

Abstract
Premature delivery occurs in 12% of all births, and accounts for nearly half of long-term neurological morbidity, and 60% to 80% of perinatal mortality. Despite advances in obstetrics and neonatology, the rate of premature delivery has increased approximately 12% since 1990. The single most common cause of spontaneous preterm birth is infection. Several lines of evidence have demonstrated the role of endothelin-1 as both a constrictor of uterine myometrial smooth muscle and a proinflammatory mediator. Endothelin-1 activates the phospholipase C pathway, leading to activation of protein kinase C and, in turn, sphingosine kinase (SphK). The inhibition of SphK has been recently shown to control the proinflammatory response associated with sepsis. We show herein, for the first time, that SphK inhibition prevents inflammation-associated preterm birth in a murine model. Rescue of pups from premature abortion with an SphK inhibitor occurs by suppression of the proinflammatory cytokines tumor necrosis factor α, Il-1β, and Il-6 and attenuation of polymorphonuclear inflammatory cells into the placental labyrinth. Moreover, we postulate that inhibition of SphK leads to suppression of endothelin-converting enzyme-1 expression, indicating the presence of an endothelin-converting enzyme 1/endothelin 1–SphK positive feedback loop. This work introduces a novel approach for the control of infection-triggered preterm labor, a condition for which there is no effective treatment.
Preterm birth (PTB), defined as any birth before 37 complete weeks of gestation, accounts for 11.1% of all deliveries worldwide, but next to congenital anomalies, accounts for most perinatal morbidity and mortality. The United States, with a rate of PTB of 11.7%, ranks among the 10 countries that have the highest rates of PTB.1 The single most common cause of spontaneous PTB is infection.2 Intrauterine bacteria interact with cell surface recognition molecules, such as Toll-like receptor-2 (Tlr2) and Tlr4,3,4 leading to release of T helper cell 1 cytokines, such as Il-1β and tumor necrosis factor α (TNFα).5–7 This inflammatory response culminates in the final steps of the parturition cascade, consisting of decreased prostaglandin metabolism, functional progesterone withdrawal, increased expression of proteases and contraction-associated proteins, and increased uterine contractile activity.8–10
We have previously shown that endothelin 1 (Edn1) is a key player in lipopolysaccharide (LPS)–induced PTB in a murine model.11–14 Specifically, we have previously reported that endothelin-converting enzyme 1 (Ece1), the enzyme that synthesizes Edn1, colocalizes with its substrate in the placenta15 and that the Ece1 inhibitor, phosphoramidon, decreases rates of PTB in a murine model of infection-associated preterm labor.11 Subsequently, we have also shown that Ece1 levels are increased in a mouse model of PTB and that rates of preterm delivery are decreased with endothelin-A receptor antagonists.12–14 Finally, we have shown that virtually complete control of PTB is achieved in this model by silencing Ece1 mRNA.13 LPS, a Gram-negative bacterial wall component that activates Tlr4, stimulates the synthesis of Edn1 by monocytes and endothelial cells in vitro and in vivo.16–18
The activation of the phospholipase C pathway by Edn1 leads to activation of protein kinase C (PKC), which acts upstream of sphingosine kinase (SphK).19,20 SphK, in turn, can activate Rho kinase, ultimately leading to contraction of myometrial tissue. SphK is the only enzyme that catalyzes the conversion of SphK to sphingosine-1-phosphate, which behaves in an autocrine manner in myometrial tissues and, in orchestration with SphK, PKC, and extracellular signal–regulated kinase, leads to the induction of cyclooxygenase-2 and to labor.21
Two human SphK isozymes, SphK1 and SphK2, have been identified,22 both of which are inhibited by sphingosine kinase inhibitor II (SKI II), the compound used in the work reported herein. We show herein, for the first time, that inhibition of SphK and consequent suppression of the SphK1-dependent inflammatory pathway prevents infection-triggered PTB.
Materials and Methods
Reagents
LPS (serotype 026:B6), polyethylene glycol 400 (PEG 400), and antibodies against TNFα, Il-1β, and Il-6 were purchased from Sigma-Aldrich (St. Louis, MO), Il-10 antibody from Epitomics (Burlingame, CA), Ece1 antibody from US Biological (Swampscott, MA), and glyceraldehyde-3-phosphate dehydrogenase antibody from Cell Signaling Technology (Danvers, MA). Lysis, loading, running, and transfer buffers as well as molecular weight standards and polyvinylidene difluoride (PVDF) membranes were purchased from Life Technologies (Grand Island, NY); skim milk powder from EMD Chemicals (Gibbstown, NJ); bicinchoninic assay reagents from VWR Scientific (Bridgeport, NJ); ECL Plus Western blotting detection system from GE Healthcare (Buckinghamshire, UK); bovine serum albumin from Fisher Scientific (Pittsburgh, PA); anti-rabbit IgG, horseradish peroxidase–linked whole antibody from GE Healthcare (Buckinghamshire, UK); autoradiographic film from Denville Scientific (Metuchen, NJ); stripping buffer from Thermo Scientific (Rockford, IL); universal labeled streptavidin-biotin2 system, horseradish peroxidase (LSAB2 System, HRP) from Dako (Carpinteria, CA); xylene from Malinckrodt Chemical Inc. (Washington, DC); and hematoxylin from VWR Scientific. The SphK inhibitor, SKI II, was purchased from Tocris Bioscience (Ellisville, MO). Histochoice mounting media was purchased from Amresco Inc. (Solon, OH). All other reagents were purchased from VWR Scientific.
Animals
All experimental protocols involving animals were approved by the St. John's University Animal Care and Use Committee of the College of Pharmacy and Health Sciences and conducted according to the guidelines in the Guide for the Care and Use of Laboratory Animals.23 C57BL/6 mice from Taconic Laboratories (Cranbury, NJ) were used for all in vivo experiments. Animals were housed in individually ventilated cages in an animal facility with controlled conditions of temperature (23°C ± 1°C), humidity (50% ± 10%), and 12:12-hour light/dark cycles. Standard laboratory chow and water were provided ad libitum.
In Vivo Studies
A total of 17 timed pregnant embryonic day (E) 15.5 mice, weighing between 28 and 35 g, were used for in vivo studies. The control group (n = 7) was injected i.p. with 50 mg/kg LPS (serotype 026:B6; Sigma-Aldrich) dissolved in 0.5 mL phosphate-buffered saline (PBS) at time (T) = 0 and 50 μL PEG 400 at T = 1 and 7 hours. The treatment group (n = 7) was also injected i.p. with the same dose of LPS and then injected with 50 mg/kg SphK inhibitor (SKI II) dissolved in PEG 400 at T = 1 and 7 hours. The sham group (n = 3) was injected i.p. with 0.5 mL PBS at T = 0 and 50 μL PEG 400 at T = 1 and 7 hours. After injections at T = 7 hours, mice were continuously observed for time of delivery and number of pups dropped. Mice that did not deliver were observed until T = 24 hours. All mice were euthanized by carbon dioxide asphyxiation and necropsied to confirm pregnancy, and the number of pups retained in utero was recorded. The retained placentas and uteri were harvested and stored at −80°C or in 10% neutral-buffered formalin at room temperature.
High-Capacity Real-Time RT-PCR
On necropsy, placental tissue was immediately harvested, incubated in RNAlater (Qiagen, Valencia, CA), and stored at −80°C until ready for use. RNA was isolated using the Qiagen RNeasy Mini Kit (Qiagen), according to the manufacturer's instructions, and the A260/A280 ratio was measured to determine nucleic acid purity. Only samples with an A260/A280 ratio of 1.8 to 2.1 were used. Reverse transcription of RNA to cDNA was then performed using Applied Biosystems/Life Technologies high-capacity cDNA reverse transcription kit. Aliquots of 1 μg of RNA were used per reaction, and cDNA was stored at 4°C until ready for use. Equimolar amounts of RNA from 10 different placentas from 10 different mice were pooled. Quantitative RT-PCR was performed on a Roche LightCycler480 (Roche Diagnostics Corporation, Indianapolis, IN). Qiagen's RT2 Profiling PCR Array (Qiagen) for 84 different genes involved in mouse Toll-like receptor signaling (including housekeeping genes) was used, following the manufacturer's instructions.
Gel Electrophoresis
Frozen placentas were allowed to thaw on ice. Tissues were homogenized in 0.25 mL ice cold lysis buffer, using a Polytron homogenizer (VWR, Bridgeport, NJ). The samples were homogenized on ice for 2 minutes at 15-minute intervals. This procedure was continued for 2 hours, and the final homogenates were centrifuged at 10,000 × g for 10 minutes. Volumes of supernatant containing 30 to 40 μg of protein based on bicinchoninic assay, using bovine serum albumin as standard, were diluted in NuPAGE LDS sample buffer (Life Technologies, Norwalk, CT), and the samples were heated at 95°C for 5 minutes. Bis-Tris gels were used for protein separation. Gel electrophoresis was performed in an XCell SureLock Mini-Cell apparatus (Life Technologies). The gels were allowed to run for 45 minutes with 3-(N-morpholino) propanesulfonic acid running buffer at 125 mA and 200 V. Proteins were then transferred to a PVDF membrane, using an XCell II Blot Module (Life Technologies). Transfer was performed for 2 hours on ice at a 170-mA current and a constant voltage of 30 V with NuPAGE transfer buffer (Life Technologies).
Western Blot Analysis
PVDF membranes were washed with Tris-buffered saline containing 0.1% Tween 20 (TBS-T; Sigma-Aldrich), pH 7.8, for 5 minutes and then blocked with 5% skim milk powder (EMD Chemicals) in TBS-T solution for 1 hour. The membranes were analyzed for different proteins using their respective primary antibodies (ie, TNFα, Il-1β, Il-6, Il-10, and Ece1) diluted in the blocker solution overnight at 4°C. The PVDF membranes were washed with TBS-T three times at intervals of 15 minutes at room temperature. They were then incubated with a 1:1000 dilution of secondary antibody, anti-rabbit IgG, horseradish peroxidase–linked whole antibody, in blocker solution for 2 hours at room temperature. The membranes were then washed with TBS-T four times at intervals of 15 minutes at room temperature. They were then treated with ECL Plus Western blotting detection system (VWR), and the chemiluminescence produced by the chemical reaction was detected by exposure to an autoradiography film. On development of the film, the membranes were washed with TBS-T and stored in TBS-T at 4°C. For reprobing, the membranes were treated with stripping buffer for 5 minutes at room temperature. They were then washed with TBS-T three times at 10-minute intervals at room temperature and incubated with primary anti– glyceraldehyde-3-phosphate dehydrogenase or anti–β-actin antibody as the gel loading control. The same procedure was repeated as above. The protein content was quantified using ImageJ software version 1.48 (NIH, Bethesda, MD). Band density was normalized to housekeeping protein glyceraldehyde-3-phosphate dehydrogenase or β-actin. Each placenta subjected to Western blot analysis came from a different dam. Western blot experiments were performed at least three times, using at least three placentas for each data point each time.
Histopathological Studies
IHC Data
Formalin-fixed, paraffin-embedded tissue sections were used for immunohistochemistry (IHC). The sections were deparaffinized by washing in xylene for 3 minutes. They were then rehydrated by washing in 100% isopropyl alcohol (two changes), 95% alcohol, 80% alcohol, 70% alcohol, and distilled water for 3 minutes each. The sections were incubated for 5 minutes with peroxidase block (3% hydrogen peroxide in water) and washed in distilled water for 3 minutes. Antigen retrieval of the sections was performed using citrate buffer (8.2 mmol/L sodium citrate and 1.8 mmol/L citric acid, pH 6.0, containing 0.01% Triton X-100) at 95°C to 98°C for 20 minutes. The slides were allowed to cool for 20 minutes and were then washed with PBS (Sigma-Aldrich) for 3 minutes. The sections were blocked with 1% bovine serum albumin in PBS for 1 hour in a humidified chamber. The sections were again washed in PBS for 2 minutes. Primary antibodies directed against TNFα, Il-1β, and Il-6, diluted in 1% bovine serum albumin, were used for incubation for 1 hour in the humidified chamber. The slides were washed in fresh PBS for 3 minutes. A biotinylated link was applied to the sections for 10 minutes, and the slides were then washed in fresh PBS for 3 minutes. Streptavidin–horseradish peroxidase was then applied to the sections for 10 minutes, and the slides were again washed in fresh PBS for 3 minutes. The 3,3′-diaminobenzidine substrate–chromogen solution was applied to the sections for 5 minutes, and the slides were washed with distilled water for 3 minutes. The sections were then immersed in hematoxylin for 2 minutes and immediately washed in distilled water for 3 minutes. The slides were wiped and mounted with Histochoice mounting media and coverslipped. The sections were observed under a Nikon Eclipse 80i light microscope (Nikon Inc., Melville, NY), and images were taken with a Nikon Digital Sight camera (Nikon Inc.). Image-Pro Premier 3,3′-diaminobenzidine analysis software version 9.0 (Media Cybernetics Manufacturing, Warrendale, PA) was used for quantification of the results. At least three placentas from three different dams were subjected to IHC analysis for each condition and each cytokine.
Histological Analysis
Placental tissues harvested during necropsy and previously stored in 10% buffered formalin were paraffin embedded, divided into sections (4 μm thick), mounted on clear glass slides, and stained with hematoxylin and eosin. Sections of the tissues were observed using a Nikon Eclipse 80i light microscope by three blinded observers (V.V., S.S., and S.M.) and graded for the extent of inflammatory cell infiltration. The slides were initially scanned at ×100 magnification to identify any areas of inflammation. The three most active fields on each slide were then used for analysis at ×400 magnification. The number of polymorphonuclear neutrophils were counted in the three fields, and the average value was recorded. On the basis of the average number of neutrophils per ×400 field recorded, the slides were assigned grades as follows: 0, 0 to 5 cells; 1, 6 to 50 cells; 2, 51 to 100 cells; and 3, >100 cells. At least three placentas from three different dams were subjected to histological analysis for each condition and each cytokine. The observers were trained by a practicing pathologist (S.E.R.) before grading began, and concordance among the three observers was confirmed with test slides. Images were captured with a Nikon Digital Sight camera.
Statistical Analysis
For in vivo studies, the effect of SKI II on the percentage of LPS-stimulated mice delivering and on the percentage of pups rescued from spontaneous abortion was evaluated with the χ2 and Fisher exact tests, respectively. Differences in rates of PTB and rates of pups delivered over time were evaluated with log-rank (Mantel-Cox) or Gehan-Breslow-Wilcoxon test. The U-test, along with analysis of variance and Tukey's multiple-comparison test, was used to evaluate differences in inflammatory infiltration in histological sections. Differences in mean expression of cytokine proteins in immunoblots and in IHC were determined using analysis of variance with Tukey's multiple-comparison test.
Results
Treatment with 50 mg/kg SKI II significantly reduced the number of mice delivering because of LPS-induced inflammation. In the LPS control group, which received LPS (T = 0 hour) and PEG 400 (T = 1 and 7 hours), five (71%) of seven mice delivered (Table 1 and Figure 1A) by the end of the 24-hour experimental period. With SKI II treatment (T = 1 and 7 hours) after LPS injection, on the other hand, only one (14%) of seven mice delivered prematurely (Table 1 and Figure 1A) by the end of 24 hours, showing a statistically significant effect of SKI II on LPS-induced PTB (P < 0.05). None of the three mice in the sham group delivered prematurely, as expected (Table 1 and Figure 1A).
Table 1.
SKI II Prevents LPS-Induced PTB and Spontaneous Abortion
| Group | LPS control | SKI II treatment | Sham |
|---|---|---|---|
| LPS (50 mg/kg) received | Yes | Yes | No |
| No. of mice | 7 | 7 | 3 |
| No. of mice delivered | 5 | 1∗ | 0 |
| Total no. of pups | 47 | 50 | 20 |
| No. of pups spontaneously aborted | 21 | 2∗∗∗∗ | 0 |
LPS, lipopolysaccharide; PTB, preterm birth; SKI II, sphingosine kinase inhibitor II.
P < 0.05, ∗∗∗∗P < 0.0001.
Figure 1.
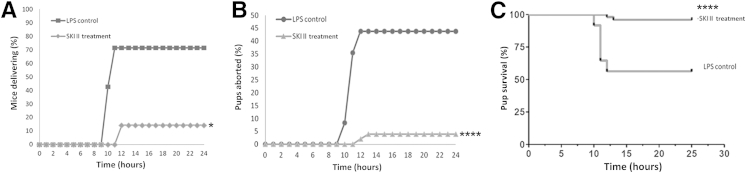
Sphingosine kinase inhibitor, sphingosine kinase inhibitor II (SKI II), rescues lipopolysaccharide (LPS)-stimulated timed pregnant embryonic day 15.5 mice from preterm birth (PTB). Mice induced with 50 mg/kg LPS i.p. were treated with 50 mg/kg SKI II or polyethylene glycol (PEG) 400. All mice were observed for PTB for a period of 24 hours. A: SKI II significantly reduces the percentage of mice delivering over time. B: SKI II significantly reduces the percentage of pups lost to spontaneous abortion over time. C: SKI II significantly increases the rate of survival of pups in LPS-treated mice, as seen from the Kaplan-Meier survival curves for pups in both groups. ∗P < 0.05, ∗∗∗∗P < 0.0001.
In addition to decreasing the number of mice delivering prematurely, SKI II treatment rescued a significant number of pups from being dropped prematurely and hence spontaneously aborted. In the LPS control group, 21 (44%) of 47 pups were dropped (Table 1 and Figure 1B) within the 24-hour experimental time. In the treatment group, on the other hand, only 2 (4%) of 50 pups were dropped prematurely in the 24-hour time period (P < 0.0001) (Table 1 and Figure 1B). In the sham group, none of the 20 pups were delivered prematurely, as expected (Table 1). Comparison of the two Kaplan-Meier survival curves by log-rank test revealed a statistically significant difference between the two groups (P < 0.0001) (Figure 1C). No significant changes in litter size or fetal growth and development were observed throughout the study, and no macroscopic congenital anomalies were noted in any of the 97 pups.
We performed real-time RT-PCR analysis of 84 Tlr4-associated mouse placental genes from E15.5 mice induced by LPS and found up-regulation of most of the genes in this panel (Figure 2, Table 2, and Supplemental Tables S1 and S2). Genes encoding TNFα, Il-1β, and Il-6, which have been shown to be elevated in amniotic fluid in the setting of PTB, were all up-regulated by LPS. In the current work, TNFα, Il-1β, and Il-6 expression were all significantly increased in placental lysates from LPS-induced mice as compared to shams (P < 0.05). Moreover, levels of all of these proteins were reduced in the SKI II–treated group when compared to the LPS control group (P < 0.05) (Figure 3, A–C). Interestingly, Il-10, an anti-inflammatory cytokine, was similarly suppressed by SKI II (Figure 3D), perhaps because its anti-inflammatory regulatory role is made less necessary as a result of SphK inhibition.
Figure 2.

Lipopolysaccharide (LPS)-induced up-regulation of Toll-like receptor 4 pathway genes in embryonic day (E) 15.5 mouse placenta. High-capacity real-time RT-PCR was performed on RNA isolated from placentas harvested from LPS-treated and normal control E15.5 mice. Equimolar amounts of RNA from 10 different placentas from 10 different mice were pooled. The gene expression heat map indicates up-regulation in red and down-regulation in green. The location of the 84 tested genes (plus housekeeping genes and controls) in the array is displayed in Supplemental Table S1. A complete list of genes represented on the array, with UniGene (http://www.ncbi.nlm.nih.gov/unigene) and GenBank (http://www.ncbi.nlm.nih.gov/genbank) identifiers, gene symbols, and descriptions of corresponding proteins is provided in Supplemental Table S2.
Table 2.
Differential Expression of Tlr4 Pathway Genes in Placentas from LPS-Induced Mice
| Down-regulated | No change | Up-regulated | ||||
|---|---|---|---|---|---|---|
| Elk1 | Fadd | Agfg1 | Hmgb1 | Irf1 | Nfkbib | Tlr1 |
| Nfkb2 | Il10 | Btk | Hras1 | Irf3 | Nfkbil1 | Tlr2 |
| Rela | Il12a | Casp8 | Hspala | Lta | Nfrkb2 | Tlr4 |
| Ly86 | Ccl2 | Ifnb1 | Ly96 | Nr2c2 | Tlr6 | |
| Mapk9 | Cd14 | Ifng | Map2k3 | Peli1 | Tlr8 | |
| Ripk2 | Cd80 | Ikbkb | Map2k4 | Pglyrp1 | Tlr9 | |
| Tlr3 | Cd86 | Il1a | Map3k1 | Ppara | Tnf | |
| Tlr5 | Cebpb | Il1b | Map3k7 | Ptgs2 | Tnfaip3 | |
| Ube2n | Chuk | Il2 | Mapk8 | Rel | Tnfrsf1a | |
| Clec4e | Il6 | Mapk8ip3 | Thk1 | Tollip | ||
| Csf2 | Il6ra | Muc13 | Ticam | Tradd | ||
| Csf3 | Irak1 | Nfkb1 | Ticam2 | Traf6 | ||
| Eif2ak2 | Irak2 | Nfkb1a | Tirap | Ube2v1 | ||
LPS, lipopolysaccharide; Tlr4, Toll-like receptor 4.
Figure 3.

Sphingosine kinase inhibitor, sphingosine kinase inhibitor II (SKI II), suppresses expression of placental inflammatory cytokines and endothelin-converting enzyme-1 (Ece1) in lipopolysaccharide (LPS)-stimulated timed pregnant embryonic day 15.5 mice. Placental tissue lysates from sham, LPS-induced, and LPS-treated mice rescued from preterm birth with SKI II were subjected to gel electrophoresis and immunoblotting. Western blots were probed with antibodies directed against tumor necrosis factor α (TNFα;) (A), Il-1β (B), Il-6 (C), Il-10 (D), and Ece1 (E). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and β-actin were used as loading controls. Western blot experiments were performed at least three times, using at least three placentas from three different dams for each data point each time. One representative immunoblot is shown for each group for each protein. Splicing of immunoblot bands is indicated by white lines. ∗P < 0.05, ∗∗P < 0.01.
On the basis of the premise that SphK acts downstream of Ece1/Edn1, one would not expect SKI II to affect Ece1 levels. However, we found that SKI II clearly decreased levels of Ece1 (Figure 3E). The suppression of Ece1 levels by SKI II introduces a novel concept to our understanding of the Ece1/Edn1-PKC-SphK pathway.
IHC analysis was used to determine the localization of cytokines in the placenta and to confirm the results obtained from Western blot analysis. Placental labyrinthine tissue from mice treated with LPS showed a significant increase in Il-1β and Il-6 levels as compared to shams (P < 0.0001) (Figure 4, C–F). Interestingly, no such increase was seen in TNFα (Figure 4, A and B). SKI II, however, significantly reduced labyrinthine levels of TNFα (P < 0.0001), Il-1β (P < 0.0001), and Il-6 (P < 0.01) (Figure 4).
Figure 4.

Sphingosine kinase inhibitor, sphingosine kinase inhibitor II (SKI II), suppresses cytokine immunoreactivity in the placental labyrinth in lipopolysaccharide (LPS)-stimulated embryonic day 15.5 mice. Sections of the placenta from sham, LPS-induced, and LPS-treated mice rescued from preterm birth with SKI II were fixed in formalin and subjected to immunohistochemistry, using antibodies directed against tumor necrosis factor α (TNFα;) (A), Il-1β (C), and Il-6 (E). Image-Pro Premier version 9.0 3,3′-diaminobenzidine analysis software was used to quantify intensity of immunoreactivity (B, D, and F). Arrows indicate immunostaining. At least three placentas from three different dams were subjected to immunohistochemical analysis for each condition and each cytokine. Representative sections are shown. ∗∗P < 0.01, ∗∗∗P < 0.005. Original magnification, ×400 (A, C, and E).
Placental labyrinthine tissue harvested from sham, LPS-induced, and LPS-treated plus SKI II rescued mice showed significant differences in inflammatory cell recruitment. Figure 5A shows a representative section of placental labyrinth from an LPS-induced mouse with a robust neutrophilic infiltrate when compared to placental labyrinthine tissue from sham mice (P < 0.01). With treatment with SKI II, the inflammatory cell infiltrate subsides significantly (P < 0.05) (Figure 5).
Figure 5.

Sphingosine kinase inhibitor, sphingosine kinase inhibitor II (SKI II), suppresses recruitment of inflammatory cells to the placental labyrinth in lipopolysaccharide (LPS)-stimulated embryonic day 15.5 mice. A: Sections of placenta from sham, LPS-induced, and LPS-treated mice rescued from preterm birth with SKI II were fixed in formalin and stained with hematoxylin and eosin. Arrows indicate neutrophils. B: Magnitude of inflammatory response was graded on a four-point scale by three blinded observers (V.V., S.S., and S.M.). At least three placentas from three different dams were subjected to histological analysis for each condition and each cytokine. Representative sections are shown. ∗P < 0.05, ∗∗P < 0.01. Original magnification, ×400 (A).
Discussion
This work presents a novel approach to controlling infection-triggered PTB, a clinical challenge accounting for a significant amount of perinatal morbidity and mortality in both the United States and around the globe. Previous studies performed by us and others have implicated the Ece1/Edn1 axis in PTB. Edn1 binds to endothelin-A receptors and causes myometrial contraction by activating the phospholipase C pathway.24 In fact, amniotic Edn1, which increases in the setting of infection, is a more potent uterine myometrial muscle constrictor than oxytocin.25,26 Our laboratory has shown that infection-associated PTB can be controlled by inhibition of Edn1 synthesis11; endothelin-A receptor blockade was performed with both BQ-123, a commercially available antagonist,13 and 1-3-6-trisubstituted-2-carboxy-quinol-4-one, a novel antagonist synthesized by our collaborator12,14; and Ece1 mRNA silencing.13 Moreover, we have shown that Ece1 knockdown decreases levels of matrix metalloproteinase-1 in mouse placenta and uterus.27 This finding is significant because matrix metalloproteinase levels are increased during amniotic infection, causing zinc-dependent catalysis of the extracellular matrix, resulting in cervical ripening, rupture of fetal membranes, and placental detachment.28 Thus, numerous studies indicate that Edn1 plays a role during both term labor and infection-associated preterm labor, suggesting that it could serve as a pharmacotherapeutic target for the prevention of PTB. However, knockout of Ece1 and Edn1, the genes encoding Ece1 and Edn1, respectively, early in gestation results in craniofacial and cardiac abnormalities, revealing the importance of Edn1 for normal embryonic development.29,30 Because Edn1 activates SphK via PKC,19 it was of interest to test whether inhibition of SphK, a downstream mediator in the same molecular pathway as Edn1, would offer a novel approach to controlling PTB without producing teratogenic effects.
The importance of Tlr4 in infection-associated PTB has been established; mice with a mutant form of Tlr4 are less likely to deliver prematurely after LPS induction.3 Genomic studies in our laboratory show concordance with the profile of cytokine expression seen in the quantitative RT-PCR performed herein31 (Gene Expression Omnibus; http://www.ncbi.nlm.nih.gov/geo; Accession number GSE49895), pointing to TNFα, Il-1β, and Il-6 as important signaling molecules in the LPS-induced inflammatory cascade. Il-6, in particular, is implicated in the pathogenesis of the fetal inflammatory syndrome, a constellation of abnormalities resulting from in utero exposure to inflammation, including periventricular leukomalacia and bronchopulmonary dysplasia.32
The data presented herein suggest that SphK inhibition may prevent PTB triggered by Gram-negative bacterial infection by suppressing several Tlr4-linked proinflammatory cytokines. Although we used a relatively small number of mice in our control and treated groups (n = 7 each), the effect of SphK inhibition is so dramatic that our study was adequately powered. The effect of SKI II on dams at risk for PTB, and particularly on pups at risk for spontaneous abortion, was significant (P < 0.05 and P < 0.001, respectively).
Although we chose a well-established model of inflammation-associated PTB for this focused work, inducing PTB with LPS does not represent all types of infection-triggered PTB. Future lines of investigation include repeating this work using peptidoglycan-polysaccharide to simulate infection with Gram-positive bacteria and polyinosinic:polycytidylic acid to simulate infection with viral pathogens. Moreover, the effect of SphK inhibition on PTB triggered by actual microorganisms, such as the human periodontal pathogens Campylobacter rectus and Porphyromonas gingivalis, should be tested before the therapeutic potential of SphK inhibitors for PTB can be evaluated.
SphK and sphingosine-1-phosphate participate in myometrial contractions in the rat19; inhibition of SphK causes a reduction in Edn1-mediated myometrial contraction. On the basis of our finding of decreased placental Ece1 in LPS-stimulated mice treated with SKI II as compared to LPS-exposed mice that were not treated, we postulate that the Ece1/Edn1-PKC-SphK pathway is regulated by positive feedback with increasing activity of SphK in the setting of infection, leading to more Edn1 synthesis and greater SphK activity (Figure 6). Cyclooxygenase-2 levels correlate with SphK levels in late gestation.21 This is the first report, to our knowledge, however, showing that inhibition of SphK controls PTB or any other in vivo model of LPS-triggered inflammation-based disorder via suppression of cytokine and inflammatory cell infiltration.
Figure 6.

Sphingosine kinase (SphK)–endothelin-converting enzyme 1 (Ece1) positive feedback loop. Synthesis of endothelin 1 (Edn1) triggers activation of phospholipase C (PLC), which leads to activation of protein kinase C (PKC) and SphK. Inhibition of SphK with sphingosine kinase inhibitor II suppresses not only several inflammatory cytokines, but also decreases levels of Ece1, which is known to act upstream of SphK. The data support the existence of a positive feedback loop between SphK and the Ece1/Edn1 axis. DAG, diacylglycerol; ET-1, endothelin 1; InsP3, inositol triphosphate; PIP2, phospholipid phosphatidyl inositol 4,5-bisphophate; S1P, sphingosine-1-phosphate.
Acknowledgments
We thank Helen Scaramell, Eileen Hussey, and the entire St. John's Animal Facility Staff for their assistance with maintaining and breeding the mice and Ernestine Middleton for preparation of microscopic slides.
V.V. performed the in vivo studies, Western blot analysis, immunohistochemistry, and some statistical analysis, prepared the figures, and helped with writing; C.R.A. helped with the conception of the original project, provided sphingosine kinase inhibitor II, and helped with writing; N.S.O. performed the high-capacity real-time RT-PCR; S.S., O.S., and S.M. graded the intensity of mouse neutrophilic infiltrates; R.P. performed statistical analysis; P.M. provided help with Western blot analysis; and S.E.R. guided all aspects of the project, wrote the manuscript, and provided funding.
Footnotes
Supported by NIH grant 1 R01 NS069577 (S.E.R.).
Disclosures: A provisional patent (61/964,127) on the use of sphingosine kinase inhibition for the treatment and prevention of obstetrical disorders has been filed by S.E.R. and C.R.A.
Supplemental Data
References
- 1.Blencowe H., Cousens S., Oestergaard M.Z., Chou D., Moller A., Narwal R., Say L. National, regional, and worldwide estimates of preterm birth rates in the year 2010 with time trends since 1990 for selected countries: a systematic analysis and implications. Lancet. 2012;379:2162–2172. doi: 10.1016/S0140-6736(12)60820-4. [DOI] [PubMed] [Google Scholar]
- 2.Epstein F.H., Goldenberg R.L., Hauth J.C., Andrews W.W. Intrauterine infection and preterm delivery. N Engl J Med. 2000;342:1500–1507. doi: 10.1056/NEJM200005183422007. [DOI] [PubMed] [Google Scholar]
- 3.Elovitz M.A., Wang Z., Chien E.K., Rychlik D.F., Phillippe M. A new model for inflammation-induced preterm birth: the role of platelet-activating factor and Toll-like receptor-4. Am J Pathol. 2003;163:2103–2111. doi: 10.1016/S0002-9440(10)63567-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abrahams V.M. Antagonizing Toll-like receptors to prevent preterm labor. Reprod Sci. 2008;15:108–109. doi: 10.1177/1933719108314574. [DOI] [PubMed] [Google Scholar]
- 5.Romero R., Mazor M., Brandt F., Sepulveda W., Avila C., Cotton D.B., Dinarello C.A. Interleukin-1 alpha and interleukin-1 beta in preterm and term human parturition. Am J Reprod Immunol. 1992;27:117–123. doi: 10.1111/j.1600-0897.1992.tb00737.x. [DOI] [PubMed] [Google Scholar]
- 6.Romero R., Mazor M., Sepulveda W., Avila C., Copeland D., Williams J. Tumor necrosis factor in preterm and term labor. Am J Obstet Gynecol. 1992;166:1576–1587. doi: 10.1016/0002-9378(92)91636-o. [DOI] [PubMed] [Google Scholar]
- 7.Dudley D.J., Collmer D., Mitchell M.D., Trautman M.S. Inflammatory cytokine mRNA in human gestational tissues: implications for term and preterm labor. J Soc Gynecol Investig. 1996;3:328–335. [PubMed] [Google Scholar]
- 8.Brown N.L., Alvi S.A., Elder M.G., Bennett P.R., Sullivan M.H. Interleukin-1beta and bacterial endotoxin change the metabolism of prostaglandins E2 and F2alpha in intact term fetal membranes. Placenta. 1998;19:625–630. doi: 10.1016/s0143-4004(98)90024-8. [DOI] [PubMed] [Google Scholar]
- 9.Challis J.R.G. Mechanism of parturition and preterm labor. Obstet Gynecol Surv. 2000;55:650–660. doi: 10.1097/00006254-200010000-00025. [DOI] [PubMed] [Google Scholar]
- 10.Hirsch E., Wang H. The molecular pathophysiology of bacterially induced preterm labor: insights from the murine model. J Soc Gynecol Investig. 2005;12:145–155. doi: 10.1016/j.jsgi.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 11.Koscica K., Sylvestre G., Reznik S.E. The effect of phosphoramidon on inflammation-mediated preterm delivery in a mouse model. Am J Obstet Gynecol. 2004;190:528–531. doi: 10.1016/j.ajog.2003.08.021. [DOI] [PubMed] [Google Scholar]
- 12.Olgun N., Patel H., Wang W., Yen H., Stephani R., Reznik S.E. The role of a 1,3,6-trisubstituted-2-carboxy-quinol-4-one, a novel putative selective ETA antagonist, in controlling preterm labor in a mouse model. Can J Physiol Pharmacol. 2008;86:571–575. doi: 10.1139/Y08-057. [DOI] [PubMed] [Google Scholar]
- 13.Wang W., Yen H., Chen C.-H., Soni R., Jasani N., Sylvestre G., Reznik S.E. The endothelin converting enzyme-1 (ECE-1)/endothelin-1 (ET-1) pathway plays a critical role in infection-associated premature delivery in a mouse model. Am J Pathol. 2008;173:1–8. doi: 10.2353/ajpath.2008.080257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olgun N.S., Stephani R.A., Patel H.J., Reznik S.E. Blockade of endothelin-1 with a novel series of 1,3,6-trisubstituted-2-carboxy-quinol-4-one’s controls infection-associated preterm birth. Am J Pathol. 2010;177:1929–1935. doi: 10.2353/ajpath.2010.100281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahmad Z., Reznik S.E. Immunohistochemical localization of ECE-1 in the human placenta. Placenta. 2000;21:226–233. doi: 10.1053/plac.1999.0454. [DOI] [PubMed] [Google Scholar]
- 16.Wahl J.R., Goetsch N.J., Young H.J., Maanen R.V., Johnson J.D., Pea A.S., Brittingham A. Murine macrophages produce endothelin-1 after microbial stimulation. Exp Biol Med. 2005;230:652–658. doi: 10.1177/153537020523000907. [DOI] [PubMed] [Google Scholar]
- 17.Waneeek M., Weitzberg E., Rudchill A., Oldner A. The endothelin system in septic and endotoxic shock. Eur J Pharmacol. 2000;407:1–15. doi: 10.1016/s0014-2999(00)00675-0. [DOI] [PubMed] [Google Scholar]
- 18.Forni M., Mazzola S., Ribeiro L.A., Pirrone F., Zannoni A., Bernardini C., Bacci M.L., Albertini M. Expression of endothelin-1 system in a pig model of endotoxic shock. Regul Pept. 2005;131:89–96. doi: 10.1016/j.regpep.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 19.Leiber D., Banno Y., Tanfin Z. Exogenous sphingosine 1-phosphate and sphingosine kinase activated by endothelin-1 induced myometrial contraction through differential mechanisms. Am J Physiol Cell Physiol. 2007;292:C240–C250. doi: 10.1152/ajpcell.00023.2006. [DOI] [PubMed] [Google Scholar]
- 20.Vyas V., Ashby C.R., Reznik S.E. Sphingosine kinase: a novel putative target for the prevention of infection-triggered preterm birth. Obstet Gynecol Int. 2013;2013:302592. doi: 10.1155/2013/302952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Serrano-Sanchez M., Tanfin Z., Leiber D. Signaling pathways involved in sphingosine kinase activation and sphingosine-1-phosphate release in rat myometrium in late pregnancy: role in the induction of cyclooxygenase 2. Endocrinology. 2008;149:4669–4679. doi: 10.1210/en.2007-1756. [DOI] [PubMed] [Google Scholar]
- 22.Liu H., Chakravarty D., Maceyka M., Milstien S., Spiegel S. Sphingosine kinases: a novel family of lipid kinases. Prog Nucleic Acid Res Mol Biol. 2002;71:493–511. doi: 10.1016/s0079-6603(02)71049-0. [DOI] [PubMed] [Google Scholar]
- 23.Committee for the Update of the Guide for the Care and Use of Laboratory Animals; National Research Council . Eighth Edition. National Academies Press; Washington, DC: 2011. Guide for the Care and Use of Laboratory Animals. [Google Scholar]
- 24.Rubanyi G., Polokoff M.L. Endothelins: molecular biology, biochemistry, pharmacology, physiology, and pathophysiology. Pharmacol Rev. 1994;46:325–415. [PubMed] [Google Scholar]
- 25.Romero R., Avila C., Edwin S.S., Mitchell M.D. Endothelin-1, 2 levels are increased in the amniotic fluid of women with preterm labor and microbial invasion of the amniotic cavity. Am J Obstet Gynecol. 1992;166:95–99. doi: 10.1016/0002-9378(92)91837-z. [DOI] [PubMed] [Google Scholar]
- 26.Wolff K., Nisell H., Modin A., Lundberg J., Lunell N., Lindblom B. Contractile effect of endothelin 1 and endothelin 3 on myometrium and small intramyometrial arteries of pregnant women at term. Gynecol Obstet Invest. 1993;36:166–171. doi: 10.1159/000292619. [DOI] [PubMed] [Google Scholar]
- 27.Wang W., Yen H., Chen C.-H., Jasani N., Soni R., Koscica K., Reznik S.E. Endothelin-1 and matrix metalloproteinase-1 function in the same molecular pathway in infection-associated preterm birth. Mol Med. 2010;16:505–512. doi: 10.2119/molmed.2010.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harirah H., Donia S.E., Hsu C. Amniotic fluid matrix metalloproteinase-9 and interleukin-6 in predicting intra-amniotic infection. Obstet Gynecol. 2002;99:80–84. doi: 10.1016/s0029-7844(01)01632-5. [DOI] [PubMed] [Google Scholar]
- 29.Clouthier D.E., Williams S.C., Yanagisawa H., Wieduwilt M., Richardson J.A., Yanagisawa M. Signaling pathways crucial for craniofacial development revealed by endothelin-A receptor deficient mice. Dev Biol. 2007;217:10–24. doi: 10.1006/dbio.1999.9527. [DOI] [PubMed] [Google Scholar]
- 30.Yanagisawa H., Yanagisawa M., Kapur R.P., Richardson J.A., Williams S.C., Clouthier D.E., Hammer R.E. Dual genetic pathways of endothelin-mediated intercellular signaling revealed by targeted disruption of endothelin converting enzyme-1 gene. Development. 1998;125:825–836. doi: 10.1242/dev.125.5.825. [DOI] [PubMed] [Google Scholar]
- 31.Sundaram S., Ashby C.R., Jr., Pekson R., Sampat V., Sitapara R., Mantell L., Chen C.H., Yen H., Abhichandani K., Munnangi S., Khadtare N., Stephani R.A., Reznik S.E. N,N-dimethylacetamide regulates the proinflammatory response associated with endotoxin and prevents preterm birth. Am J Pathol. 2013;183:422–430. doi: 10.1016/j.ajpath.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gomez R., Romero R., Ghezzi F., Yoon B.H., Mazor M., Berry S.M. The fetal inflammatory response syndrome. Am J Obstet Gynecol. 1998;179:194–202. doi: 10.1016/s0002-9378(98)70272-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.