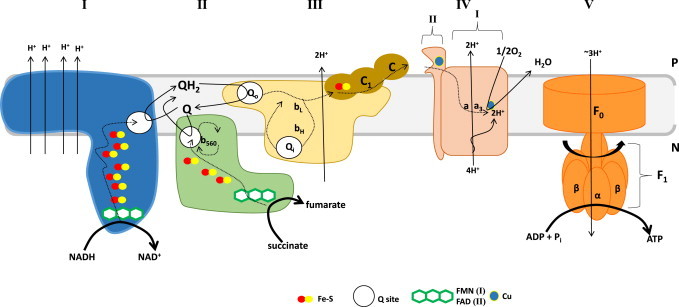
The electron transport chain and oxidative phosphorylation. Following the genesis of NADH or succinate by the Krebs cycle, both electron carriers are oxidized by complex I (NADH:ubiquinone oxidoreductase) and complex II (succinate:ubiquinone oxidoreductase or succinate dehydrogenase). Oxidation of NADH by complex I yields NAD which returns to the Krebs cycle and two electrons which reduce FMN and are then systematically passed through 7–8 Fe−S clusters to the quinone binding site reducing Q to QH2. Due to the large difference in E°′ between NADH and Q, electron transfer induces changes in the membrane module of complex I resulting in the pumping of four protons into the IMS. Similarly succinate oxidation by complex II yields fumaric acid and the liberated electrons are passed through FAD and three Fe–S clusters reducing Q to QH2. Note that complex II is a Krebs cycle enzyme providing a direct link between Krebs cycle flux and electron transfer in the respiratory chain. Complex II is also embedded in the MIM but is not a transmembrane protein. Unlike complexes I, III, and IV, complex II does not couple electron transfer to proton translocation into the IMS. This is attributed to the low Gibbs free energy change for electron transfer in complex II to Q (refer to text for E°′ values for FAD and Q). Complex II also harbors a heme (b560) which is thought to be involved in electron recycling. QH2 is then oxidized by complex III in the Qo (quinone outer membrane) binding site resulting in the transfer of one electron through the Rieske Fe−S protein and cytochrome C1 to cytochrome C. Note that C1 and C can only accept one electron at a time. This generates a semiquinone radical in the Qo site. The second electron is then passed through heme groups bL and bH and utilized to re-reduce Q in the Qi (quinone inner membrane) binding site. This is referred to as the Q-cycle and is required to recycle electrons during aerobic respiration. Electron movement from QH2 to C is coupled to the transfer of two protons into the IMS. C then binds subunit II on complex IV where electrons are passed systematically through two copper moieties and two heme groups (a and a3) resulting in the reduction of oxygen to water at subunit I. For two electrons transferred only one oxygen atom is reduced which also requires an input of two protons. This process is coupled to the pumping of two additional protons into the IMS. Considering, that the the full reduction of di-oxygen (O2) requires four electrons that means four protons are actually pumped into the IMS (for every two electrons from NADH or succinate two protons are pumped out). Electron transfer and proton pumping creates are transmembrane potential of protons called the protonmotive force (pmf) which is utilized by complex V to drive ATP synthesis. Note that proton re-uptake by complex V is coupled to the rotation of the stalk and F1 portion of complex V resulting in the biosynthesis of ATP. P; positive side; IMS and N; negative side; matrix.
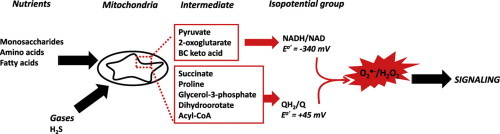
It is well documented that mitochondria can serve as important sites for ROS production [4,25–27]. However, ROS is a broad term that includes all oxyradicals such as singlet and doublet oxyradicals and nonradicals like H2O2 [28]. Different ROS vary tremendously in reactivity, half-life, abundance, and production. In addition, some ROS actually serve as important signaling molecules rather than by-products that indiscriminately damage cell constituents. Thus, using the term “mitochondrial ROS” is inappropriate considering it fails to define which oxyradical species is being produced. O2−• is the proximal ROS generated by mitochondria and is the result of the premature univalent reduction of O2 by various enzymes in the Krebs cycle and respiratory chain. O2−• is then rapidly dismutated by superoxide dismutase, either in the matrix (MnSOD) or intermembrane space (Cu/ZnSOD), producing H2O2 which can then be further degraded to O2 and H2O by various enzymes [12] (Fig. 4). It is important to critically evaluate how mitochondria maintain O2−•/H2O2 homeostasis considering that both molecules are required for cellular signaling and, when generated at sufficient amounts, can induce oxidative stress, cellular damage, and death. Indeed, overproduction of O2−•/H2O2 by mitochondria is associated with a myriad of diseases including neurological deficits, heart disorders, and metabolic diseases like obesity and type 2 diabetes and has even been suggested to be the cause of aging [14,29–31]. The mechanisms by which O2−•/H2O2 are degraded in mitochondria and the cell have been reviewed extensively (Fig. 4) [12,32,33]. In addition, the contributions of Complex I and III to production of O2−•/H2O2 has been heavily investigated and discussed in several reviews [4,32,34–36]. Intriguingly a number of other potential O2−•/H2O2 production sites have also been identified in mitochondria. In fact, a total of 11 sources of O2−•/H2O2 have been identified in mitochondria (Fig. 5) [37,38]. Recent studies have also shown sites other than Complex I and III namely, 2-oxoglutarate dehydrogenase (Odh), pyruvate dehydrogenase (Pdh), and Sdh serve as highly significant sources of mitochondrial O2−•/H2O2 [39–42]. The other sites do not make a substantial contribution to O2−•/H2O2 production [16,17,43,44]. In the present review, current knowledge of mitochondrial O2−•/H2O2 production will be surveyed along with the proposed classification of the different suggested sources for mitochondrial O2−•/H2O2 production into two different isopotential groups, NADH/NAD isopotential group and QH2/Q isopotential group, as originally suggested by Brand and colleagues [39]. The influence of different nutritional states and redox signaling on rates O2−•/H2O2 production in physiology and disease are discussed. Finally, emerging technologies that can be utilized to accurately quantify O2−•/H2O2 production in isolated mitochondria, live cells, and in vivo in the context of health and disease will also be reviewed. It should also be noted that ROS production by mitochondria is referred to as O2−•/H2O2 considering the rapid dismutation of O2−• to H2O2 and the lack of sensitive techniques for quantitative measurement of O2−•.
Fig. 4.
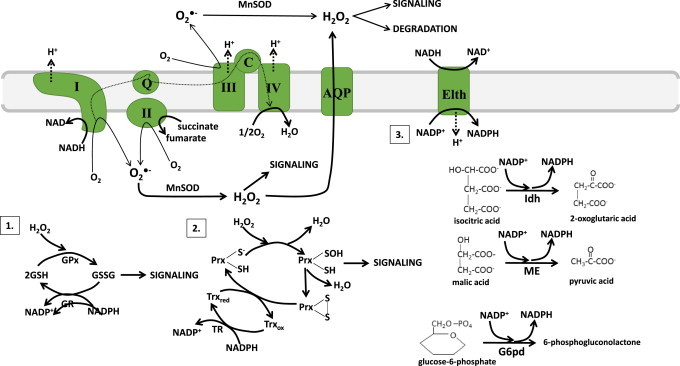
Anti-oxidant defense systems in mitochondria: Mitochondria can be a major source of reactive oxygen species (ROS) and production which depends on the metabolic state and redox poise of mitochondria. Metabolic state refers to the efficiency of electron transfer from nutrients to O2 whereas redox poise is associated with the anti-oxidant capacity, maintenance of a reductive environment by reduced glutathione (GSH; normally in the mitochondrial matrix, the glutathione pool is highly reduced with the ratio of 2GSH to GSSG (2GSH/GSSG) ~100 giving E°′=−320 mV) and the redox state of anti-oxidant enzymes Prx and Trx. The proximal ROS O2−• is dismutated rapidly by MnSOD or Cu/ZnSOD in the matrix or intermembrane space, respectively, to H2O2 which is then used for signaling via oxidation of protein cysteine thiols (see Fig. 8 for thiol based reactions with oxidants and reductants). Note that H2O2 can also diffuse passively from one side of the mitochondrial membrane to the next with the aid of aquaporins (AQP). H2O2 levels are continuously monitored by endogenous anti-oxidant systems. The far most efficient systems utilized to quench H2O2 are 1. Glutathione peroxidase (GPx)/glutathione reductase (GR) and 2. Peroxiredoxin (Prx)/thioredoxin (Trx)/Thioredoxin Reductase (TR) system. Mitochondria contain two GPx and two Prx isozymes; GPx1 and GPx4; Prx3 and Prx5. Although all four enzymes quench H2O2, GPx1 and Prx3 have a higher affinity for H2O2 while GPx4 and Prx5 metabolize lipid hydroperoxides more efficiently [12]. Systems 1 and 2 are supported by system 3 which produces NADPH, the reductive power required to rejuvenate anti-oxidant systems after a round of H2O2 sequestration. Note that NADPH is either generated from the metabolism of isocitrate, malate, or glucose-6-phosphate by isocitrate dehydrogenase (Idh), malic enzyme (ME), or glucose-6-phosphate dehydrogenase (G6pd) or via conversion of NADH into NADPH by energy liberating transhydrogenase (Elth).
Fig. 5.
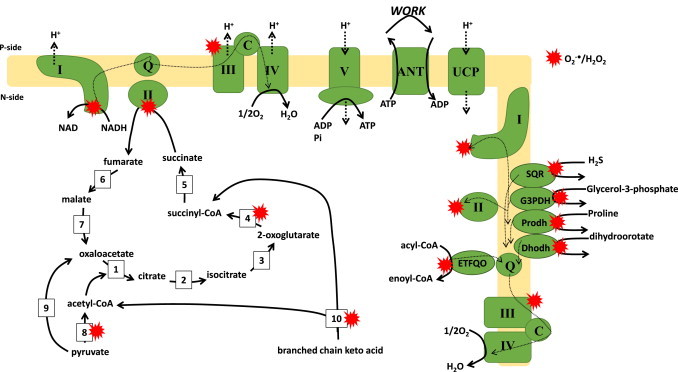
Overview of the 11 different sources for O2−•/H2O2 production. Mitochondrial ATP and O2−•/H2O2 are intimately linked by electron transfer from nutrients to di-oxygen (O2). Nutrients (glucose, fatty acids, amino acids) are enzymatically converted to common intermediates (acetyl-CoA, oxaloacetate, pyruvate) which enter the Krebs cycle to undergo further oxidation. Metabolite oxidation is coupled to the evolution of carbon dioxide (CO2) and the production of NADH and succinate which are then oxidized by complexes I and II respectively. Electron flow through the respiratory complexes through ubiquinone (Q) and cytochrome C (C) and the reduction of O2 to H2O is coupled to the formation of a transmembrane potential of proteins across the mitochondrial inner membrane (MIM) which is then utilized to drive ATP synthesis by complex V. ATP is then transported out of mitochondria in exchange for ADP by ATP:ADP exchanger (ANT). The proton gradient can also be mildly uncoupled by uncoupling proteins (UCP) 2 and 3 which are utilized to control O2−•/H2O2 production. Electron transfer flavoprotein oxidoreductase (ETFQO), dihydroorotate dehydrogenase (Dhodh), proline dehydrogenase (Prodh), succinate:quinone reductase (SQR), sn-glycerol-3-phosphate dehydrogenase (G3PDH) can also feed electrons into the Q pool following oxidation of their cognate substrates. Red stars indicate that 11 potential sources of O2−•/H2O2. Dotted lines represent flow of electrons. Bold dotted lines indicate flow of protons (H+). (1) Citrate synthase, (2) aconitase, (3) NAD(P)+-isocitrate dehydrogenase, (4) 2-oxoglutarate dehydrogenase, (5) succinyl-CoA synthase, (6) fumarase, (7) malate dehydrogenase, (8) pyruvate dehydrogenase, (9) pyruvate carboxylase, (10) branched chain keto acid dehydrogenase.
Classification of mitochondrial sources of O2−•/H2O2
Chemiosmotic coupling and mitochondrial production of ATP fundamentally relies on the transfer of electrons between different redox carriers embedded in proteins. Electron transfer reactions in mitochondria are often viewed as a simple movement of electrons through a defined pathway from a donor to acceptor molecule [45]. However, electron movement in mitochondria is far more complicated considering that different redox centers in mitochondrial enzymes, especially the respiratory complexes, are separated by polypeptide chains with most carriers buried deep in proteins within the lipid bilayer of the MIM [45]. Thus, electron transfer cannot be as simple as the donation and acceptance of an electron(s) between two different ions in aqueous solution [45]. Rather, electron movement between prosthetic groups proceeds via electron tunneling [45]. Essentially, electron tunneling predicts the probability of whether or not an electron will move from a donor to an acceptor molecule. Tunneling between donor and acceptor molecules is heavily influenced by distance between the centers, difference in redox potential, and response of electron carriers to changes in charge on donor or acceptor molecules [45]. In the respiratory chain efficiency of electron transfer between carriers varies considerably according to distance between donor and acceptor molecules. Electron transfer occurs at a maximum distance of 14 Å which provides a relative electron transfer rate of ~104 s−1 [45]. As the distance between centers decreases there is an exponential increase in the rate of electron transfer [4]. Likewise, if redox centers are separated by more than 14 Å electron transfer most likely does not occur since rate of transfer is too slow [4]. Discussing the principles of electron transfer reactions in mitochondria is important considering that formation of O2−• is most likely influenced by the same factors; distance, redox difference, and response to changes in charge on donor and acceptor molecules [46].
Complex I and III are chief sites for mitochondrial O2−• production in mitochondria. O2−• production from either complex fulfills various signaling functions including hypoxic signaling, adipocyte regulation, immune cell function, satiety signaling in the hypothalamus, and responsiveness to insulin [47–49]. It is now known that mitochondria harbor up to 11 different sources of O2−•/H2O2 which can be divided into two different subgroups based on redox potential at which they produce O2−•/H2O2; (1) NADH/NAD isopotential group and (2) QH2/Q isopotential group [39]. The NADH/NAD isopotential group consists of Complex I, Odh, Pdh, and Bckdh while the QH2/Q isopotential group is made up of 7 other enzymes; Complex III, Sdh, ETFQO, proline dehydrogenase, dihydroorotate dehydrogenase, SQR, and sn-G3PDH. In group 1, O2−•/H2O2 is reliant on the concentration of NADH whereas group 2 requires reduction of Q to QH2. Notably, in the QH2/Q group the major sources of O2−•/H2O2 are Complex I, II, and III and thus most of the O2−•/H2O2 is attributed to reverse electron transport (RET) (Fig. 5) [16,17,39,44]. Classification of O2−•/H2O2 producing enzymes in mitochondria into different isopotential subgroups was recently suggested by Brand and colleagues [37,39]. Indeed, it has been shown in a series of studies that other O2−•/H2O2 sources like Odh, Pdh, and Sdh, can also make substantial contributions to the overall production of O2−•/H2O2 production by mitochondria which could contribute to mitochondrial O2−•/H2O2 signaling [18,39]. Rates of production from the different enzymes in each isopotential group is highly dependent on nutrient status, mitochondrial redox poise, and availability of ADP. In addition, most of these enzymes harbor thiol residues that are close to or adjacent to O2−•/H2O2 producing centers suggesting that redox signaling serves as mechanism that controls O2−•/H2O2 production [6]. For example, Odh produces both O2−•/H2O2 however; these molecules can also feedback and deactivate Odh thus controlling mitochondrial O2−•/H2O2 production [50,51]. Intriguingly a recent study also provided evidence that O2−•/H2O2 from the two different isopotential groups can have different effects on signaling since the different groups specifically target very specific proteins for regulation by redox signaling [52]. Other factors including mitochondrial fission and fusion and assembly of Krebs cycle enzymes and respiratory complexes into metabolon and respirasomes also likely influence O2−•/H2O2 production from these sites [53,54].
NADH/NAD isopotential group – Odh and Pdh are major sources of O2−•/H2O2
As mentioned above the NADH/NAD isopotential group consists of four different enzymes; Complex I, Odh, Pdh, and Bckdh which produce O2−•/H2O2 in response to fluctuations in NADH levels. All four enzymes harbor a flavin prosthetic group, FMN in Complex I or FAD in the other three enzymes, which are able to produce O2−• and/or H2O2 [14,51]. While Complex I generates only O2−•, the other enzymes generate both O2−•/H2O2 with H2O2 making up ~75% of the production (e.g. Odh) [51,55]. It has been known for a long time that flavins can produce either O2−• or H2O2 in O2 saturated aqueous solution [56]. Rates of production by free flavins are dependent on genesis of flavin hydroperoxides, flavin radicals, or flavin ion intermediates which can generate either O2−• and H2O2 at rates that vary between 250 and 5×108 M−1 s−1. Importantly rates of O2−•/H2O2 production can be substantially enhanced in a polypeptide environment [56]. For example the neutral flavin radical of glucose oxidase produces O2−• at a rate of 1×109 M−1 s−1 in comparison to free neutral flavin radical which generates O2−• at a rate of ~104 M−1 s−1 [56]. Production of O2−• or O2−•/H2O2 by either enzyme in this isopotential group is highly dependent on the concentration of NADH [39]. An increase in NADH/NAD leads to sharp increase in O2−•/H2O2 production by all enzymes in this isopotential group but in particular Complex I, Odh, and Pdh. In a recent publication Quinlan et al. provided evidence that FAD in Odh and Pdh and to a lesser extent Bckdh produce more O2−•/H2O2 than FMN in Complex I [39]. In fact based on their calculations the authors estimated that Odh produces 8 times more O2−•/H2O2 than Complex I [39]. Overall, it would appear that the hierarchy for O2−•/H2O2 production in the NADH/NAD isopotential group is Odh>Pdh>Bckdh>Complex I organized from highest rate to lowest. However, measurements were conducted only on skeletal muscle mitochondria. It is well known that sites for mitochondrial O2−•/H2O2 can vary substantially from tissue to tissue. It would thus be crucial to apply a similar methodology to other tissues and profile the different sites for mitochondrial O2−•/H2O2 production.
Odh and Pdh serve as major regulatory hubs for modulation of metabolite entry, exit, and flux into and through the Krebs cycle. Various allosteric activators and inhibitors including CoASH, NADH, NAD, and ATP converge upon these two enzymes to modulate their activity (Fig. 6). Allosteric regulation is required for rapid modulation of 2-oxoglutarate and pyruvate oxidation in response to changing energy demands and alterations in metabolic flux [57,58]. Odh and Pdh are also modulated by mitochondrial calcium uptake during cardiac and skeletal muscle contraction and relaxation (Fig. 6) [59]. In addition, both enzymes are also subjected to a range of covalent modifications, including phosphorylation, which is required to modulate Krebs cycle flux and the entry and exit of nutrients and metabolites from the cycle. Both Odh and Pdh are also important redox sensors and are modulated by changes in mitochondrial H2O2. Indeed, Odh and Pdh harbor a dihydrolipoamide moiety located in the E2 subunit which plays a critical role in the genesis of succinyl-CoA and acetyl-CoA and NADH [60,61]. The vicinal thiols (SH) on dihydrolipoamide are highly amenable towards oxidation by H2O2 forming highly reactive sulfenic acid (SOH) residues [62,63]. In fact, the vicinal thiols on dihydrolipoamide react quickly with low micromolar (~5 µM) amounts of H2O2 [64]. Odh and Pdh contain three subunits E1, E2, and E3 which transfer electrons from the substrate through dihydrolipamide in the E2 subunit reducing FAD to FADH2 in E3 which then produces NADH [61,65]. Considering that Odh and Pdh are (1) important sites for O2−•/H2O2 production, (2) are highly sensitive to deactivation by H2O2, and (3) are important regulatory hubs for the Krebs cycle, it would appear that both enzymes serve as central sites for modulation of mitochondrial redox signaling (Fig. 7). Indeed, an increase in mitochondrial O2−•/H2O2 produced by Odh and Pdh is the result of an increase in mitochondrial NADH which would mean that Complex I-mediated NADH oxidation has decreased. The burst in O2−•/H2O2 feeds back to inhibit Odh and Pdh at the level of dihydrolipoamide preventing further electron transfer to FAD thus limiting the further production of O2−•/H2O2.
Fig. 6.
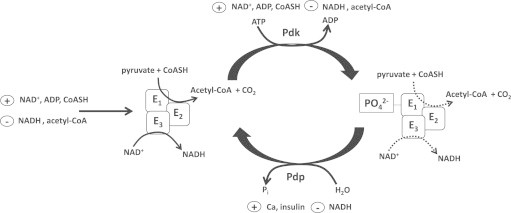
Modulation of Pdh by allosteric regulation and phosphorylation. The enzymatic products acetyl-CoA and NADH serve as allosteric inhibitors whereas NAD+, ADP, and CoASH serve as activators of Pdh activity. Thus, Pdh efficiency is reliant on the oxidation of NADH by complex I, condensation of acetyl-CoA with oxaloacetate, and turnover of ATP in the cell. Note that these allosteric modulators also control pyruvate dehydrogenase kinase (Pdk) and pyruvate dehydrogenase phosphatase (Pdp) which phosphorylate and dephosphorylate, respectively, the E1 subunit to modulate Pdh activity. Phosphorylation inhibits Pdh activity whereas dephosphorylation has the opposite effect. In addition, hormonal signaling cascades like insulin signaling also play a part in modulating Pdh activity in response to whole body changes in nutrition and energy state. Odh is also modulated by allosteric regulators, calcium, and phosphorylation in a similar manner.
Fig. 7.
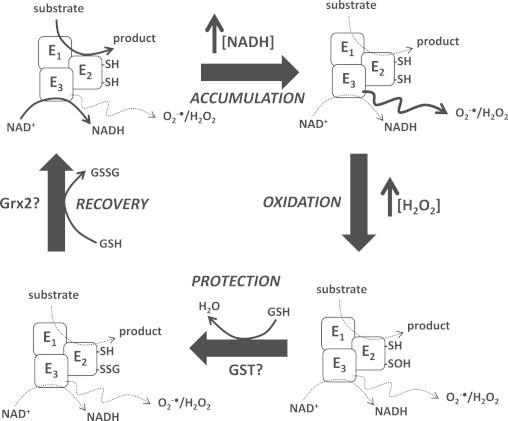
Hypothetical mechanism for the regulation of Odh and Pdh by reversible S-glutathionylation is required to modulate mitochondrial O2−•/H2O2. Odh and Pdh couple oxidation of 2-oxoglutarate or pyruvate to formation of either acetyl-CoA or succinyl-CoA and the production of NADH. O2−•/H2O2 production is minimal due to the rapid oxidation of NADH to rejuvenate NAD pools. PHASE 1, ACCUMULATION: NADH oxidation slows most likely due to a decrease in complex I activity resulting in an increase in NADH levels. This prompts increased O2−•/H2O2 production by the E3 subunit of either enzyme. Substrate oxidation also slows diminishing NADH formation. PHASE 2, OXIDATION: vicinal thiols (-SH) on E2 subunit are oxidized by the increase in H2O2 levels yielding highly reactive sulfenic acids (-SOH) deactivating the enzyme complex. Although O2−•/H2O2 emission is decreased by this modification the –SOH renders the enzyme complex amenable to irreversible oxidation. PHASE 3, PROTECTION: sulfenic acids are modified by S-glutathionylation by conjugation to glutathione (GSH), a reaction potentially catalyzed by glutathione S-transferase (GST). This effectively protects Odh and Pdh from further oxidation. Enzyme remains inactive during this phase of regulation which also prevents O2−•/H2O2 production. PHASE 4, RECOVERY: the glutathionyl moiety is removed potentially by glutaredoxin-2 (Grx2) yielding a fully active enzyme complex. Odh and Pdh are now fully active coupling substrate oxidation to NADH formation. Grx2-mediated deglutathionylation generates GSSG which is reduced back to GSH by glutathione reductase and NADPH.
Numerous studies have established that Odh and Pdh sense mitochondrial H2O2 levels which effectively curtails O2−•/H2O2 production by mitochondria [66–69]. However, for Odh and Pdh to serve as redox sensors the modification must be reversible. Indeed, at sufficient quantities H2O2 can oxidize SOH to sulfinic (SO2H) and sulfonic (SO3H) acids, irreversible redox modifications that render Odh and Pdh inactive (Fig 8). Oxidative stress often leads to the deactivation of Odh and Pdh which has been associated with cardiomyopathy, ischemia-reperfusion injury in cardiac and brain tissue, obesity, heavy metal toxicity, and cardiovascular disease [70]. It has also been predicted that SOH groups have a pKa ~6 and thus ionize readily to form a highly nucleophilic sulfenate anion (SO−) [71,72]. Sulfenates can react rapidly to form covalent adducts with a number of molecules but in particular when O2−•/H2O2 production is high SO− can by further oxidized to SO3H or react with 4-hydroxy-2-nonenal (4-HNE), an end product of lipid damage, to form 4-HNE protein adducts [73,74]. Importantly, formation of 4-HNE adducts on the dihydrolipoamide of Odh and Pdh is associated with cardiac and neurological disease and treatment of mitochondria with 4-HNE deactivates both enzymes leading to an energy deficit [73–75]. Thus, in order for Odh and Pdh to serve as a redox sensor for O2−•/H2O2 SOH must be protected to prevent further oxidation. One mechanism that has been documented to protect dihydrolipoamide residues in Odh and possibly Pdh from further oxidation is S-glutathionylation (Fig. 7). S-glutathionylation is a redox sensitive covalent modification that involves formation of a disulfide bond between an available glutathione moiety and a protein cysteine thiol (Fig. 8) [6]. This results in the formation of a protein glutathione mixed disulfide (PSSG). Notably the modification is reversible and proceeds enzymatically with glutathione S-transferases (GST) catalyzing the forward reaction and glutaredoxins (Grx), a class of thiol oxidoreductases that part of the thioredoxin superfamily, catalyzing both S-glutathionylation and deglutathionylation (Fig. 8) [76,77]. In regard to protecting Odh, SOH formation results in immediate S-glutathionylation, possibly catalyzed by mitochondrial GST isoforms or mitochondrial glutaredoxin (Grx2), which effectively protects dihydrolipoamide in Odh from further oxidation [64,78]. The glutathionyl moiety is then removed by Grx2 which restores the activity of Odh [64,78]. It is important to note that it has not been shown whether or not mitochondrial Grx isoform, Grx2, is able to deglutathionylate Odh however; considering that purified Grx1 can catalyze this reaction in vitro and that Grx1 can supplement for Grx2 for deglutathionylation reactions it stands to reason that Grx2 likely deglutathionylates Odh. Thus, reversible S-glutathionylation plays a critical role in Odh redox sensing (Figs. 7 and 8).
Fig. 8.
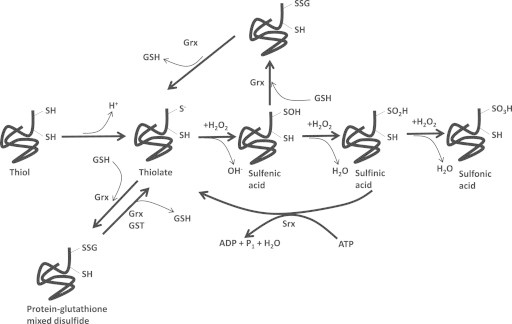
Regulation of protein function by S-glutathionylation. Redox signaling refers to the control of protein function via site specific oxidation of protein cysteine thiols in response to redox fluctuations in the surrounding cellular environment. Although there are a number of redox modifications that are known to modulate cellular protein functions [6], S-glutathionylation and the formation of protein glutathione mixed disulfides is highly specific, sensitive to redox fluctuations and mediated enzymatically and thus the most relevant redox modification. The reactivity of a thiol towards glutathione depends on its ability to ionize and form a reactive thiolate anion. Ionization is heavily influenced by the chemistry of the surrounding protein environment. In the presence of sufficient quantities of H2O2, the thiolate anion nucleophilically attacks H2O2 yielding an oxidized sulfur residue (SO−; sulfenic acid). If H2O2 is in high enough amounts, the sulfenic acid can be further oxidized to sulfinic (SO2H) and sulfonic acid (SO3H). Note that sulfinic acid can be reduced back to a thiolate by the action of sulfiredoxin (Srx) which requires ATP. Sulfonic acids, however, are an irreversible type of oxidation associated with oxidative stress. Proteins can either be S-glutathionylated at the level of the thiolate or sulfenic acid. In terms of the former, S-glutathionylation is driven by glutaredoxin-2 in mitochondria (Grx2) or glutaredoxin-1 (Grx1) in the cytosol and intermembrane space. S-glutathionylation can be quickly reversed by Grx. Importantly, protein S-glutathionylation is highly sensitive to fluctuations in reduced and oxidized glutathione levels and the circumstances by which a protein is S-glutathionylated varies according to the type of protein and the environment surrounding the protein cysteine thiol (reviewed in [6,12,23]). In terms of the latter, S-glutathionylation of sulfenic acids is required to protect cysteine thiols from further oxidation when H2O2 is at higher concentrations.
According to Quinlan et al., Pdh produces two times less O2−•/H2O2 than Odh but is still a significant source nonetheless [39]. Intriguingly it still remains to be determined if Pdh can be protected from further oxidation by S-glutathionylation. It has been suggested however in a recent report that glutathione directly binds to Pdh forming disulfide bonds with dihydrolipoamide [40]. It is important to point out here that S-glutathionylation proceeds enzymatically given the poor reactivity of reduced glutathione towards other thiols [79]. In addition, Grx2 has been shown to catalyze S-glutathionylation reactions at rates that are 250-fold faster nonenzymatic reactions [76,80]. Also, with the recent identification of S-glutathionylation motifs it is more probable that reversible S-glutathionylation of Pdh is enzymatically catalyzed, most likely by GST or Grx2 [81]. Considering that Pdh is deactivated by H2O2 it stands to reason that it is also subjected to reversible S-glutathionylation which most likely maintains its redox sensing properties. Bckdh, is predicted to produce four times less O2−•/H2O2 than Odh [39]. Although Bckdh has not been shown to be modulated by redox signaling cascades like S-glutathionylation, the enzyme upstream to Bckdh that commits branched chain amino acids to degradation, branched chain aminotransferase, can be modulated by S-glutathionylation [82]. Notably, though Bckdh activity also relies on dihydrolipoate and thus may be modulated in a fashion similar to Odh and perhaps Pdh. Thus, the FAD-dependent dihydrolipoamide enzymes Odh, Pdh, and Bckdh serve as important sources of O2−•/H2O2 and also serve as redox sensors modulating mitochondrial O2−•/H2O2 emission in response to ROS fluctuations, a process dependent on S-glutathionylation.
QH2/Q isopotential group
Q sits at a major electron transfer junction for aerobic respiration accepting electrons from Complex I and II following NADH and succinate oxidation [83]. As shown in Fig. 5, other metabolic enzymes such as ETFQO, SQR, sn-G3PDH, Dhodh, and Prodh also couple oxidation of acyl-CoA, H2S, glycerol-3-phosphate, dihydroorotate, and proline to reduction of Q to QH2. It is assumed that not all these enzymes simultaneously supply electrons to Q electron carriers in mitochondria. In addition, some of these enzymes display tissue and species specific expression. It has been documented for some time that some of these enzymes produce O2−•/H2O2 [20,44,84,85]. However, several recent studies have established that most of the O2−•/H2O2 generated by these enzymes is indirect and produced mostly from Complex I, II, III, and in some cases Odh [16,17,44]. For example, sn-G3PDH is found in brain, skeletal muscle, and heart tissue but is most heavily expressed brown adipose tissue [43]. Studies have now shown that sn-G3PDH can generate high amounts of O2−•/H2O2 [20,86]. However, a recent report has provided evidence sn-G3PDH is indirectly responsible for O2−•/H2O2 with a vast majority being generated in brain, heart, and skeletal muscle by reverse electron flow (RET) to FAD in Complex II [43]. The only exception is brown fat mitochondria where sn-G3PDH accounts for a large fraction of the O2−•/H2O2 generation [20]. Prodh is expressed in some cancer cells and found in flight muscle mitochondria of insects like Drosophila melanogaster [87,88]. Studies have shown that proline oxidation can produce O2−•/H2O2 but like sn-G3PDH most of the production is attributed to other sites; for breast cancer cell lines Complex I and II and insect flight muscle, Complex I and Odh. Again, in the case of Prodh, most of the O2−•/H2O2 is produced as a consequence of RET but also the accumulation of NADH due to proline oxidation which eventually generates 2-oxoglutarate [16]. Most of the O2−•/H2O2 resulting from palmitoyl-carnitine oxidation by ETFQO is produced by Complex I, II, and III with the relative contributions of each site varying according to the presence or absence of malate and/or l-carnitine [44]. Thus, although a number of substrates can feed electrons into the Q pool of mitochondria, the ultimate sites of O2−•/H2O2 production are still Complex I and III and intriguingly Complex II and Odh. The enzymes Prodh, Drodh, SQR, ETFQO, and sn-G3PDH do produce small amounts of O2−•/H2O2 though and if inhibited can produce a lot more indicating that under special circumstances these enzymes can be significant sources. However physiologically, it should be revised that mitochondria can produce O2−•/H2O2 from 11 potential substrates; pyruvate, 2-oxoglutarate, NADH, succinate, H2S, branched chain keto acids, acyl-CoA, dihydroorotate, QH2, proline, and glycerol-3-phosphate, whereas the actual source for most of the mitochondrial O2−•/H2O2 seems to be reserved to a handful of enzymes across both isopotential groups; Complexes I, II, III, Odh, and Pdh.
Complex III serves as a major source of mitochondrial O2−•/H2O2. Emission of O2−•/H2O2 from the Qo site of Complex III into the intermembrane space has been viewed as a major signaling platform for ROS mediated communication between mitochondria and the rest of the cell [89,90]. It has been shown that Complex III can be targeted for covalent modification, such as phosphorylation however; it remains unclear whether or not posttranslational modifications can modulate electron flow and O2−•/H2O2 production by Complex III. O2−•/H2O2 production from Complex III arises from the accumulation of semi-ubiquinone (QH−●) in the Qo site of Complex III [14,32]. Indeed, QH2 can only donate one electron at a time to cytochrome C1 and cytochrome C. This yields QH−● which then enters the Q cycle where it is recycled by b cytochromes to regenerate Q and QH2 for another round of oxidation (reviewed in 91). The Q cycle is highly efficient but the rate of QH−● recycling is dependent on polarity of Δμm. As Δμm increases this creates a significant protonic back pressure that slows the Q cycle prompting the accumulation of QH−● which elevates O2−•/H2O2 production (Fig. 9) [92]. It is well documented that there is a non-Ohmic relationship between Δμm and mitochondrial O2−•/H2O2 production where small increases in membrane potential can substantially increase ROS production [93]. The reverse is also true where “mild uncoupling” of Δμm can substantially limit mitochondrial O2−•/H2O2 production [94]. Mitochondria from a myriad of tissues contain proteins embedded in the MIM that ferry protons from the IMS to the matrix which controls mitochondrial O2−•/H2O2 production [95]. The uncoupling protein family (UCPs), specifically UCP2 and UCP3, have been shown to be required for the protection of various tissues from oxidative stress via the control of mitochondrial O2−•/H2O2 production by induction of mild uncoupling of Δμm [32]. In addition, it has now been established that both proteins are controlled by reversible S-glutathionylation where H2O2 is required to deglutathionylate and activate both proteins by an as of yet unidentified enzyme [96–98]. Subsequent reglutathionylation of UCP2 and UCP3 deactivates proton leaks. Recent work has also identified UCP3 as a target for the thiol oxidoreductase activity of Grx2 which catalyzes the protein S-glutathionylation of UCP3 [96]. Thus, proton leaks are sensitive to redox signaling which is required to control mitochondrial O2−•/H2O2 production. The control of leaks through UCP2 and UCP3 by reversible S-glutathionylation also has important physiological implications. UCP2 is more ubiquitously expressed while UCP3 is found almost exclusively in skeletal muscle (Fig. 9). Reversible S-glutathionylation of UCP2 has been found to play on important role in modulating glucose stimulated insulin release from pancreatic β cells [97]. Control of leaks through UCP3 on the other hand is required to maintain efficient glucose and fatty acid oxidation in skeletal muscle which is most likely important for maintaining insulin sensitivity and contractility [99,100]. Adenine nucleotide translocator (ANT), which catalyzes the anti-port of ADP and ATP across the MIM also catalyzes inducible proton leaks in various tissues including brain, heart, skeletal muscle and liver. Intriguingly it is also a major site for redox-mediated regulation particularly by S-glutathionylation [101]. Although it has not been investigated, it would be important to ascertain if S-glutathionylation reactions also modulate leaks through ANT. It is also critical to point out that leaks through ANT are also activated by covalent modification by nitrolipids and 4-HNE [102,103]. Thus, O2−•/H2O2 production by Complex III can be indirectly controlled by redox signaling, mainly through the activation and deactivation of proton leak mechanisms.
Fig. 9.
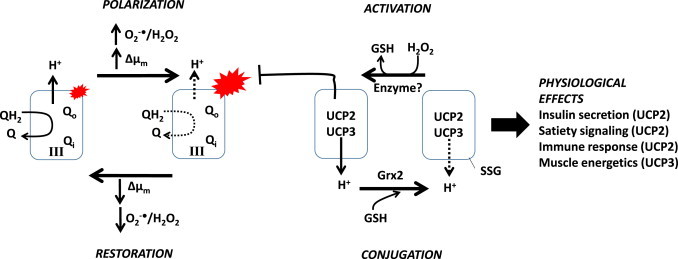
Hypothetical mechanism for the modulation of O2−•/H2O2 by complex III via S-glutathionylation controlled proton leaks. Complex III systematically oxidizes QH2 donating one electron at a time to cytochrome C. The resulting QH−• is recycled via the Q-cycle for another round of oxidation. Fully oxidized Q returns to the Q pool to be reduced by complex I, II, or other electron donors (see Fig. 1). PHASE 1, POLARIZATION: electrochemical transmembrane potential of protons (ΔΨm) increasing membrane polarity which limits QH2 and QH−● oxidation. QH−● accumulates in the Qo site augmenting mitochondrial O2−•/H2O2 production. PHASE 2, ACTIVATION: the rise in mitochondrial O2−•/H2O2 production and ΔΨm results in the deglutathionylation and activation of UCP2 and UCP3. Note that UCP2 and UCP3 are expressed in different tissues and activation can have different physiological consequences. Deglutathionylation is mediated by low µM increase in H2O2. The reaction is catalyzed by an as of yet unidentified enzyme [97,98]. Activation induces mild uncoupling of the mitochondrial inner membrane decreasing protonic pressure on complex III thus limiting O2−•/H2O2 production. PHASE 3, CONJUGATION: the decrease O2−•/H2O2 production and ΔΨm results in the reglutathionylation and deactivation of UCP2 and UCP3. The reaction is catalyzed by Grx2. PHASE 4, RESTORATION: with the ΔΨm brought back down QH2 oxidation by complex III resumes with efficient recovery of electrons from QH−● in the Q-cycle. Note that in this diagram ANT was omitted for clarity however; as indicated in the text it also plays an important role in inducible proton leaks and is targeted for S-glutathionylation.
Numerous studies have also focused on Complex I which can produce high amounts of O2−•/H2O2 via electron back flow from various substrates that are involved in direct reduction of Q to QH2 as described above. It has also been well documented that Complex I serves as a major hub for redox signaling [104]. Several subunits on Complex I including Ndusf1 and Ndufv1, which form part of the NADH binding site and are required to coordinate several Fe–S clusters, are targets for reversible S-glutathionylation which is mediated by Grx2 [104,105]. Deregulation of S-glutathionylation of these subunits on Complex I not only disrupts its activity but also increases mitochondrial O2−•/H2O2 production which is associated with heart disease (decompensated hypertrophy), ischemia reperfusion injury, doxorubicin toxicity, Parkinson’s disease, and potentially liver disease [96,105–108]. ND3 subunit, which forms part of the Q binding site, can also be targeted for modification by S-nitrosylation and potentially S-glutathionylation [23]. Recent work has established pharmacological induction of S-nitrosylation Cys39 of ND3 prevents ischemia-reperfusion injury and reduces myocardial infarct size [109]. Thus, Complex I is a key site for redox regulation of mitochondrial processes and can also serve as an important pharmacological target in prevention of disease. As mentioned in the NADH/NAD isopotential section, Quinlan et al. found that when NADH is abundant Odh produces 8 times more O2−•/H2O2 Complex I [39]. However, in the same study, it was shown that reverse electron flow through the quinone pool has the opposite effect – Complex I serves as one of the major sites [39]. In fact, as mentioned above, Complex I serves as a major source of O2−•/H2O2 when substrates that feed electrons directly into the Q pool are being oxidized. Most of studies that have shown that RET produces a large amount of O2−•/H2O2 from Complex I utilize supraphysiological concentrations of substrate, most commonly succinate (5–10 mM). Considering that under normal conditions succinate occurs in much lower concentrations in mitochondria the role of RET in Complex I-mediated O2−•/H2O2 has been questioned. However, a very recent report has shown that succinate accumulates in the ischemic myocardium and produces a high amount of O2−•/H2O2 following reperfusion leading to development of heart disease and formation of myocardial infarcts [42]. Intriguingly, dimethyl malonate, a membrane permeable Complex II inhibitor, decreased myocardial infarct size [42]. Thus, RET from succinate to Complex I may not be a physiologically relevant source of O2−•/H2O2 but would appear to be a major source during ischemia-reperfusion injury and heart disease.
Acyl-CoA is another intriguing source O2−•/H2O2 produced by RET since fatty acids serve as a major energy source for several tissues including exercised skeletal muscle, liver, and the myocardium. In particular, ~70–90% of the ATP generated by mitochondria in cardiomyocytes is provided by fatty acid oxidation [110]. At rest the human heart turns over ~30 kg of ATP per day and ~90% of that ATP is provided by mitochondria meaning that ~18.9–24.3 kg of this ATP is produced by fatty acid oxidation [111]. Studies have shown that high fat diet does induce diabetic cardiomyopathy potentially through increased mitochondrial O2−•/H2O2 production [112]. The fact that acyl-CoA, in particular palmitoyl-carnitine, can produce O2−•/H2O2 by RET under specific nutritional states indicates that fatty acid oxidation and RET could contribute to mitochondrial O2−•/H2O2 in the myocardium in physiological and pathological states. The amount of O2−•/H2O2 produced is also proportional to efficiency of electron flow to O2 at Complex IV and the polarity of the MIM. Intriguingly Perevoshchikova et al. was also able to show that FAD in Complex II also serves as an important site for O2−•/H2O2 when rat skeletal muscle mitochondria are metabolizing either palmitoyl-carnitine alone or palmitoyl-carnitine + carnitine [44]. Thus, RET-mediated O2−•/H2O2 production can also occur at sites other than Complex I (e.g. Complex II). Several studies have shown that Complex II can produce O2−•/H2O2 [41,113]. In fact, Complex II generates a substantial amount of O2−•/H2O2 especially when FADH2 oxidation is prevented by Q binding site inhibitors [41]. It has been established that the source of O2−•/H2O2 in Complex II is FAD in the SdhA subunit of the Complex [41]. Siebels and Drӧse also established using submitochondrial particles from bovine heart that Complex II generated the most O2−•/H2O2 when only 100 µM succinate was added to reaction chambers [41]. This is intriguing since it shows that physiologically relevant levels of succinate can prompt O2−•/H2O2 from Complex II. In addition, Complex II is sensitive to redox modifications such as S-glutathionylation [114]. In fact, when maintained in an S-glutathionylated state Complex II efficiently metabolizes succinate and produces little O2−•/H2O2 in cardiac tissue [114]. However, following ischemia-reperfusion injury to the myocardium Complex II adopts a deglutathionylated state and produces higher amounts of O2−•/H2O2. This also leads to the accumulation of succinate (as high as 1 mM) which can interact with orphan G-protein coupled receptor 91, SUCNR1, leading to further development of cardiovascular disease, disruption of myocardial function, and inflammation [115]. It is clear that RET can produce O2−•/H2O2 following oxidation of different substrates and that several respiratory complexes can partake in its production which has strong implications for O2−•/H2O2 production in health and disease.
Measuring mitochondrial O2−•/H2O2 production
Methodologies that measure O2−• directly
Accurate quantification of O2−• production by mitochondria is of importance considering it is the proximal ROS generated by mitochondria and over production is associated with various pathologies [116]. O2−• production in mitochondria is always favorable given the superior concentration of O2 in comparison to O2−• ([O2]=3−30 µM in mitochondria in vivo, [O2−•] ~pM range) [117]. The low concentration and short half-life of O2−• is attributed to SOD which dismutates O2−• to H2O2 very rapidly (1.8×109 M−1 s−1) [12]. This presents a significant hurdle in quantification of O2−• levels in mitochondria since O2−• does not last very long in solution and concentrations are extremely low. It is also ideal to attempt to quantify O2−• production in intact cell systems or potentially in vivo considering it provides a biological context for mitochondrial O2−• production in health and disease. The most popular chemical probe utilized to measure O2−• in live cells is MitoSOX which consists of a hydroethidine molecule (HE) covalently bound to triphenylphosphonium ion (TPP+) (Fig. 10) [118]. HE is an O2−• sensitive probe that fluoresces following a reaction with O2−•. TPP+ is a lipophilic cation that promotes uptake of HE by mitochondria which is based on the charge difference between the matrix and intermembrane space (Fig. 10) [119]. A number of studies have utilized MitoSOX to measure mitochondrial O2−• production using plate methods including kinetic reads or end-point measures, flow cytometry, HPLC analysis of MitoSOX oxidation, and live cell imaging [120]. However, there are a number of considerable drawbacks associated with the use of MitoSOX. The first is the fact that HE can form non-fluorescent dimers as a result of oxidation [120]. To properly quantify both the red fluorescent product and the non-fluorescent dimer HPLC techniques can be utilized however; it requires cell disruption thus rendering any live cell imaging impossible. This impediment can be overcome by measuring HE fluorescence at excitation wavelength 396 nm which specifically excites HE that has reacted with O2−• but has not intercalated with DNA [121]. Second, the calculated rate of oxidation of MitoSOX by O2−• is 3.9×106 M−1 s−1 which, although rapid, is several orders of magnitude slower than SOD [121]. This would mean that (1) mitochondria would need to generate a considerable amount of O2−• to afford proper fluorescence detection and (2) enough MitoSOX would need to be utilized to compete with SOD. Considering the high concentration (~10 µM) and rapid kinetics of MnSOD this presents a significant challenge in accurate quantification of O2−• production by mitochondria. In addition most assays are conducted at ambient oxygen ([O2]atmosphere=~200 µM) whereas the [O2] experienced by mitochondria in vivo is far less [122]. This may lead to over-estimation of mitochondrial O2−• production. Finally, cell lines and primary cells are usually cultured in highly artificial systems such as a humidified atmosphere containing 5% CO2, 20% O2 in media supplemented with ample amounts of energy substrates and serum factors. In addition cell density can affect the quality of the measurements leading to over or under estimation of mitochondrial O2−• production. Thus, a considerable amount of assay optimization must be undertaken to ensure that O2−• production by mitochondria is accurately measured. Most importantly, intact cell systems lack context for in vivo production of O2−• considering the conditions are artificial, the high O2 tension, and the reactivity of MitoSOX. Interestingly, Henderson et al. recently developed a sensitive method for measuring real-time production of O2−• by mitochondria which utilizes a cytochrome C functionalized amperometric sensor [123]. The same group recently showed that this technique can also be utilized to accurately measure O2−• production in intact cell systems and even successfully employed this technique to illustrate that mitochondrial O2−• produced from Complex II plays an important role in melanogenesis and cellular pigmentation [124]. Another promising chemical method for measuring O2−• is the probe 4,5-dimethoxy-2-nitro-benzenesulfonyl tetrafluorofluorescein (BESSo) which following a nucleophilic substitution reaction with O2−• yields a highly fluorescent tetrafluorofluorescein product [125].
Fig. 10.
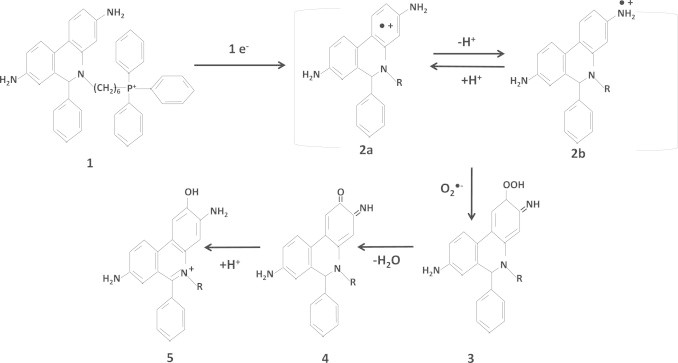
Proposed mechanism for the reaction of MitoSOX with O2•− (modified from [121] and [120]). MitoSOX is composed of hydroethidine which reacts with O2•− and triphenylphosphonium (R-group) which prompts accumulation in mitochondria (1). The structure is univalently reduced by one electron generate a chemical structure that resonates between a hydroethidine radical with the lone electron delocalized to throughout the ring (2a) and an amide radical (2b). Electron delocalization between the ring and amide aids in stabilizing this reactive intermediate. This is followed by an interaction with O2•− which results in the production of a perhydroxyl intermediate (3) which is then dehydrated to yield a carbonyl derivative (4) that is then protonated to generate 2-hydroxy-ethidine or 2-hydroxy-MitoSOX (5).
The lack of an in vivo context with intact cell systems and MitoSOX prompted investigations into cyclic permuted yellow fluorescent protein (cpYFP). Unlike MitoSOX this probe can be expressed stably within either cell systems or in vivo [126]. Further, cpYFP can be selectively targeted to mitochondria by mitochondrial localization sequences. Initially this protein based probe showed a lot of promise and appeared to selectively detect fluctuations in mitochondrial O2−• production in heart tissue from transgenic mice expressing cpYFP or in live cells transiently expressing the probe (Fig. 11) [126,127]. In addition, the observations by the same group and others were extended pointing to a role for stochastic O2−• flashes in mitochondria in the opening of the mitochondrial permeability transition pore and modulation of other physiological processes [128,129]. However, detection of spatiotemporal changes in O2−• production would mean that cpYFP would have to be highly selective and would have to react with kinetics that are at very least similar to SOD. The O2−• mediated changes in cpYFP fluorescence were attributed to oxidation of Cys171 and Cys193 residues in the fluorescent protein. It is unlikely that Cys residues partake in the detection of O2−• considering the slow reaction kinetics of Cys with O2−• [130]. Indeed, O2−• is both a weak oxidant and reductant and thus does not react rapidly with a number of molecules except for Fe−S clusters [131]. In addition, a recent publication by Schwartzlander et al. showed that Cys171 and Cys193 in cpYFP are not accessible to solutes [132]. Thus, given its charged nature, O2−• cannot gain access to the two Cys residues [132]. In addition, unlike other ROS detecting molecules, the mechanism by which cpYFP changes its fluorescence in response to O2−• has not been adequately delineated. With this in mind, it is improbable that cpYFP reacts with O2−• since (1) Cys171 and Cys193 are inaccessible and (2) reaction kinetics are far too slow and thus cannot compete with SOD.
Fig. 11.
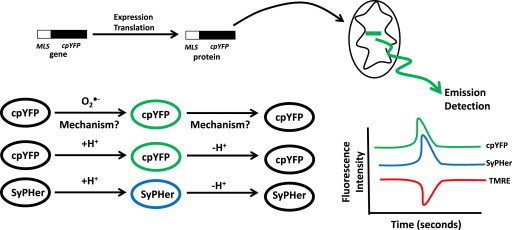
cpYFP is not a mitochondrial O2•− indicator but pH detector. Cyclic permuted YFP (cpYFP) gene can be tagged with a mitochondrial localization sequence (MLS) and stably expressed in cells or animal tissues. Note that cpYFP gene can also be placed under control of tissue specific promoters to allow for site specific expression of cpYFP. Following gene transcription and translation the MLS selectively targets cpYFP for uptake and accumulation in the matrix of mitochondria where cpYFP detects transient changes in mitochondrial metabolism and physiology. Although cpYFP has been utilized in numerous studies to measure “stochastic” changes in mitochondrial O2•− emission the mechanism by which it does so remains elusive but also seems chemically improbable. Rather, given the features of cpYFP and its sensitivity to changes in matrix pH it is far more likely that cpYFP serves as a protein-based pH sensor akin to SyPHer. As indicated in the text, it has been well documented that transient shifts in cpYFP fluorescence are sensitive to changes in the concentration of H+ in the matrix. In addition, shifts in cpYFP fluorescence correlate strongly with changes in Δμm as indicated by fluctuations in TMRE fluorescence.
Several studies have employed cpYFP to measure “stochastic O2−• flashes” in mitochondria. It is clear that a spatiotemporal change in cpYFP fluorescence is being recorded. Considering it is chemically implausible for O2−• to react with cpYFP what biological process is being measured? Intriguingly, the pH-sensitive fluorescence probe SyPHer also contains cpYFP [130]. Schwartzlander et al. established that cpYFP actually detects fluctuations in mitochondrial pH rather than O2−• [130] (Fig. 11). Indeed, changes in cpYFP fluorescence correlate strongly with changes in ΔΨm [133]. It was also found that cpYFP responds poorly to xanthine/xanthine oxidase, an enzyme system that generates high amounts of O2−• [132]. Further, SyPHer probe, which detects pH fluctuations, is also a cpYFP [130] (Fig. 11). It is very important to note that ΔΨm changes rapidly in response to various cellular stimuli, ATP demand, nutrient supply, electron transfer efficiency, and inducible proton leaks which will inevitably alter the overall pH of the matrix environment. Thus, cpYFP is more likely to be an appropriate probe utilized for measurement of pH changes in mitochondria rather than O2−•.
H2O2 detection
H2O2 is the result of O2−• dismutation. Unlike O2−•, H2O2 is more stable and thus much easier to quantify. H2O2 can also freely diffuse through membranes and also reacts readily with protein cysteine thiols [134]. It is therefore critical to properly quantify H2O2 levels in physiological and pathological states since it plays a key role in redox signaling and can also induce oxidative damage at high concentrations. Also, considering the rapidity of O2−• dismutation measurement of H2O2 production can serve as a proxy measure for O2−• production. Unlike O2−• detection a number of chemical and protein based probes that allow the reliable quantification of H2O2 have been developed. These probes are highly sensitive, selective, and have provided important information on H2O2 production and flux in isolated mitochondria, live cells and in vivo. For isolated mitochondria, the Amplex Red assay is the most commonly employed method utilized for accurate measurement of H2O2 (Fig. 12). This assay relies on oxidation of non-fluorescent Amplex Red by H2O2 producing fluorescent Resorufin, a reaction that requires the presence of horseradish peroxidase [135]. It should be noted that not all O2−• is converted to H2O2 by endogenous SOD since O2−• can also react with NO to form peroxynitrite. To maximize O2−• dismutation, exogenous SOD is also added to reaction mixtures. Given the fact that exogenous enzymes need to be added, Amplex Red assays cannot be carried out using intact cell systems. However, Amplex Red assays can be applied to tissues and cells with permeabilized plasma membranes. Another intriguing chemical method that can be employed for the detection of mitochondrial H2O2 is the MitoB method [136]. This method relies of the oxidization of arylboronic acid by H2O2 to a corresponding phenol which can be detected by fluorescence (Fig. 12). To ensure mitochondrial localization, the arylboronic acid moiety is tagged to TPP+ and is thus referred to as MitoB [136]. Both MitoB and MitoP can also be quantified by ratiometric mass spectrometry where fluctuations in matrix H2O2 are reflected by ratiometric changes in MitoB to MitoP (e.g. low H2O2 MitoP/MitoB is low whereas high H2O2 MitoP/MitoB is high) (Fig. 12) [136]. Ratiometric spectroscopic detection substantially enhances the sensitivity of the assay. This method has been utilized to successfully quantify matrix H2O2 in vivo in D. melanogaster and in mtDNA mutator mice and also to show that matrix H2O2 levels increase with age [136–138]. Of note though is that arylboronic acid compounds also react with peroxynitrite which can compromise the direct measure of H2O2.
Fig. 12.
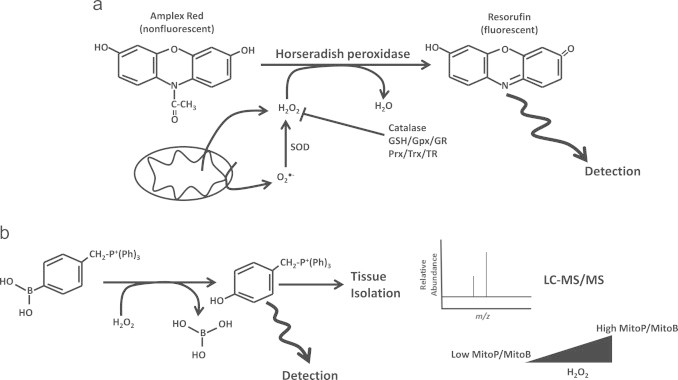
Detection of mitochondrial H2O2 with Amplex Red and MitoB. a. In the presence of H2O2, horseradish peroxidase catalyzes the oxidation of nonfluorescent Amplex red forming fluorescent product resorufin. The fluorescent signal is directly proportion to the amount of H2O2 present in the sample. H2O2 can be generated within mitochondria following dismutation of O2•− which is exported by aquaporins for detection. Alternatively H2O2 can be produced outside the matrix environment if O2•− is produced in the intermembrane space. To ensure maximal conversion of O2•− to H2O2, exogenous SOD can be added which provides a proxy measure of both O2•−/H2O2. Control reactions may include addition of exogenous catalase. Note that presence of endogenous antioxidant system such as GSH/Gpx/GR and Prx/Trx/TR quench H2O2. Thus, Gpx or Trx inhibitors can be utilized to afford accurate H2O2 quantification. b. The arylboronic acid moiety is tagged with a triphenylphosphonium ion (CH2−P+(Ph)3) group which prompts matrix accumulation of the detector allowing for more accurate quantification of matrix H2O2. Following its interaction with H2O2 with MitoB, the corresponding phenol can be detected by fluorescence. Alternatively, the tissue can be isolated and MitoP and MitoB levels can be subjected to LC-MS/MS analysis. Detection of MitoP and MitoB levels by LC-MS/MS and calculation of the MitoP/MitoB ratio provides a highly quantitative measure of matrix H2O2 levels.
The ultimate goal is to perform quantitative measures of matrix H2O2 in vivo so one can ascertain the function of mitochondrial O2−•/H2O2 production in health and disease. Amplex Red cannot be utilized for in vivo measurements and although MitoB does provide quantitative information on matrix H2O2 in an in vivo context it must be injected into specimens. TPP+ does not allow selective accumulation of compounds in specific organs however; it does accumulate the most in tissues that are rich in mitochondria like heart, liver, and skeletal muscle. TPP+ has also been documented to depolarize the MIM and could compromise mitochondrial bioenergetics [139]. In addition, tissues must be isolated and then pulverized so MitoB and MitoP can be extracted for measurement meaning that MitoB can only be utilized for end-point analysis and cannot detect spatiotemporal changes in H2O2 in live animals. To overcome these limitations, several groups have developed protein based probes that can be selectively targeted to mitochondria for H2O2 detection. With these probes it is also possible to develop transgenic animals that have tissue-specific expression of these protein based H2O2 probes which would allow highly quantitative measurement of matrix H2O2 in specific organs. Three highly sensitive fluorescent proteins have been developed that allow the direct and indirect quantification of matrix H2O2; HyPer, roGFP-Orp1, and Grx1-roGFP (Fig. 13) [140,141]. HyPer was first developed by Belousov et al. and is composed of cpYFP conjugated to prokaryotic H2O2-sensing protein OxyR. Changes in HyPer fluorescence are reliant on Cys199 and Cys208 on OxyR (Fig. 13) [140]. Oxidation of Cys199 in OxyR leads to formation of SOH which then rapidly reacts with Cys208 to form an intramolecule disulfide linkage [142]. H2O2-mediated oxidation of OxyR induces changes in cpYFP fluorescence (Fig. 13). It has been shown in several studies that HyPer is highly sensitive to changes in H2O2 which is attributed to the rapid reaction of Cys199 with H2O2 [143]. In addition, HyPer is sensitive to changes in H2O2 in the low µM range making it very advantageous for the quantification of physiological shifts in H2O2 production [140]. Another notable attribute of HyPer is that the disulfide linkages formed following H2O2 mediated oxidation can be reduced endogenously possibly by NADPH and thioredoxin [144]. This means that HyPer can be recovered for another round of H2O2 detection allowing spatiotemporal measurement of changes in matrix and cytosolic H2O2. HyPer is now used routinely for measurement of dynamic changes in H2O2 in live cells including during cellular signaling and induction of apoptosis however; it has not yet been applied in vivo [140,145]. It should also be noted that recent reports have shown that HyPer is also sensitive to changes in pH which is likely due to the cpYFP [146]. By contrast, roGFP2-Orp1 and Grx1-roGFP2 have both been utilized for the successful measurement of spatiotemporal changes in minute amounts of H2O2 in fruit flies (Fig. 13) [141]. roGFP2-Orp1 detects H2O2 directly whereas Grx1-roGFP2 provides an indirect measure by reacting with glutathione disulfide (GSSG), which is the result of the enzyme-mediated oxidation of two reduced glutathione molecules with H2O2 [141]. For roGFP2-Orp1, a thiolate anion on Orp1 is oxidized by H2O2 forming a SOH which then reacts with a neighboring SH on Orp1 forming a disulfide bridge (Fig. 13) [141]. Through a disulfide exchange reaction, the two thiols on roGFP2 then become oxidized changing the fluorescence intensity of the probe. Grx1-roGFP2 operates via a similar mechanism except a small increase in GSSG prompts a thiol disulfide exchange reaction with Grx1 generating an S-glutathionylated-Grx1-roGFP2 intermediate (Fig. 13) [147]. Through another series of thiol disulfide exchange reactions the glutathionyl moiety is transferred to roGFP2 which then prompts the formation of a disulfide bridge and a change in roGFP2 fluorescence. Both probes have been used for the sensitive quantification of spatiotemporal changes in small amounts of H2O2 and GSSG in the cytosol and mitochondria of mid-gut enterocytes in Drosophila [141]. Based on sensitive time based measurements Albrecht et al. was able to show that cells in vivo harbor natural redox gradients in mitochondria and that these gradients display substantial shifts with age [141]. Although these probes are new and have not been used as extensively collectively these probes show the most promise in terms of detecting spatiotemporal changes in H2O2 in live animals.
Fig. 13.
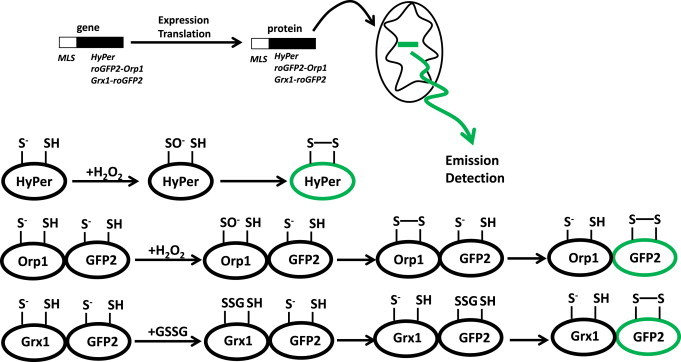
Protein-based mitochondrial H2O2 detectors. Three separate protein based probes can be employed to detect mitochondrial H2O2 levels, HyPer (OxyR-cpYFP) and roGFP2-Orp1 which directly detect fluctuations in H2O2 and Grx1-roGFP2 which indirectly detects H2O2 via interactions with GSSG (produced by glutathione peroxidase which catalyzes the sequestration of H2O2 oxidizing two GSH generating GSSG). Mitochondrial targeting of the different probes can be achieved by tagging the protein gene sequence with a mitochondrial localization signal (MLS).
Conclusions and perspectives
Mitochondrial O2−•/H2O2 plays a key role in cell communication at low amounts but can be a detriment at higher concentrations. Whether or not O2−•/H2O2 serves as a tool utilized to maintain cell function or is a harbinger of death depends on its production and degradation. O2−•/H2O2 can serve as intrinsic mitochondrial signaling molecules that modulate nutrient metabolism and bioenergetics as well as other processes including mitochondrial ultrastructure, protein import, and assembly of respirasomes. Mitochondria can also emit H2O2 which plays a key role in modulating various cellular functions and can even be utilized in intercellular signaling. Thus, it is important to critically evaluate how mitochondria generate O2−•/H2O2 in the presence of different nutrients and following changes in redox environment considering that mitochondrial ROS emission is important in cell communication. Here, the 11 potential substrates for mitochondrial O2−•/H2O2 have been critically reviewed. Based on the accumulated evidence, five out of the 11 enzymes, namely Complex I, II, III, Odh, and Pdh serve as the major sources of mitochondrial O2−•/H2O2. The other 6 enzymes can produce O2−•/H2O2 but only in small amounts which can be increased in the presence of high concentrations of inhibitors and substrate. A strong exception to this is G3PDH in brown fat mitochondria which illustrates the highly unique properties of this tissue. In addition, as originally put forth by Brand and colleagues, the 11 potential sites can be classified according to isopotential group. Thus, mitochondria can generate O2−•/H2O2 from 11 different substrate sources with contributions from the different major sites listed above varying according to nutrient and redox status in mitochondria. The major sites are also subjected to heavy regulation, either directly by redox signaling or indirectly by redox signaling-mediated control of proton leaks, which adjusts mitochondrial O2−•/H2O2 production in response to fluctuations in local redox environment. The fact that 11 sources can contribute to mitochondrial O2−•/H2O2 production illustrates the complexities associated with understanding the contribution of mitochondrial O2−•/H2O2 in health and disease. Indeed, quantitative measurement of mitochondrial O2−•/H2O2 production in vivo has proven to be the most significant hurdle in advancing our understanding of how mitochondrial O2−•/H2O2 influences the cellular environment. Although quantitative measurement of O2−• in vivo still remains out of reach new methods that accurately measure H2O2 are currently being utilized. These new quantitative methods will substantially advance our understanding of mitochondrial O2−•/H2O2 tissue function in vivo.
References
- 1.Halliwell B. Reactive species and antioxidants. Redox biology is a fundamental theme of aerobic life. Plant Physiology. 2006;141(2):312–322. doi: 10.1104/pp.106.077073. 16760481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crowe S.A., Døssing L.N., Beukes N.J., Bau M., Kruger S.J., Frei R., Canfield D.E. Atmospheric oxygenation three billion years ago. Nature. 2013;501(7468):535–538. doi: 10.1038/nature12426. 24067713 [DOI] [PubMed] [Google Scholar]
- 3.Ilbert M., Bonnefoy V. Insight into the evolution of the iron oxidation pathways. Biochimica et Biophysica Acta. 2013;1827(2):161–175. doi: 10.1016/j.bbabio.2012.10.001. 23044392 [DOI] [PubMed] [Google Scholar]
- 4.Murphy M.P. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009;417(1):1–13. doi: 10.1042/BJ20081386. 19061483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gill S.S., Tuteja N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiology and Biochemistry. 2010;48(12):909–930. doi: 10.1016/j.plaphy.2010.08.016. 20870416 [DOI] [PubMed] [Google Scholar]
- 6.Mailloux R.J., Jin X., Willmore W.G. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biology. 2014;2:123–139. doi: 10.1016/j.redox.2013.12.011. 24455476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harel A., Bromberg Y., Falkowski P.G., Bhattacharya D. Evolutionary history of redox metal-binding domains across the tree of life. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(19):7042–7047. doi: 10.1073/pnas.1403676111. 24778258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lane N. Bioenergetic constraints on the evolution of complex life. Cold Spring Harbor Perspectives in Biology. 2014;6(5):a015982. doi: 10.1101/cshperspect.a015982. 24789818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wallace D.C. Colloquium paper: bioenergetics, the origins of complexity, and the ascent of man. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(Suppl. 2):S8947–S8953. doi: 10.1073/pnas.0914635107. 20445102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fernie A.R., Carrari F., Sweetlove L.J. Respiratory metabolism: glycolysis, the TCA cycle and mitochondrial electron transport. Current Opinion in Plant Biology. 2004;7(3):254–261. doi: 10.1016/j.pbi.2004.03.007. 15134745 [DOI] [PubMed] [Google Scholar]
- 11.Lane N., Martin W. The energetics of genome complexity. Nature. 2010;467(7318):929–934. doi: 10.1038/nature09486. 20962839 [DOI] [PubMed] [Google Scholar]
- 12.Mailloux R.J., McBride S.L., Harper M.E. Unearthing the secrets of mitochondrial ROS and glutathione in bioenergetics. Trends in Biochemical Sciences. 2013;38(12):592–602. doi: 10.1016/j.tibs.2013.09.001. 24120033 [DOI] [PubMed] [Google Scholar]
- 13.Rosca M.G., Tandler B., Hoppel C.L. Mitochondria in cardiac hypertrophy and heart failure. Journal of Molecular and Cellular Cardiology. 2013;55:31–41. doi: 10.1016/j.yjmcc.2012.09.002. 22982369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y.R., Zweier J.L. Cardiac mitochondria and reactive oxygen species generation. Circulation Research. 2014;114(3):524–537. doi: 10.1161/CIRCRESAHA.114.300559. 24481843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nicholls D.G. Mitochondrial ion circuits. Essays in Biochemistry. 2010;47:25–35. doi: 10.1042/bse0470025. 20533898 [DOI] [PubMed] [Google Scholar]
- 16.Goncalves R.L., Rothschild D.E., Quinlan C.L., Scott G.K., Benz C.C., Brand M.D. Sources of superoxide/H2O2 during mitochondrial proline oxidation. Redox Biology. 2014;2:901–909. doi: 10.1016/j.redox.2014.07.003. 25184115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hey-Mogensen M., Goncalves R.L., Orr A.L., Brand M.D. Production of superoxide/H2O2 by dihydroorotate dehydrogenase in rat skeletal muscle mitochondria. Free Radical Biology and Medicine. 2014;72:149–155. doi: 10.1016/j.freeradbiomed.2014.04.007. 24746616 [DOI] [PubMed] [Google Scholar]
- 18.Quinlan C.L., Orr A.L., Perevoshchikova I.V., Treberg J.R., Ackrell B.A., Brand M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. Journal of Biological Chemistry. 2012;287(32):27255–27264. doi: 10.1074/jbc.M112.374629. 22689576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang J., Frerman F.E., Kim J.J. Structure of electron transfer flavoprotein-ubiquinone oxidoreductase and electron transfer to the mitochondrial ubiquinone pool. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(44):16212–16217. doi: 10.1073/pnas.0604567103. 17050691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shabalina I.G., Vrbacký M., Pecinová A., Kalinovich A.V., Drahota Z., Houštěk J., Mráček T., Cannon B., Nedergaard J. ROS production in brown adipose tissue mitochondria: the question of UCP1-dependence. Biochimica et Biophysica Acta. 2014;1837:2017–2030. doi: 10.1016/j.bbabio.2014.04.005. 24769119 [DOI] [PubMed] [Google Scholar]
- 21.Lagoutte E., Mimoun S., Andriamihaja M., Chaumontet C., Blachier F., Bouillaud F. Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochimica et Biophysica Acta. 2010;1797(8):1500–1511. doi: 10.1016/j.bbabio.2010.04.004. 20398623 [DOI] [PubMed] [Google Scholar]
- 22.Watt I.N., Montgomery M.G., Runswick M.J., Leslie A.G., Walker J.E. Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(39):16823–16827. doi: 10.1073/pnas.1011099107. 20847295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mailloux R.J., Willmore W.G. S-glutathionylation reactions in mitochondrial function and disease. Frontiers in Cell and Developmental Biology. 2014;2:68. doi: 10.3389/fcell.2014.00068. 25453035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rees D.M., Leslie A.G., Walker J.E. The structure of the membrane extrinsic region of bovine ATP synthase. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(51):21597–21601. doi: 10.1073/pnas.0910365106. 19995987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boveris A., Oshino N., Chance B. The cellular production of hydrogen peroxide. Biochemical Journal. 1972;128(3):617–630. doi: 10.1042/bj1280617. 4404507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boveris A., Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochemical Journal. 1973;134(3):707–716. doi: 10.1042/bj1340707. 4749271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brand M.D., Nicholls D.G. Assessing mitochondrial dysfunction in cells. Biochemical Journal. 2011;435(2):297–312. doi: 10.1042/BJ20110162. 21726199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mailloux R.J., Harper M.E. Mitochondrial proticity and ROS signaling: lessons from the uncoupling proteins. Trends in Endocrinology and Metabolism. 2012;23(9):451–458. doi: 10.1016/j.tem.2012.04.004. 22591987 [DOI] [PubMed] [Google Scholar]
- 29.Jones Q.R., Warford J., Rupasinghe H.P., Robertson G.S. Target-based selection of flavonoids for neurodegenerative disorders. Trends in Pharmacological Sciences. 2012;33(11):602–610. doi: 10.1016/j.tips.2012.08.002. 22980637 [DOI] [PubMed] [Google Scholar]
- 30.Brand M.D., Esteves T.C. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metabolism. 2005;2(2):85–93. doi: 10.1016/j.cmet.2005.06.002. 16098826 [DOI] [PubMed] [Google Scholar]
- 31.Long Y.C., Tan T.M., Takao I., Tang B.L. The biochemistry and cell biology of aging: metabolic regulation through mitochondrial signaling. The American Journal of Physiology – Endocrinology and Metabolism. 2014;306(6):E581–E591. doi: 10.1152/ajpendo.00665.2013. 24452454 [DOI] [PubMed] [Google Scholar]
- 32.Mailloux R.J., Harper M.E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radical Biology and Medicine. 2011;51(6):1106–1115. doi: 10.1016/j.freeradbiomed.2011.06.022. 21762777 [DOI] [PubMed] [Google Scholar]
- 33.Murphy M.P. Mitochondrial thiols in antioxidant protection and redox signaling: distinct roles for glutathionylation and other thiol modifications. Antioxidants and Redox Signaling. 2012;16(6):476–495. doi: 10.1089/ars.2011.4289. 21954972 [DOI] [PubMed] [Google Scholar]
- 34.Muller F.L., Liu Y., Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. Journal of Biological Chemistry. 2004;279(47):49064–49073. doi: 10.1074/jbc.M407715200. 15317809 [DOI] [PubMed] [Google Scholar]
- 35.Treberg J.R., Quinlan C.L., Brand M.D. Evidence for two sites of superoxide production by mitochondrial NADH-ubiquinone oxidoreductase (complex I) Journal of Biological Chemistry. 2011;286(31):27103–27110. doi: 10.1074/jbc.M111.252502. 21659507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quinlan C.L., Treberg J.R., Perevoshchikova I.V., Orr A.L., Brand M.D. Native rates of superoxide production from multiple sites in isolated mitochondria measured using endogenous reporters. Free Radical Biology and Medicine. 2012;53(9):1807–1817. doi: 10.1016/j.freeradbiomed.2012.08.015. 22940066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quinlan C.L., Perevoshchikova I.V., Hey-Mogensen M., Orr A.L., Brand M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biology. 2013;1:304–312. doi: 10.1016/j.redox.2013.04.005. 24024165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eghbal M.A., Pennefather P.S., O’Brien P.J. H2S cytotoxicity mechanism involves reactive oxygen species formation and mitochondrial depolarisation. Toxicology. 2004;203(1–3):69–76. doi: 10.1016/j.tox.2004.05.020. 15363583 [DOI] [PubMed] [Google Scholar]
- 39.Quinlan C.L., Goncalves R.L., Hey-Mogensen M., Yadava N., Bunik V.I., Brand M.D. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. Journal of Biological Chemistry. 2014;289(12):8312–8325. doi: 10.1074/jbc.M113.545301. 24515115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fisher-Wellman K.H., Gilliam L.A., Lin C.T., Cathey B.L., Lark D.S., Neufer P.D. Mitochondrial glutathione depletion reveals a novel role for the pyruvate dehydrogenase complex as a key H2O2-emitting source under conditions of nutrient overload. Free Radical Biology and Medicine. 2013;65:1201–1208. doi: 10.1016/j.freeradbiomed.2013.09.008. 24056031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Siebels I., Dröse S. Q-site inhibitor induced ROS production of mitochondrial complex II is attenuated by TCA cycle dicarboxylates. Biochimica et Biophysica Acta. 2013;1827(10):1156–1164. doi: 10.1016/j.bbabio.2013.06.005. 23800966 [DOI] [PubMed] [Google Scholar]
- 42.Chouchani E.T., Pell V.R., Gaude E., Aksentijević D., Sundier S.Y., Robb E.L., Logan A., Nadtochiy S.M., Ord E.N., Smith A.C., Eyassu F., Shirley R., Hu C.H., Dare A.J., James A.M., Rogatti S., Hartley R.C., Eaton S., Costa A.S., Brookes P.S., Davidson S.M., Duchen M.R., Saeb-Parsy K., Shattock M.J., Robinson A.J., Work L.M., Frezza C., Krieg T., Murphy M.P. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909. 25383517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Orr A.L., Quinlan C.L., Perevoshchikova I.V., Brand M.D. A refined analysis of superoxide production by mitochondrial sn-glycerol 3-phosphate dehydrogenase. Journal of Biological Chemistry. 2012;287(51):42921–42935. doi: 10.1074/jbc.M112.397828. 23124204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perevoshchikova I.V., Quinlan C.L., Orr A.L., Gerencser A.A., Brand M.D. Sites of superoxide and hydrogen peroxide production during fatty acid oxidation in rat skeletal muscle mitochondria. Free Radical Biology and Medicine. 2013;61:298–309. doi: 10.1016/j.freeradbiomed.2013.04.006. 23583329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.D.G. Nicholls, S.J. Ferguson, Bio energetics (third ed.), 2002.
- 46.Klinman J.P. How do enzymes activate oxygen without inactivating themselves? Accounts of Chemical Research. 2007;40(5):325–333. doi: 10.1021/ar6000507. 17474709 [DOI] [PubMed] [Google Scholar]
- 47.Waypa G.B., Marks J.D., Guzy R.D., Mungai P.T., Schriewer J.M., Dokic D., Ball M.K., Schumacker P.T. Superoxide generated at mitochondrial complex III triggers acute responses to hypoxia in the pulmonary circulation. American Journal of Respiratory and Critical Care Medicine. 2013;187(4):424–432. doi: 10.1164/rccm.201207-1294OC. 23328522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tormos K.V., Anso E., Hamanaka R.B., Eisenbart J., Joseph J., Kalyanaraman B., Chandel N.S. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metabolism. 2011;14(4):537–544. doi: 10.1016/j.cmet.2011.08.007. 21982713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gaignard P., Menezes M., Schiff M., Bayot A., Rak M., Ogier de Baulny H., Su C.H., Gilleron M., Lombes A., Abida H., Tzagoloff A., Riley L., Cooper S.T., Mina K., Sivadorai P., Davis M.R., Allcock R.J., Kresoje N., Laing N.G., Thorburn D.R., Slama A., Christodoulou J., Rustin P. Mutations in CYC1, encoding cytochrome c1 subunit of respiratory chain complex III, cause insulin-responsive hyperglycemia. The American Journal of Human Genetics. 2013;93(2):384–389. doi: 10.1016/j.ajhg.2013.06.015. 23910460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tretter L., Adam-Vizi V. Inhibition of alpha-ketoglutarate dehydrogenase due to H2O2-induced oxidative stress in nerve terminals. Annals of the New York Academy of Sciences. 1999;893:412–416. doi: 10.1111/j.1749-6632.1999.tb07867.x. 10672279 [DOI] [PubMed] [Google Scholar]
- 51.Starkov A.A., Fiskum G., Chinopoulos C., Lorenzo B.J., Browne S.E., Patel M.S., Beal M.F. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. Journal of Neuroscience. 2004;24(36):7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. 15356189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bleier L., Wittig I., Heide H., Steger M., Brandt U., Dröse S. Generator-specific targets of mitochondrial reactive oxygen species. Free Radical Biology and Medicine. 2015;78:1–10. doi: 10.1016/j.freeradbiomed.2014.10.511. 25451644 [DOI] [PubMed] [Google Scholar]
- 53.Yu T., Robotham J.L., Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(8):2653–2658. doi: 10.1073/pnas.0511154103. 16477035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Winge D.R. Sealing the mitochondrial respirasome. Molecular and Cellular Biology. 2012;32(14):2647–2652. doi: 10.1128/MCB.00573-12. 22586278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nishikawa T., Edelstein D., Du X.L., Yamagishi S., Matsumura T., Kaneda Y., Yorek M.A., Beebe D., Oates P.J., Hammes H.P., Giardino I., Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404(6779):787–790. doi: 10.1038/35008121. 10783895 [DOI] [PubMed] [Google Scholar]
- 56.Massey V. Activation of molecular oxygen by flavins and flavoproteins. Journal of Biological Chemistry. 1994;269(36):22459–22462. 8077188 [PubMed] [Google Scholar]
- 57.Williamson J.R., Cooper R.H. Regulation of the citric acid cycle in mammalian systems. FEBS Letters. 1980;117(Suppl.):K73–K85. doi: 10.1016/0014-5793(80)80572-2. 6998729 [DOI] [PubMed] [Google Scholar]
- 58.Gibson G.E., Park L.C., Sheu K.F., Blass J.P., Calingasan N.Y. The alpha-ketoglutarate dehydrogenase complex in neurodegeneration. Neurochemistry International. 2000;36(2):97–112. doi: 10.1016/s0197-0186(99)00114-x. 10676873 [DOI] [PubMed] [Google Scholar]
- 59.Sun W., Liu Q., Leng J., Zheng Y., Li J. The role of pyruvate dehydrogenase complex in cardiovascular diseases. Life Sciences. 2015;121:97–103. doi: 10.1016/j.lfs.2014.11.030. [DOI] [PubMed] [Google Scholar]
- 60.Brown J.P., Perham R.N. Selective inactivation of the transacylase components of the 2-oxo acid dehydrogenase multienzyme complexes of Escherichia coli. Biochemical Journal. 1976;155(2):419–427. doi: 10.1042/bj1550419. 180985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qi F., Pradhan R.K., Dash R.K., Beard D.A. Detailed kinetics and regulation of mammalian 2-oxoglutarate dehydrogenase. BMC Biochemistry. 2011;12:53. doi: 10.1186/1471-2091-12-53. 21943256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McLain A.L., Szweda P.A., Szweda L.I. Alpha-ketoglutarate dehydrogenase: a mitochondrial redox sensor. Free Radical Research. 2011;45(1):29–36. doi: 10.3109/10715762.2010.534163. 21110783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tabatabaie T., Potts J.D., Floyd R.A. Reactive oxygen species-mediated inactivation of pyruvate dehydrogenase. Archives of Biochemistry and Biophysics. 1996;336(2):290–296. doi: 10.1006/abbi.1996.0560. 8954577 [DOI] [PubMed] [Google Scholar]
- 64.Applegate M.A., Humphries K.M., Szweda L.I. Reversible inhibition of alpha-ketoglutarate dehydrogenase by hydrogen peroxide: glutathionylation and protection of lipoic acid. Biochemistry. 2008;47(1):473–478. doi: 10.1021/bi7017464. 18081316 [DOI] [PubMed] [Google Scholar]
- 65.Ambrose-Griffin M.C., Danson M.J., Griffin W.G., Hale G., Perham R.N. Kinetic analysis of the role of lipoic acid residues in the pyruvate dehydrogenase multienzyme complex of Escherichia coli. Biochemical Journal. 1980;187(2):393–401. doi: 10.1042/bj1870393. 6772160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Samikkannu T., Chen C.H., Yih L.H., Wang A.S., Lin S.Y., Chen T.C., Jan K.Y. Reactive oxygen species are involved in arsenic trioxide inhibition of pyruvate dehydrogenase activity. Chemical Research in Toxicology. 2003;16(3):409–414. doi: 10.1021/tx025615j. 12641442 [DOI] [PubMed] [Google Scholar]
- 67.Crane D., Häussinger D., Graf P., Sies H. Decreased flux through pyruvate dehydrogenase by thiol oxidation during t-butyl hydroperoxide metabolism in perfused rat liver. Hoppe-Seyler's Zeitschrift fur Physiologische Chemie. 1983;364(8):977–987. doi: 10.1515/bchm2.1983.364.2.977. 6629333 [DOI] [PubMed] [Google Scholar]
- 68.Nulton-Persson A.C., Starke D.W., Mieyal J.J., Szweda L.I. Reversible inactivation of alpha-ketoglutarate dehydrogenase in response to alterations in the mitochondrial glutathione status. Biochemistry. 2003;42(14):4235–4242. doi: 10.1021/bi027370f. 12680778 [DOI] [PubMed] [Google Scholar]
- 69.Rokutan K., Kawai K., Asada K. Inactivation of 2-oxoglutarate dehydrogenase in rat liver mitochondria by its substrate and t-butyl hydroperoxide. Journal of Biochemistry. 1987;101(2):415–422. doi: 10.1093/oxfordjournals.jbchem.a121926. 3584093 [DOI] [PubMed] [Google Scholar]
- 70.Sims N.R., Anderson M.F., Hobbs L.M., Kong J.Y., Phillips S., Powell J.A., Zaidan E. Impairment of brain mitochondrial function by hydrogen peroxide. Brain Research. Molecular Brain Research. 2000;77(2):176–184. doi: 10.1016/s0169-328x(00)00049-8. 10837913 [DOI] [PubMed] [Google Scholar]
- 71.Enami S., Hoffmann M.R., Colussi A.J. Simultaneous detection of cysteine sulfenate, sulfinate, and sulfonate during cysteine interfacial ozonolysis. The Journal of Physical Chemistry B. 2009;113(28):9356–9358. doi: 10.1021/jp904316n. 19537744 [DOI] [PubMed] [Google Scholar]
- 72.McGrath A.J., Garrett G.E., Valgimigli L., Pratt D.A. The redox chemistry of sulfenic acids. Journal of the American Chemical Society. 2010;132(47):16759–16761. doi: 10.1021/ja1083046. 21049943 [DOI] [PubMed] [Google Scholar]
- 73.Humphries K.M., Yoo Y., Szweda L.I. Inhibition of NADH-linked mitochondrial respiration by 4-hydroxy-2-nonenal. Biochemistry. 1998;37(2):552–557. doi: 10.1021/bi971958i. 9425076 [DOI] [PubMed] [Google Scholar]
- 74.Humphries K.M., Szweda L.I. Selective inactivation of alpha-ketoglutarate dehydrogenase and pyruvate dehydrogenase: reaction of lipoic acid with 4-hydroxy-2-nonenal. Biochemistry. 1998;37(45):15835–15841. doi: 10.1021/bi981512h. 9843389 [DOI] [PubMed] [Google Scholar]
- 75.Lucas D.T., Szweda L.I. Declines in mitochondrial respiration during cardiac reperfusion: age-dependent inactivation of alpha-ketoglutarate dehydrogenase. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(12):6689–6693. doi: 10.1073/pnas.96.12.6689. 10359773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gallogly M.M., Starke D.W., Mieyal J.J. Mechanistic and kinetic details of catalysis of thiol-disulfide exchange by glutaredoxins and potential mechanisms of regulation. Antioxidants and Redox Signaling. 2009;11(5):1059–1081. doi: 10.1089/ars.2008.2291. 19119916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Grek C.L., Zhang J., Manevich Y., Townsend D.M., Tew K.D. Causes and consequences of cysteine S-glutathionylation. Journal of Biological Chemistry. 2013;288(37):26497–26504. doi: 10.1074/jbc.R113.461368. 23861399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McLain A.L., Cormier P.J., Kinter M., Szweda L.I. Glutathionylation of alpha-ketoglutarate dehydrogenase: the chemical nature and relative susceptibility of the cofactor lipoic acid to modification. Free Radical Biology and Medicine. 2013;61:161–169. doi: 10.1016/j.freeradbiomed.2013.03.020. 23567190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Berndt C., Lillig C.H., Flohé L. Redox regulation by glutathione needs enzymes. Frontiers in Pharmacology. 2014;5:168. doi: 10.3389/fphar.2014.00168. 25100998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jensen K.S., Pedersen J.T., Winther J.R., Teilum K. The pKa value and accessibility of cysteine residues are key determinants for protein substrate discrimination by glutaredoxin. Biochemistry. 2014;53(15):2533–2540. doi: 10.1021/bi4016633. 24673564 [DOI] [PubMed] [Google Scholar]
- 81.Chen Y.J., Lu C.T., Lee T.Y., Chen Y.J. dbGSH: a database of S-glutathionylation. BioInformatics. 2014;30(16):2386–2388. doi: 10.1093/bioinformatics/btu301. 24790154 [DOI] [PubMed] [Google Scholar]
- 82.Coles S.J., Easton P., Sharrod H., Hutson S.M., Hancock J., Patel V.B., Conway M.E. S-Nitrosoglutathione inactivation of the mitochondrial and cytosolic BCAT proteins: S-nitrosation and S-thiolation. Biochemistry. 2009;48(3):645–656. doi: 10.1021/bi801805h. 19119849 [DOI] [PubMed] [Google Scholar]
- 83.Gnaiger E. Capacity of oxidative phosphorylation in human skeletal muscle: new perspectives of mitochondrial physiology. The International Journal of Biochemistry & Cell Biology. 2009;41(10):1837–1845. doi: 10.1016/j.biocel.2009.03.013. 19467914 [DOI] [PubMed] [Google Scholar]
- 84.Hu C.A., Donald S.P., Yu J., Lin W.W., Liu Z., Steel G., Obie C., Valle D., Phang J.M. Overexpression of proline oxidase induces proline-dependent and mitochondria-mediated apoptosis. Molecular and Cellular Biochemistry. 2007;295(1–2):85–92. doi: 10.1007/s11010-006-9276-6. 16874462 [DOI] [PubMed] [Google Scholar]
- 85.Forman H.J., Kennedy J. Superoxide production and electron transport in mitochondrial oxidation of dihydroorotic acid. Journal of Biological Chemistry. 1975;250(11):4322–4326. 165196 [PubMed] [Google Scholar]
- 86.Mráček T., Holzerová E., Drahota Z., Kovářová N., Vrbacký M., Ješina P., Houštěk J. ROS generation and multiple forms of mammalian mitochondrial glycerol-3-phosphate dehydrogenase. Biochimica et Biophysica Acta. 2014;1837(1):98–111. doi: 10.1016/j.bbabio.2013.08.007. 23999537 [DOI] [PubMed] [Google Scholar]
- 87.Polyak K., Xia Y., Zweier J.L., Kinzler K.W., Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389(6648):300–305. doi: 10.1038/38525. 9305847 [DOI] [PubMed] [Google Scholar]
- 88.Sacktor B. Biochemical adaptations for flight in the insect. Biochemical Society Symposium. 1976:111–131. 788715 [PubMed] [Google Scholar]
- 89.Viola H.M., Hool L.C. Qo site of mitochondrial complex III is the source of increased superoxide after transient exposure to hydrogen peroxide. Journal of Molecular and Cellular Cardiology. 2010;49(5):875–885. doi: 10.1016/j.yjmcc.2010.07.015. 20688078 [DOI] [PubMed] [Google Scholar]
- 90.Bleier L., Dröse S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. Biochimica et Biophysica Acta. 2013;1827(11–12):1320–1331. doi: 10.1016/j.bbabio.2012.12.002. 23269318 [DOI] [PubMed] [Google Scholar]
- 91.Crofts A.R., Holland J.T., Victoria D., Kolling D.R., Dikanov S.A., Gilbreth R., Lhee S., Kuras R., Kuras M.G. The Q-cycle reviewed: how well does a monomeric mechanism of the bc(1) complex account for the function of a dimeric complex? Biochimica et Biophysica Acta. 2008;1777(7–8):1001–1019. doi: 10.1016/j.bbabio.2008.04.037. 18501698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sabharwal S.S., Schumacker P.T. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nature Reviews Cancer. 2014;14(11):709–721. doi: 10.1038/nrc3803. 25342630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Korshunov S.S., Skulachev V.P., Starkov A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Letters. 1997;416(1):15–18. doi: 10.1016/s0014-5793(97)01159-9. 9369223 [DOI] [PubMed] [Google Scholar]
- 94.Harper M.E., Green K., Brand M.D. The efficiency of cellular energy transduction and its implications for obesity. Annual Review of Nutrition. 2008;28:13–33. doi: 10.1146/annurev.nutr.28.061807.155357. 18407744 [DOI] [PubMed] [Google Scholar]
- 95.Divakaruni A.S., Brand M.D. The regulation and physiology of mitochondrial proton leak. Physiology (Bethesda) 2011;26(3):192–205. doi: 10.1152/physiol.00046.2010. 21670165 [DOI] [PubMed] [Google Scholar]
- 96.Mailloux R.J., Xuan J.Y., Beauchamp B., Jui L., Lou M., Harper M.E. Glutaredoxin-2 is required to control proton leak through uncoupling protein-3. Journal of Biological Chemistry. 2013;288(12):8365–8379. doi: 10.1074/jbc.M112.442905. 23335511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mailloux R.J., Fu A., Robson-Doucette C., Allister E.M., Wheeler M.B., Screaton R., Harper M.E. Glutathionylation state of uncoupling protein-2 and the control of glucose-stimulated insulin secretion. Journal of Biological Chemistry. 2012;287(47):39673–39685. doi: 10.1074/jbc.M112.393538. 23035124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mailloux R.J., Seifert E.L., Bouillaud F., Aguer C., Collins S., Harper M.E. Glutathionylation acts as a control switch for uncoupling proteins UCP2 and UCP3. Journal of Biological Chemistry. 2011;286(24):21865–21875. doi: 10.1074/jbc.M111.240242. 21515686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mailloux R.J., Dumouchel T., Aguer C., deKemp R., Beanlands R., Harper M.E. Hexokinase II acts through UCP3 to suppress mitochondrial reactive oxygen species production and maintain aerobic respiration. Biochemical Journal. 2011;437(2):301–311. doi: 10.1042/BJ20110571. 21554247 [DOI] [PubMed] [Google Scholar]
- 100.Anderson E.J., Yamazaki H., Neufer P.D. Induction of endogenous uncoupling protein 3 suppresses mitochondrial oxidant emission during fatty acid-supported respiration. Journal of Biological Chemistry. 2007;282(43):31257–31266. doi: 10.1074/jbc.M706129200. 17761668 [DOI] [PubMed] [Google Scholar]
- 101.Queiroga C.S., Almeida A.S., Martel C., Brenner C., Alves P.M., Vieira H.L. Glutathionylation of adenine nucleotide translocase induced by carbon monoxide prevents mitochondrial membrane permeabilization and apoptosis. Journal of Biological Chemistry. 2010;285(22):17077–17088. doi: 10.1074/jbc.M109.065052. 20348099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nadtochiy S.M., Baker P.R., Freeman B.A., Brookes P.S. Mitochondrial nitroalkene formation and mild uncoupling in ischaemic preconditioning: implications for cardioprotection. Cardiovascular Research. 2009;82(2):333–340. doi: 10.1093/cvr/cvn323. 19050010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Parker N., Vidal-Puig A., Brand M.D. Stimulation of mitochondrial proton conductance by hydroxynonenal requires a high membrane potential. Bioscience Reports. 2008;28(2):83–88. doi: 10.1042/BSR20080002. 18384278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dröse S., Brandt U., Wittig I. Mitochondrial respiratory chain complexes as sources and targets of thiol-based redox-regulation. Biochimica et Biophysica Acta. 2014;1844(8):1344–1354. doi: 10.1016/j.bbapap.2014.02.006. 24561273 [DOI] [PubMed] [Google Scholar]
- 105.Mailloux R.J., Xuan J.Y., McBride S., Maharsy W., Thorn S., Holterman C.E., Kennedy C.R., Rippstein P., deKemp R., da Silva J., Nemer M., Lou M., Harper M.E. Glutaredoxin-2 is required to control oxidative phosphorylation in cardiac muscle by mediating deglutathionylation reactions. Journal of Biological Chemistry. 2014;289(21):14812–14828. doi: 10.1074/jbc.M114.550574. 24727547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nagy N., Malik G., Tosaki A., Ho Y.S., Maulik N., Das D.K. Overexpression of glutaredoxin-2 reduces myocardial cell death by preventing both apoptosis and necrosis. Journal of Molecular and Cellular Cardiology. 2008;44(2):252–260. doi: 10.1016/j.yjmcc.2007.08.021. 18076901 [DOI] [PubMed] [Google Scholar]
- 107.Diotte N.M., Xiong Y., Gao J., Chua B.H., Ho Y.S. Attenuation of doxorubicin-induced cardiac injury by mitochondrial glutaredoxin 2. Biochimica et Biophysica Acta. 2009;1793(2):427–438. doi: 10.1016/j.bbamcr.2008.10.014. 19038292 [DOI] [PubMed] [Google Scholar]
- 108.Lee D.W., Kaur D., Chinta S.J., Rajagopalan S., Andersen J.K. A disruption in iron–sulfur center biogenesis via inhibition of mitochondrial dithiol glutaredoxin 2 may contribute to mitochondrial and cellular iron dysregulation in mammalian glutathione-depleted dopaminergic cells: implications for Parkinson’s disease. Antioxidants and Redox Signaling. 2009;11(9):2083–2094. doi: 10.1089/ars.2009.2489. 19290777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chouchani E.T., Methner C., Nadtochiy S.M., Logan A., Pell V.R., Ding S., James A.M., Cochemé H.M., Reinhold J., Lilley K.S., Partridge L., Fearnley I.M., Robinson A.J., Hartley R.C., Smith R.A., Krieg T., Brookes P.S., Murphy M.P. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nature Medicine. 2013;19(6):753–759. doi: 10.1038/nm.3212. 23708290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rosca M.G., Hoppel C.L. New aspects of impaired mitochondrial function in heart failure. Journal of Bioenergetics and Biomembranes. 2009;41(2):107–112. doi: 10.1007/s10863-009-9215-9. 19347572 [DOI] [PubMed] [Google Scholar]
- 111.Lemieux H., Hoppel C.L. Mitochondria in the human heart. Journal of Bioenergetics and Biomembranes. 2009;41(2):99–106. doi: 10.1007/s10863-009-9211-0. 19353253 [DOI] [PubMed] [Google Scholar]
- 112.Bayeva M., Sawicki K.T., Ardehali H. Taking diabetes to heart − deregulation of myocardial lipid metabolism in diabetic cardiomyopathy. Journal of the American Heart Association. 2013;2(6):e000433. doi: 10.1161/JAHA.113.000433. 24275630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ishii T., Miyazawa M., Onouchi H., Yasuda K., Hartman P.S., Ishii N. Model animals for the study of oxidative stress from complex II. Biochimica et Biophysica Acta. 2013;1827(5):588–597. doi: 10.1016/j.bbabio.2012.10.016. 23142169 [DOI] [PubMed] [Google Scholar]
- 114.Chen Y.R., Chen C.L., Pfeiffer D.R., Zweier J.L. Mitochondrial complex II in the post-ischemic heart: oxidative injury and the role of protein S-glutathionylation. Journal of Biological Chemistry. 2007;282(45):32640–32654. doi: 10.1074/jbc.M702294200. 17848555 [DOI] [PubMed] [Google Scholar]
- 115.Deen P.M., Robben J.H. Succinate receptors in the kidney. Journal of the American Society of Nephrology. 2011;22(8):1416–1422. doi: 10.1681/ASN.2010050481. 21803970 [DOI] [PubMed] [Google Scholar]
- 116.James A.M., Collins Y., Logan A., Murphy M.P. Mitochondrial oxidative stress and the metabolic syndrome. Trends in Endocrinology and Metabolism. 2012;23(9):429–434. doi: 10.1016/j.tem.2012.06.008. 22831852 [DOI] [PubMed] [Google Scholar]
- 117.Turrens J.F. Mitochondrial formation of reactive oxygen species. Journal of Physiology. 2003;552(2):335–344. doi: 10.1113/jphysiol.2003.049478. 14561818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kalyanaraman B. Oxidative chemistry of fluorescent dyes: implications in the detection of reactive oxygen and nitrogen species. Biochemical Society Transactions. 2011;39(5):1221–1225. doi: 10.1042/BST0391221. 21936793 [DOI] [PubMed] [Google Scholar]
- 119.Ross M.F., Kelso G.F., Blaikie F.H., James A.M., Cochemé H.M., Filipovska A., Da Ros T., Hurd T.R., Smith R.A., Murphy M.P. Lipophilic triphenylphosphonium cations as tools in mitochondrial bioenergetics and free radical biology. Biochemistry Moscow. 2005;70(2):222–230. doi: 10.1007/s10541-005-0104-5. 15807662 [DOI] [PubMed] [Google Scholar]
- 120.Zielonka J., Kalyanaraman B. Hydroethidine- and MitoSOX-derived red fluorescence is not a reliable indicator of intracellular superoxide formation: another inconvenient truth. Free Radical Biology and Medicine. 2010;48(8):983–1001. doi: 10.1016/j.freeradbiomed.2010.01.028. 20116425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Robinson K.M., Janes M.S., Pehar M., Monette J.S., Ross M.F., Hagen T.M., Murphy M.P., Beckman J.S. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(41):15038–15043. doi: 10.1073/pnas.0601945103. 17015830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Reynafarje B., Costa L.E., Lehninger A.L. O2 solubility in aqueous media determined by a kinetic method. Analytical Biochemistry. 1985;145(2):406–418. doi: 10.1016/0003-2697(85)90381-1. 4014672 [DOI] [PubMed] [Google Scholar]
- 123.Henderson J.R., Swalwell H., Boulton S., Manning P., McNeil C.J., Birch-Machin M.A. Direct, real-time monitoring of superoxide generation in isolated mitochondria. Free Radical Research. 2009;43(9):796–802. doi: 10.1080/10715760903062895. 19562601 [DOI] [PubMed] [Google Scholar]
- 124.Boulton S.J., Birch-Machin M.A. Impact of hyperpigmentation on superoxide flux and melanoma cell metabolism at mitochondrial complex II. FASEB Journal. 2015;29(1):346–353. doi: 10.1096/fj.14-261982. 25351989 [DOI] [PubMed] [Google Scholar]
- 125.Maeda H., Yamamoto K., Kohno I., Hafsi L., Itoh N., Nakagawa S., Kanagawa N., Suzuki K., Uno T. Design of a practical fluorescent probe for superoxide based on protection-deprotection chemistry of fluoresceins with benzenesulfonyl protecting groups. Chemistry. 2007;13(7):1946–1954. doi: 10.1002/chem.200600522. 17136791 [DOI] [PubMed] [Google Scholar]
- 126.Wang W., Fang H., Groom L., Cheng A., Zhang W., Liu J., Wang X., Li K., Han P., Zheng M., Yin J., Wang W., Mattson M.P., Kao J.P., Lakatta E.G., Sheu S.S., Ouyang K., Chen J., Dirksen R.T., Cheng H. Superoxide flashes in single mitochondria. Cell. 2008;134(2):279–290. doi: 10.1016/j.cell.2008.06.017. 18662543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sheu S.S., Wang W., Cheng H., Dirksen R.T. Superoxide flashes: illuminating new insights into cardiac ischemia/reperfusion injury. Future Cardiology. 2008;4(6):551–554. doi: 10.2217/14796678.4.6.551. 19649173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fang H., Chen M., Ding Y., Shang W., Xu J., Zhang X., Zhang W., Li K., Xiao Y., Gao F., Shang S., Li J.C., Tian X.L., Wang S.Q., Zhou J., Weisleder N., Ma J., Ouyang K., Chen J., Wang X., Zheng M., Wang W., Cheng H. Imaging superoxide flash and metabolism-coupled mitochondrial permeability transition in living animals. Cell Research. 2011;21(9):1295–1304. doi: 10.1038/cr.2011.81. 21556035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Pouvreau S. Superoxide flashes in mouse skeletal muscle are produced by discrete arrays of active mitochondria operating coherently. PLoS One. 2010;5(9) doi: 10.1371/journal.pone.0013035. 20927399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Schwarzländer M., Murphy M.P., Duchen M.R., Logan D.C., Fricker M.D., Halestrap A.P., Müller F.L., Rizzuto R., Dick T.P., Meyer A.J., Sweetlove L.J. Mitochondrial ‘flashes’: a radical concept repHined. Trends in Cell Biology. 2012;22(10):503–508. doi: 10.1016/j.tcb.2012.07.007. 22917552 [DOI] [PubMed] [Google Scholar]
- 131.Imlay J.A. Pathways of oxidative damage. Annual Review of Microbiology. 2003;57:395–418. doi: 10.1146/annurev.micro.57.030502.090938. 14527285 [DOI] [PubMed] [Google Scholar]
- 132.Schwarzländer M., Wagner S., Ermakova Y.G., Belousov V.V., Radi R., Beckman J.S., Buettner G.R., Demaurex N., Duchen M.R., Forman H.J., Fricker M.D., Gems D., Halestrap A.P., Halliwell B., Jakob U., Johnston I.G., Jones N.S., Logan D.C., Morgan B., Müller F.L., Nicholls D.G., Remington S.J., Schumacker P.T., Winterbourn C.C., Sweetlove L.J., Meyer A.J., Dick T.P., Murphy M.P. The ‘mitoflash’ probe cpYFP does not respond to superoxide. Nature. 2014;514(7523):E12–E14. doi: 10.1038/nature13858. 25341790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Schwarzländer M., Logan D.C., Fricker M.D., Sweetlove L.J. The circularly permuted yellow fluorescent protein cpYFP that has been used as a superoxide probe is highly responsive to pH but not superoxide in mitochondria: implications for the existence of superoxide ‘flashes’. Biochemical Journal. 2011;437(3):381–387. doi: 10.1042/BJ20110883. 21631430 [DOI] [PubMed] [Google Scholar]
- 134.Finkel T. From sulfenylation to sulfhydration: what a thiolate needs to tolerate. Science Signalling. 2012;5(215):e10. doi: 10.1126/scisignal.2002943. 22416275 [DOI] [PubMed] [Google Scholar]
- 135.Starkov A.A. Measurement of mitochondrial ROS production. Methods in Molecular Biology. 2010;648:245–255. doi: 10.1007/978-1-60761-756-3_16. 20700717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cochemé H.M., Quin C., McQuaker S.J., Cabreiro F., Logan A., Prime T.A., Abakumova I., Patel J.V., Fearnley I.M., James A.M., Porteous C.M., Smith R.A., Saeed S., Carré J.E., Singer M., Gems D., Hartley R.C., Partridge L., Murphy M.P. Measurement of H2O2 within living drosophila during aging using a ratiometric mass spectrometry probe targeted to the mitochondrial matrix. Cell Metabolism. 2011;13(3):340–350. doi: 10.1016/j.cmet.2011.02.003. 21356523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cochemé H.M., Logan A., Prime T.A., Abakumova I., Quin C., McQuaker S.J., Patel J.V., Fearnley I.M., James A.M., Porteous C.M., Smith R.A., Hartley R.C., Partridge L., Murphy M.P. Using the mitochondria-targeted ratiometric mass spectrometry probe MitoB to measure H2O2 in living drosophila. Nature Protocol. 2012;7(5):946–958. doi: 10.1038/nprot.2012.035. 22517261 [DOI] [PubMed] [Google Scholar]
- 138.Logan A., Shabalina I.G., Prime T.A., Rogatti S., Kalinovich A.V., Hartley R.C., Budd R.C., Cannon B., Murphy M.P. In vivo levels of mitochondrial hydrogen peroxide increase with age in mtDNA mutator mice. Aging Cell. 2014;13(4):765–768. doi: 10.1111/acel.12212. 24621297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Reily C., Mitchell T., Chacko B.K., Benavides G., Murphy M.P., Darley-Usmar V. Mitochondrially targeted compounds and their impact on cellular bioenergetics. Redox Biology. 2013;1(1):86–93. doi: 10.1016/j.redox.2012.11.009. 23667828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Belousov V.V., Fradkov A.F., Lukyanov K.A., Staroverov D.B., Shakhbazov K.S., Terskikh A.V., Lukyanov S. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nature Methods. 2006;3(4):281–286. doi: 10.1038/nmeth866. 16554833 [DOI] [PubMed] [Google Scholar]
- 141.Albrecht S.C., Barata A.G., Grosshans J., Teleman A.A., Dick T.P. In vivo mapping of hydrogen peroxide and oxidized glutathione reveals chemical and regional specificity of redox homeostasis. Cell Metabolism. 2011;14(6):819–829. doi: 10.1016/j.cmet.2011.10.010. 22100409 [DOI] [PubMed] [Google Scholar]
- 142.Claiborne A., Miller H., Parsonage D., Ross R.P. Protein-sulfenic acid stabilization and function in enzyme catalysis and gene regulation. FASEB Journal. 1993;7(15):1483–1490. doi: 10.1096/fasebj.7.15.8262333. 8262333 [DOI] [PubMed] [Google Scholar]
- 143.Aslund F., Zheng M., Beckwith J., Storz G. Regulation of the OxyR transcription factor by hydrogen peroxide and the cellular thiol-disulfide status. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(11):6161–6165. doi: 10.1073/pnas.96.11.6161. 10339558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Malinouski M., Zhou Y., Belousov V.V., Hatfield D.L., Gladyshev V.N. Hydrogen peroxide probes directed to different cellular compartments. PLoS One. 2011;6(1):e14564. doi: 10.1371/journal.pone.0014564. 21283738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ermakova Y.G., Bilan D.S., Matlashov M.E., Mishina N.M., Markvicheva K.N., Subach O.M., Subach F.V., Bogeski I., Hoth M., Enikolopov G., Belousov V.V. Red fluorescent genetically encoded indicator for intracellular hydrogen peroxide. Nature Communications. 2014;5:5222. doi: 10.1038/ncomms6222. 25330925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Roma L.P., Duprez J., Takahashi H.K., Gilon P., Wiederkehr A., Jonas J.C. Dynamic measurements of mitochondrial hydrogen peroxide concentration and glutathione redox state in rat pancreatic beta-cells using ratiometric fluorescent proteins: confounding effects of pH with HyPer but not roGFP1. Biochemical Journal. 2012;441(3):971–978. doi: 10.1042/BJ20111770. 22050124 [DOI] [PubMed] [Google Scholar]
- 147.Morgan B., Sobotta M.C., Dick T.P. Measuring E(GSH) and H2O2 with roGFP2-based redox probes. Free Radical Biology and Medicine. 2011;51(11):1943–1951. doi: 10.1016/j.freeradbiomed.2011.08.035. 21964034 [DOI] [PubMed] [Google Scholar]