

Abstract
Enhanced mitochondrial generation of oxidants, including hydrogen peroxide (H2O2), is related to a large number of pathological conditions, including diet-induced obesity and steatohepatosis. Indeed, we have previously shown that high fat diets increase the generation of H2O2 in liver mitochondria energized by activated fatty acids. Here, we further study fatty-acid induced H2O2 release in liver mitochondria, and determine the characteristics that regulate it. We find that this production of H2O2 is independent of mitochondrial inner membrane integrity and insensitive to purine nucleotides. On the other hand, palmitate-induced H2O2 production is strongly enhanced by high fat diets and is pH-sensitive, with a peak at a matrix pH of ~8.5. Using recombinantly expressed human very long chain acyl-CoA dehydrogenase, we are able to demonstrate that palmitate-induced H2O2 release may be ascribed to the activity of this enzyme alone, acting as an oxidase. Our results add to a number of findings indicating that sources outside of the electron transport chain can generate significant, physiopathologically relevant, amounts of oxidants in mitochondria.
Keywords: Mitochondria, Hydrogen peroxide, Fatty acid oxidation, Acyl-CoA
Graphical abstract
Highlights
-
●
High fat diets increase H2O2 in palmitoyl CoA-energized liver mitochondria.
-
●
H2O2 release is pH-sensitive, with a peak at a matrix pH of ~8.5.
-
●
Recombinantly expressed purified human VLCAD releases H2O2, acting as an oxidase.
-
●
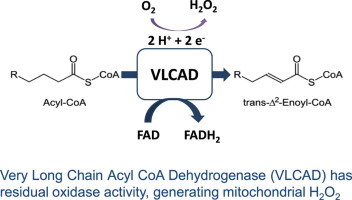
VLCAD presents residual oxidase activity, generating significant H2O2 in mitochondria.
Introduction
Oxidants, including superoxide radicals and hydrogen peroxide, are continuously produced intracellularly as byproducts of energetic metabolism [1–3]. These oxidants have been increasingly recognized as participants in both signaling and damaging processes [4–7]. Indeed, dietary habits and pathological conditions are closely associated with changes in oxidant production rates, types of oxidants generated and their levels within different tissues, resulting in changes in oxidative modifications to biomolecules that may lead to cell damage and disease [7–11].
Mitochondria are recognized as the most quantitatively relevant source of oxidants in most cells, and are particularly important when considering oxidants produced as a result of energetic metabolism [7,12–14]. Mitochondrial oxidant production and release is highly variable depending on the tissue, age, diet and antioxidant status and is regulated by respiratory state and substrates, among other characteristics [7,12–14]. This production of mitochondrial oxidants is most often ascribed to the electron transport chain, in which monoelectronic reduction of oxygen may occur at the levels of complexes I, II and III, generating superoxide radicals and other oxidants derived from it. However, more recent work has demonstrated that other sources of mitochondrial oxidants not only exist, but are quantitatively sizable. These alternative mitochondrial oxidant sources include the monoaminoxidase, α-ketoglutarate dehydrogenase and glycerol phosphate dehydrogenase [7,13,15].
Recently, using a diet-induced steatosis model, we demonstrated that high fat ingestion strongly increases liver mitochondrial H2O2 release supported by activated fatty acids [16]. Experiments modulating electron transport chain activity suggested that the production of H2O2 did not involve the generation of superoxide radicals at the level of the respiratory chain, but rather occurred in a manner dependent only on fatty-acid oxidation pathways. Based on these results, and the comparison of the effects of different sizes of fatty acids, we suggested that the production of H2O2 observed stemmed from flavoproteins involved in the metabolism of very long chain acyl-CoAs. Here, we study the regulation of fatty-acid-supported H2O2 release from mitochondria and provide direct evidence that the very long chain acyl-CoA dehydrogenase, a component of the β-oxidation pathway, has a residual oxidase activity, generating H2O2 within mitochondria.
Materials and methods
Animals and diet
Experiments involving isolated mitochondria were performed with samples prepared from 6 weeks old female Swiss mice. During 1 week prior to the experiment, control and high fat diet (HFD) mice were fed with standard diets, but the water source of the HFD group was supplemented with soy oil (30% v/v) in emulsion with 9 g L−1 sodium stearoyl lactylate [16]. All experiments were approved by the local animal care and use committee (Comissão de Ética no Uso de Animais) which is overseen by the National committee on research animal use (Conselho Nacional de Controle de Experimentação Animal – CONCEA) and has standards compatible with the NIH Guide for the Care and Use of Laboratory Animals.
Mitochondrial isolation
Mice were sacrificed by cervical dislocation and the livers were placed in cold isolation buffer containing 300 mM sucrose, 1 mM EGTA, 10 mM HEPES and 0.1% BSA, pH 7.2. The tissue was minced with surgical scissors and then extracts were obtained using a potter homogenizer. The extract was centrifuged at 4 °C for 5 min at 600g. The supernatant was then centrifuged at 4 °C for 5 min at 12,000g. The remaining pellet, containing mitochondria, was resuspended in the same buffer [17]. To promote membrane permeabilization, samples were frozen at −20 °C and then thawed just prior to the experiments.
Hydrogen peroxide generation
H2O2 generation was monitored fluorimetrically by following the oxidation of Amplex Red to resorufin, at 37 °C, ex=563 nm and em=595 nm. Mitochondria permeabilized by freezing and thawing (0.2 mg mL−1) were incubated in experimental buffer (150 mM KCl, 2 mM MgCl2, 2 mM EGTA, 2 mM KH2PO4 and 10 mM HEPES, pH 7.4) in presence of 5 µM Amplex Red, 1 U mL−1 horseradish peroxidase (HRP), 50 µM palmitoyl-CoA and varying concentrations of AMP, ADP, ATP, GDP and GTP [16]. For the pH assay, mitochondria were incubated in 100 mM KCl, 2 mM EGTA, 2 mM MgCl2, 50 µM palmitoyl-CoA, 2 mM KH2PO4, 10 mM acetate, 10 mM MES, 10 mM HEPES, 10 mM tricine, 10 mM TRIS, and 10 mM glycine, and the pH was adjusted from 4.5 to 10 using K+ and Cl− salts. Purified VLCAD was incubated in phosphate buffer (100 mM KH2PO4, 0.1 mM EDTA, pH 7.2) at 37 °C for 45 min in presence of Amplex Red, HRP (as described above), and varying concentrations of palmitoil-CoA (0–400 µM). Calibration curves were constructed using known H2O2 concentrations for each pH in which measurements were made.
VLCAD (ΔEx3) expression in Escherichia coli and purification
The plasmid for the expression of human VLCAD was kindly donated by professors Jung-Ja Kim (Medical College of Wisconsin, Milwaukee, WI) and Jerry Vockley (University of Pittsburgh, Children's Hospital of Pittsburgh, Pittsburgh, PA). It consists of the human VLCAD sequence devoid of exon 3 by alternative splicing of the N-terminal region [VLCAD (ΔEx3)] inserted into a pET21a plasmid. pET21A-VLCAD (ΔEx3) was inserted by heat shock into competent XL1Blue E. coli (obtained by the CaCl2 method) and grown overnight at 37 °C. The expanded construct was then purified with the Promega Wizard® Plus SV Minipreps DNA Purification System and eluted in nuclease-free water. The VLCAD (ΔEx3) sequence was amplified by PCR, separated in an agarose gel and purified with the Promega Wizard® PCR Gel and PCR Clean Up kit. The purified PCR product was inserted in the pLATE51 plasmid using an aLICator Thermo Scientific® kit, which adds a 6× histidine tail at the N-terminal region. pLATE51-VLCAD (ΔEx3) was then inserted by heat shock into competent Nova Blue E. coli (also obtained by the CaCl2 method) and a pre-inoculum was prepared in Luria-Bertani (LB) medium containing 50 µg mL−1 ampicillin and grown overnight at 37 °C. Two hundred microliters were inoculated in fresh LB (200 mL) containing ampicillin (50 µg mL−1) and grown to an OD600 of 0.6–0.8. Isopropyl β-d-1-thiogalactopyranoside (IPTG, 0.4 mM) was added and the culture was grown overnight at 20 °C. The culture was centrifuged (14,000g, 10 min) and the pellet was resuspended in lysis buffer (200 mM KH2PO4, 200 mM NaCl, 20 mM imidazole, 10% glycerol, pH 7.8) and sonicated (3 times, 15 s) over ice. The sample was centrifuged (14,000g, 10 min) and 200 µL of Ni-NTA Agarose resin were added, followed by 15 min incubation with agitation. The resin was washed five times with phosphate buffer (200 mM KH2PO4, 200 mM NaCl, 20 mM imidazole, pH 7.8) and VLCAD (ΔEx3) was eluted in phosphate buffer containing a higher concentration of imidazole (200 mM KH2PO4, 200 mM NaCl, 500 mM imidazole, pH 7.8) initial.
Western blotting
One milliliter of the culture was centrifuged (14,000g, 5 min). The pellets (induced or not) were resuspended in 200 and 100 µL, respectively, of sample buffer (4% glycerol, 10 mM dithiothreitol, 2% SDS, 0.05% bromophenol blue, 40 mM Tris–HCl, pH 6.8) and boiled for 5 min. Ten microliters of the purified enzyme were diluted in sample buffer and boiled for 5 min. Ten microliters of resuspended culture in sample buffer were then added to each well of an SDS-PAGE gel. After 1 h of separation at 200 V, the proteins were transferred to a membrane which was later developed using primary Santa Cruz® anti-VLCAD overnight and secondary anti-rabbit for 1 h. A second membrane was developed using primary Qiagen® HRP conjugated anti-PentaHis overnight. Both systems were visualized directly on the membrane with the BioRad® Opti-4CN Substrate kit.
VLCAD enzymatic activity
The purified enzyme (0.7±0.3 µg) was incubated in phosphate buffer (100 mM KH2PO4 and 0.1 mM EDTA, pH 7.2) at 37 °C for 45 min in presence of 150 µM ferricenium hexafluorophosphate and palmitoyl-CoA at varying concentrations (0–1400 µM). For pH experiments, VLCAD and palmitoyl-CoA (200 µM) was incubated in buffer containing 2 mM KH2PO4, 10 mM acetate, 10 mM MES, 10 mM HEPES, 10 mM tricine, 10 mM TRIS, 10 mM glycine and 2 mM EGTA, and the pH was adjusted from 4.5 to 10. The decrease in absorbance of ferricenium due its reduction by VLCAD-linked FADH2 was accompanied at 300 nm. The molar extinction coefficient used was 4.3 mM cm−1 [18].
Protein quantification
Experiments were normalized by protein concentration using the Bio-Rad Bradford reagent protein assay and bovine serum albumin as standard. The absorbance was measured at 595 nm.
Statistics
Data analysis was conducted using OriginLab and GraphPad Prism software using T tests and ANOVA. Data shown are averages±standard deviations of 3–6 experiments.
Results
In a prior publication [16], we demonstrated that a short-term (1 week) high fat diet resulted in largely increased H2O2 release in liver mitochondria energized by palmitoyl-CoA (palm-CoA), but not other substrates such as pyruvate or succinate. These results suggest that H2O2 was produced as a result of fatty acid oxidation, and not at the level of the mitochondrial electron transport chain. Indeed, as observed in Fig. 1A, we see a consistently increased mitochondrial H2O2 production in the presence of palm-CoA in liver mitochondria from animals fed a high fat diet (full bars) relative to animals on standard laboratory chow alone (open bars). These experiments were conducted in mitochondrial samples in which membrane integrity was disrupted by freeze–thawing, thus avoiding the accumulation of β-oxidation and reducing intermediates, again suggesting that H2O2 was being produced by reactions directly coupled to palm-CoA oxidation.
Fig. 1.
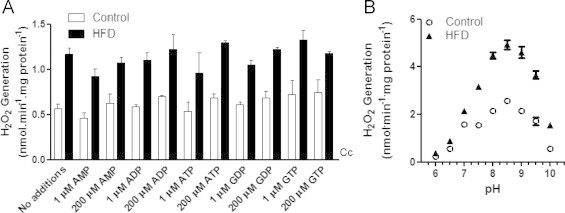
Palm-CoA-induced H2O2 release is modulated by diet and pH, but not purine nucleotides. H2O2 release from freeze-permeabilized mitochondria was measured in using 5 µM Amplex Red and 1 U mL−1 HRP. Panel A: H2O2 release in the presence of varying AMP, ADP, ATP, GDP or GTP concentrations, as indicated; n=4. Panel B: H2O2 release was measured at varying pH, as indicated. The detection was calibrated separately with known H2O2 concentrations for each pH. n=6 per group.
Given the large increase in oxidant release observed, we sought to uncover potential regulatory stimuli for high fat diet-stimulated, palm-CoA-induced, H2O2 release in mitochondria. Interestingly, we found out that, while high fat diet samples (Fig. 1A, full bars) consistently generated higher H2O2 levels, classical metabolic regulators AMP, ADP, ATP, GDP and GTP had no significant influence on this effect. On the other hand, palm-CoA-induced mitochondrial H2O2 release was strongly pH sensitive (Fig. 1B) in both control (open symbols) and high fat diet (closed symbols) samples. The peak of H2O2 release was observed at a pH of ~8.5, which is close to the physiological mitochondrial matrix pH (~7.8 [19,20]), and suggests this H2O2 production may be sensitive to fluctuations in ATP synthesis, which is accompanied by mitochondrial matrix acidification.
H2O2 generation promoted by palm-CoA but independent of electron transport chain activity may occur at multiple points of mitochondrial fatty acid oxidation pathways. Most probable sources of oxidants are flavoenzymes, many of which have been previously demonstrated to be significant mitochondrial sources of oxidants [7,13,15]. Indeed, we find that the short-term high fat diet used significantly increases the expression of many liver mitochondrial flavoenzymes involved in fatty acid oxidation, including various isoforms of acyl-CoA dehydrogenases and electron transferring flavoproteins [16]. Experiments using different sizes of activated fatty acids as substrates suggested that the probable source of H2O2 under these conditions was the very long chain acyl CoA dehydrogenase (VLCAD).
Since the mitochondrial microenvironment is highly rich in electron-transferring enzymes, in order to directly verify if VLCAD can be, in itself, a source of mitochondrial oxidants, we decided to express the recombinant enzyme and study its redox activity (Fig. 2). Human VLCAD [21] with a His tag was successfully expressed in E. coli, and could be detected with a molecular mass of ~74 kDa after induction using both anti-VLCAD (Fig. 2A, upper blot) or anti-His (Fig. 2A, lower blot). Purity was ensured by using an affinity column. The enzyme was also fully active, and presented a saturable, palm-CoA-dependent reducing activity (Fig. 2B and C). Furthermore, the activity of the enzyme was pH dependent, with a peak at ~7.9 (Fig. 2D).
Fig. 2.
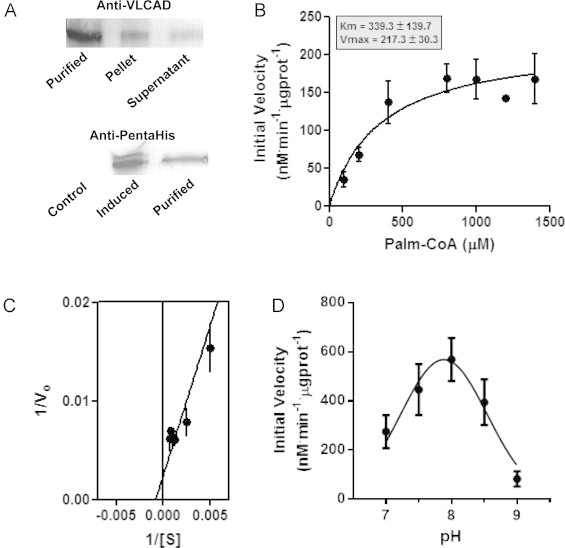
Expression and purification of active VLCAD (ΔEx3). Panel A: Western blot of Nova blue E. coli. The upper blot was developed using Santa Cruz® anti-VLCAD; the lower blot with Qiagen® anti-PentaHis. “Purified” refers to the eluted enzyme in phosphate buffer (pH 7.8), and “induced” refers to cultures incubated with 0.4 mM IPTG. Panels B and C: the activity of the purified recombinant VLCAD (ΔEx3) was measured as described in [18] in phosphate buffer, pH 7.2, at 37 °C, for 30 min, in presence of 150 µM hexafluorophosphate ferricenium and varying concentrations of palm-CoA (100–1500 µM). Panel D: The activity of the purified recombinant VLCAD (ΔEx3) was measured as described in [18] in the buffer described in Section Materials and methods adjusted to the range of pH shown in presence of 150 µM hexafluorophosphate ferricenium and 200 µM palm-CoA.
Interestingly, the purified recombinant VLCAD generated detectable levels of H2O2 (Fig. 3A), in a manner dependent on the concentration of palm-CoA (Fig. 3B). Because these experiments were conducted in the absence of superoxide dismutase, the presence of detectable H2O2 release indicates that this oxidant is produced directly by VLCAD, acting as an oxidase. Comparisons between the dehydrogenase activity (Fig. 2) and oxidase activity (Fig. 3) indicate that between 3.5% and 5.7% of electrons derived from palm-CoA by VLCAD are destined to the oxidase activity.
Fig. 3.
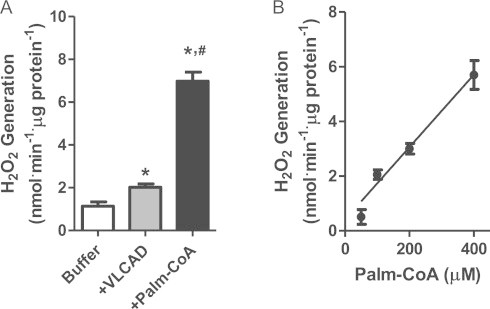
H2O2 generation by purified VLCAD (ΔEx3). Panel A: H2O2 was detected using 5 µM Amplex Red and 1 U mL−1 HRP in phosphate buffer, pH 7.2, containing purified recombinant VLCAD in the presence of 400 µM palm-CoA, at 37 °C, for 30 min; ⁎p<0.001 relative to buffer; #p<0.001 relative to VLCAD. Panel B: H2O2 release measured under the conditions of Panel A in the presence of varying palm-CoA concentrations (0–400 µM).
Discussion
Mitochondrially-originated oxidants, including H2O2, are implicated in the pathology and progression of a variety of conditions, including metabolic diseases. Specifically in the liver, mitochondrially-generated oxidants may contribute to the development of both non-alcoholic and alcoholic steatohepatosis, cytolytic hepatitis, cirrhosis and hepatocarcinoma [22,23]. Indeed, we have previously found that non-alcoholic steatohepatosis induced by a short-term high fat diet leads to increases in tissue oxidative damage markers associated with increased mitochondrial H2O2 release [16]. Since these increased H2O2 levels were only observed in the presence of palm-CoA, were not dependent on mitochondrial respiration and were inhibited by increasing concentrations of NAD+, we hypothesized that the production of this oxidant occurred upstream of the respiratory chain, most probably at the level of VLCAD.
Here, by using recombinant, purified, human VLCAD, we provide unequivocal evidence that this enzyme can, indeed, generate H2O2 directly, in a manner stimulated by its substrate and electron donor, palm-CoA. Since H2O2 could be detected in the absence of added superoxide dismutase, these data also show that VLCAD generates H2O2, and not superoxide radicals, primarily. VLCAD is thus acting as an acyl-CoA oxidase. The oxidase activity of VLCAD is partial, comprising less than 6% of its maximal activity under our experimental conditions, an expected result since the primary metabolic function of this enzyme within β-oxidation is to act as a dehydrogenase. However, although partial, this oxidase activity is continuous and thus expected to have quite a sizable overall contribution, particularly under conditions, such as high fat diets, in which the expression of VLCAD increases significantly [16].
VLCAD thus joins a growing family of mitochondrial enzymes capable of generating detectable quantities of oxidants under biologically relevant conditions, including electron transfer chain components (Complexes I, II and III), pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, glycerol phosphate dehydrogenase, electron transfer flavoprotein (ETF) and ETF-oxidoreductase and monoamine oxidase [1,7,10,12–14]. Overall, our data and that from other laboratories indicate that mitochondrial sources of oxidants are numerous, not restricted to the electron transport chain and present variable regulatory characteristics. In particular, mitochondrial flavoenzymes have been increasingly recognized as intracellular sources of oxidants. Many of these flavoenzymes (including pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, glycerol phosphate, ETF and ETF-oxidoreductase) appear to generate superoxide radicals as the primary oxidant. Superoxide radicals are then efficiently converted to H2O2 due to the abundance of superoxide dismutase in mitochondria [7]. On the other hand, we find that VLCAD, similarly to the monoamine oxidase [1,24,25], generates H2O2 directly.
The partial oxidase activity of VLCAD measured here may be justified by its structural characteristics. All mitochondrial acyl-CoA dehydrogenases present structural similarities to peroxisomal acyl-CoA oxidases [26], but are able to exclude oxygen from the catalytic site, thus maintaining a primary dehydrogenase activity. VLCAD, predictably, has the largest hydrophobic substrate binging cavity of the acyl-CoA dehydrogenases [27], a characteristic which may facilitate a partial oxidase activity. Indeed, when structural characteristics are compared side-by-side, the main difference between oxidases and their corresponding dehydrogenases is the shielding of the FAD from the solvent in dehydrogenases [28]. This shielding is expected to be more difficult to achieve in VLCAD due to the size of the substrate side chain, thus making this enzyme the acyl-CoA dehydrogenase most prone to act as a partial oxidase.
Overall, our data show that VLCAD is a sizable source of H2O2 in mitochondria, capable of generating this oxidant in the nmol min−1 mg−1 protein range, in a manner sensitive to matrix pH (therefore stimulated by conditions of low oxidative phosphorylation) and very significantly stimulated by high fat diets. The understanding of the mechanisms leading to the production of this oxidant are particularly important given the prevalence of high fat diets in modern society and the large impact on liver mitochondrial H2O2 release, even when these diets are adopted short-term.
Acknowledgements
The authors would like to thank Camille Cristine Caldeira da Silva, Edson Alves Gomes and Doris D. Araújo for the outstanding technical support. This work was supported by grants from the Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP) (grants: 2012/50500-7 and 2010/519016), Conselho Nacional de Desenvolvimento Científico Tecnológico (CNPq) (grant: 471162/2012-4) and Pro-Reitoria. PAHBK, ARC and AJK are members of the INCT Redoxoma (FAPESP/CNPq/CAPES), the NAP Redoxoma (PRPUSP) and the CEPID Redoxoma (FAPESP, grant: 2013/07937-8). SRM and AJK are staff members of the Departamento de Bioquímica – IQUSP and CNPq research fellows.
References
- 1.Cadenas E., Davies K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radical Biology and Medicine. 2000;29(3–4):222–230. doi: 10.1016/s0891-5849(00)00317-8. 11035250 [DOI] [PubMed] [Google Scholar]
- 2.Dröge W. Free radicals in the physiological control of cell function. Physiological Reviews. 2002;82(1):47–95. doi: 10.1152/physrev.00018.2001. 11773609 [DOI] [PubMed] [Google Scholar]
- 3.Wallace D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annual Review of Genetics. 2005;39(1):359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suzuki Y.J., Forman H.J., Sevanian A. Oxidants as stimulators of signal transduction. Free Radical Biology and Medicine. 1997;22(1–2):269–285. doi: 10.1016/s0891-5849(96)00275-4. 8958153 [DOI] [PubMed] [Google Scholar]
- 5.Chandel N.S., Maltepe E., Goldwasser E., Mathieu C.E., Simon M.C., Schumacker P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(20):11715–11720. doi: 10.1073/pnas.95.20.11715. 9751731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giorgio M., Trinei M., Migliaccio E., Pelicci P.G. Hydrogen peroxide: a metabolic by-product or a common mediator of ageing signals? Nature Reviews Molecular Cell Biology. 2007;8(9):722–728. doi: 10.1038/nrm2240. 17700625 [DOI] [PubMed] [Google Scholar]
- 7.Figueira T.R., Barros M.H., Camargo A.A., Castilho R.F., Ferreira J.C.B., Kowaltowski A.J., Sluse F.E., Souza-Pinto N.C., Vercesi A.E. Mitochondria as a source of reactive oxygen and nitrogen species: from molecular mechanisms to human health. Antioxidants & Redox Signaling. 2013;18:2029–2074. doi: 10.1089/ars.2012.4729. [DOI] [PubMed] [Google Scholar]
- 8.Sohal R.S., Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273(5271):59–63. doi: 10.1126/science.273.5271.59. 8658196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barja G. Free radicals and aging. Trends in Neurosciences. 2004;27(10):595–600. doi: 10.1016/j.tins.2004.07.005. 15374670 [DOI] [PubMed] [Google Scholar]
- 10.Balaban R.S., Nemoto S., Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120(4):483–495. doi: 10.1016/j.cell.2005.02.001. 15734681 [DOI] [PubMed] [Google Scholar]
- 11.Valko M., Leibfritz D., Moncol J., Cronin M.T., Mazur M., Telser J. Free radicals and antioxidants in normal physiological functions and human disease. International Journal of Biochemistry & Cell Biology. 2007;39(1):44–84. doi: 10.1016/j.biocel.2006.07.001. 16978905 [DOI] [PubMed] [Google Scholar]
- 12.Turrens J.F. Mitochondrial formation of reactive oxygen species. Journal of Physiology. 2003;552(2):335–344. doi: 10.1113/jphysiol.2003.049478. 14561818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adam-Vizi V., Chinopoulos C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends in Pharmacological Sciences. 2006;27(12):639–645. doi: 10.1016/j.tips.2006.10.005. 17056127 [DOI] [PubMed] [Google Scholar]
- 14.Murphy M.P. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009;417(1):1–13. doi: 10.1042/BJ20081386. 19061483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lambert A.J., Brand M.D. Reactive oxygen species production by mitochondria. Methods in Molecular Biology. 2009;554:165–181. doi: 10.1007/978-1-59745-521-3_11. 19513674 [DOI] [PubMed] [Google Scholar]
- 16.Cardoso A.R., Kakimoto P.A., Kowaltowski A.J. Diet-sensitive sources of reactive oxygen species in liver mitochondria: role of very long chain acyl-CoA dehydrogenases. PLOS One. 2013;8(10):e77088. doi: 10.1371/journal.pone.0077088. 24116206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tahara E.B., Navarete F.D., Kowaltowski A.J. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radical Biology and Medicine. 2009;46(9):1283–1297. doi: 10.1016/j.freeradbiomed.2009.02.008. 19245829 [DOI] [PubMed] [Google Scholar]
- 18.Doulias P.T., Tenopoulou M., Greene J.L., Raju K., Ischiropoulos H. Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Science Signaling. 2013;6(256):rs1. doi: 10.1126/scisignal.2003252. 23281369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Llopis J., McCaffery J.M., Miyawaki A., Farquhar M.G., Tsien R.Y. Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(12):6803–6808. doi: 10.1073/pnas.95.12.6803. 9618493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Porcelli A.M., Ghelli A., Zanna C., Pinton P., Rizzuto R., Rugolo M. pH difference across the outer mitochondrial membrane measured with a green fluorescent protein mutant. Biochemical and Biophysical Research Communications. 2005;326(4):799–804. doi: 10.1016/j.bbrc.2004.11.105. 15607740 [DOI] [PubMed] [Google Scholar]
- 21.Goetzman E.S., Wang Y., He M., Mohsen A.W., Ninness B.K., Vockley J. Expression and characterization of mutations in human very long-chain acyl-CoA dehydrogenase using a prokaryotic system. Molecular Genetics and Metabolism. 2007;91(2):138–147. doi: 10.1016/j.ymgme.2007.01.013. 17374501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pessayre D., Berson A., Fromenty B., Mansouri A. Mitochondria in steatohepatitis. Seminars in Liver Disease. 2001;21(1):57–69. doi: 10.1055/s-2001-12929. 11296697 [DOI] [PubMed] [Google Scholar]
- 23.Jaeschke H., Gores G.J., Cederbaum A.I., Hinson J.A., Pessayre D., Lemasters J.J. Mechanisms of hepatotoxicity. Toxicological Sciences. 2002;65(2):166–176. doi: 10.1093/toxsci/65.2.166. 11812920 [DOI] [PubMed] [Google Scholar]
- 24.Sandri G., Panfili E., Ernster L. Hydrogen peroxide production by monoamine oxidase in isolated rat-brain mitochondria: its effect on glutathione levels and Ca2+ efflux. Biochimica et Biophysica Acta. 1990;1035(3):300–305. doi: 10.1016/0304-4165(90)90092-b. 2207125 [DOI] [PubMed] [Google Scholar]
- 25.Kaludercic N., Mialet-Perez J., Paolocci N., Parini A., Di Lisa F. Monoamine oxidases as sources of oxidants in the heart. Journal of Molecular and Cellular Cardiology. 2014;73:34–42. doi: 10.1016/j.yjmcc.2013.12.032. 24412580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim J.J., Miura R. Acyl-CoA dehydrogenases and acyl-CoA oxidases. Structural basis for mechanistic similarities and differences. European Journal of Biochemistry. 2004;271(3):483–493. doi: 10.1046/j.1432-1033.2003.03948.x. 14728675 [DOI] [PubMed] [Google Scholar]
- 27.McAndrew R.P., Wang Y., Mohsen A.W., He M., Vockley J., Kim J.J. Structural basis for substrate fatty acyl chain specificity: crystal structure of human very-long-chain acyl-CoA dehydrogenase. Journal of Biological Chemistry. 2008;283(14):9435–9443. doi: 10.1074/jbc.M709135200. 18227065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakajima Y., Miyahara I., Hirotsu K., Nishina Y., Shiga K., Setoyama C., Tamaoki H., Miura R. Three-dimensional structure of the flavoenzyme acyl-CoA oxidase-II from rat liver, the peroxisomal counterpart of mitochondrial acyl-CoA dehydrogenase. Journal of Biochemistry. 2002;131(3):365–374. doi: 10.1093/oxfordjournals.jbchem.a003111. 11872165 [DOI] [PubMed] [Google Scholar]