

Abstract
The C/EBP homologous protein CHOP is normally present at low levels in cells but increases rapidly after insults such as DNA damage or endoplasmatic reticulum stress where it contributes to cellular homeostasis and apoptosis. By forming heterodimers with other transcription factors, CHOP can either act as a dominant-negative regulator of gene expression or to induce the expression of target genes. Recent work demonstrated that seizure-induced hippocampal damage is significantly worse in mice lacking CHOP and these animals go on to develop an aggravated epileptic phenotype. To identify novel CHOP-controlled target genes which potentially influence the epileptic phenotype, we performed a bioinformatics analysis of tissue microarrays from chop-deficient mice after prolonged seizures. GO analysis revealed genes associated with biological membranes were prominent among those in the chop-deficient array dataset and we identified myelin-associated genes to be particularly de-repressed. These data suggest CHOP might act as an inhibitor of myelin-associated processes in the brain and could be targeted to influence axonal regeneration or reorganisation.
Keywords: CHOP, epilepsy, mRNA microarray, myelin basic protein, status epilepticus
Introduction
CHOP (C/EBP homologous protein), also known as GADD153 (growth arrest and DNA damage 153) or DDIT3 (DNA damage-induced transcript 3), belongs to the C/EBP and jun/fos family of transcription factors containing a conserved basic-leucine zipper domain at the C-terminus (BZIP) which is involved in dimerization and DNA binding [1]. In contrast to other members of the C/EBP family, CHOP does not form homodimers, but instead forms heterodimers sequestering transcription factor partners in the nucleus as well as in the cytoplasm to either block or increase transcription of target genes [1,2]. First identified as being induced by growth arrest and DNA damage [3], CHOP expression has since been shown to be increased by various stress stimuli, including hypoxia and starvation but stress of the endoplasmatic reticulum (ER) has been described as the main CHOP activating pathway [4-6]. The ER is the site of synthesis and folding of secretory proteins and perturbations of ER homeostasis lead to ER-stress which is sensed by three upstream signalling proteins activating the ER-specific unfolded protein response (UPR) [5]. CHOP expression can be induced by all three arms of the UPR; namely IRE1 (inositol-requiring protein 1), PERK (protein kinase (PRK)-like ER kinase) and ATF6 (activating transcription factor-6) [5] with the PERK pathway being the most prominent CHOP inducer [5]. In addition to expressional changes, CHOP activity can also be increased by its phosphorylation through the mitogen-activated protein kinase p38 [7].
CHOP activation is involved in a wide array of different functions which range from cell proliferation to cell differentiation [5,8,9], however the main focus and effort to identify CHOP-regulated target genes has been put on processes related to cell death such as DOCs (down-stream of CHOP) [10], Bcl-2 (B-cell lymphoma) [10], TRB3 (tribbles-related protein3) [11], GADD34 [12] and the BH3-only family members Bim and Puma [13,14].
CHOP has been implicated in a wide array of diseases of the CNS [5] including Alzheimer’s disease [15] and in acute insults to the brain such as ischemia [16] and more recently epilepsy [17]. Whereas most studies propose a neuroprotective contribution of CHOP inactivation/depletion to disease pathology [8], more recent studies suggest a protective role of CHOP under certain circumstances [17,18]. For example, chop deletion reduced life span and increased cell death in a mouse model of Pelizaeus-Merzbacher disease [19] and provided neuroprotection during hypoxia [20]. CHOP also seems to play an important role during normal physiology, as chop-deficient mice showed increased hippocampal apoptosis and diminished memory function [21]. We recently reported that mice lacking chop showed increased seizure-induced cell death [17] which was apparently caused by CHOP-targeting of the E3 ubiquitin ligase MDM2 which in turn increased the levels of the proapoptotic transcription factor p53 resulting in altered p53 signalling [17]. A complete analysis of our microarray data was not performed and it is possible that other processes may be changed. Here, we identified myelin-associated genes to be targeted by CHOP where CHOP seems to carry out a transcriptional repressing role after seizures. These results demonstrate the context specific role of CHOP during cellular stress and normal physiology and show the importance of identifying potential and new CHOP-regulated target genes to fully understand its functions during pathology and normal physiology.
Material and methods
Mouse seizure model
All animal experiments were performed in accordance with the principles of the European Communities Council Directive (86/609/EEC), and procedures were approved by the Research Ethics Committee of the Royal College of Surgeons in Ireland. Experiments were performed as previously reported [17], using adult C57BL/6 wild-type (Harlan, Oxon, UK) and chop-/- mice which were originally back-crossed to C57BL/6 background for at least five generations (Jackson Laboratory, Maine, USA; B6.129S-Ddit3tm1Dron/J; [22]). First, mice were anaesthetized using isoflurane and placed in a stereotaxic frame. Then, three partial craniectomies were performed to affix cortical skull-mounted EEG electrodes (Plastics One, Bilaney Consultants Ltd, Kent, UK), and EEG was recorded using a Grass Comet digital EEG. A guide cannula was affixed for intra-amygdala targeting, and the skull assembly was fixed in place with dental cement. After baseline EEG, kainic acid (0.3 μg in 0.2 μl PBS) (Sigma-Aldrich, Dublin, Ireland) was microinjected into the basolateral amygdala. Non-seizure control mice received 0.2 μl of intra-amygdala vehicle. Mice received intraperitoneal lorazepam (6 mg/kg) 40 minutes after KA injection.
To record from the hippocampus and cortex at the same time, mice were implanted with a bipolar electrode (Plastics One, Bilaney Consultants Ltd, Kent, UK), into the dorsal CA3 subfield of the hippocampus (Coordinates from Bregma; AP = -2.25 mm, L = -0.9 mm, V = -1.94 mm) [23]. Epilepsy monitoring was performed via implanted EEG telemetry units, as previously described [17]. EEG data were acquired with EEG transmitters (Model: F20-EET, Data Systems International, St. Paul, MN, USA) configured to record 2-channel EEG that were skull-affixed over dorsal hippocampi/temporal cortex as described before [17]. Mice were euthanized at various time points after status epilepticus (1, 4, 8, 24 h or 72 h, and 14 days). Animals were given a pentobarbital overdose and perfused with ice-cold saline to remove intravascular blood components. Brains for molecular and biochemical work were microdissected to obtain whole hippocampus [17]. For histology, mice were either perfusion-fixed with paraformaldehyde (4%) or brains fresh-frozen in 2-methylbutane (at -30°C).
Histopathology
Fresh-frozen brains were processed on a cryostat in the coronal plane and 12 μm sections were collected at the level of dorsal hippocampus. Irreversible neuronal injury was assessed using Fluoro-Jade B (FJB) staining as described before [17]. Briefly, sections were post-fixed, incubated in 0.006% potassium permanganate, rinsed and transferred to 0.001% FJB solution (Chemicon Europe Ltd., Chandlers Ford, UK). Sections were then rinsed, dried, cleared and mounted in DPX (Sigma-Aldrich, Dublin, Ireland).
Immunohistochemistry
Mice were perfused with 4% paraformaldehyde, brains extracted, post-fixed and cryoprotected in 30% sucrose solution, and 30 μm sagittal sections were cut on a Leica cryostat. Next, brain sections were pretreated for 1 h with 1% bovine serum albumin, 5% fetal bovine serum and 0.2% Triton™ X-100 and then incubated with Neuropeptide Y (NPY) (Sigma-Aldrich, Dublin, Ireland) primary antibody. Finally, brain sections were incubated in avidin-biotin complex using the Elite® VECTASTAIN® kit (Vector Laboratories, Peterborough, UK). Chromogen reactions were performed with diaminobenzidine (Sigma-Aldrich, Dublin, Ireland) and 0.003% hydrogen peroxide for 10 min. Sections were coverslipped with Fluorosave and imaged using a Nikon 2000 s epifluorescence microscope with a Hamamatsu Orca 285 camera (Micron-Optical).
RNA extraction and real-time quantitative polymerase chain reaction
RNA extraction was undertaken as previously described using TRIzol® (Invitrogen, Marseille, France) [24]. Briefly, 1 μg of total RNA was used to generate complementary DNA by reverse transcription using SuperScript® II reverse transcriptase enzyme (Invitrogen, Marseille, France). Quantitative real-time PCR was performed using a LightCycler 1.5 (Roche Diagnostics, Sussex, UK) in combination with QuantiTect® SYBR® Green PCR kit (Qiagen, Sussex, UK) as per manufacturer’s protocol, and 1.25 μM of primer pair was used. Data were analysed by LightCycler 1.5 software, data were normalized to expression of β-actin and represented as relative quantification values. Primers were designed using Primer3 software (http://frodo.wi.mit.edu) and were verified by basic local alignment search tool BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Primers sequences: chop: forward 5’-gaataacagccggaacctga-3’, reverse 5’-cgtttcctggggatgagata-3’; c-fos: forward 5’ggaattaacctggtgctgga, reverse 3’cattcagaccacctcgacaa, mbp (against all isoforms): forward 5’cacgctggagatatgtgtgg, reverse 3’accatgagaagtggccagag and β-actin: forward 5’gggtgtgatggtgggaatgg, reverse 3’ggttggccttagggttcagg.
Microarray analysis
Microarray studies and data were reported previously [17] and undertaken at an Affymetrix authorized service provider (University College Dublin, Dublin, Ireland) [17,24]. Briefly, total RNA was extracted from wild-type and chop-/- mice 6 h after status epilepticus and was hybridized to the Mouse Genome 430 2.0 Genechip array. Affymetrix GeneChip image files were analysed by robust multichip analysis using RMAExpress 0.5 (http://rmaexpress.bmbolstad.com). Data were log transformed, and the threshold for significant regulation was set at 1.5-fold to retain genes that exhibit a biologically meaningful level of regulation, but not exclude certain genes that, because of high constitutive expression, may show lower degrees of change. Gene ontology and function were assigned by interrogating the database for annotation, visualization and integrated discovery (http://david.abcc.ncifcrf.gov/), Cytoscape (http://www.cytoscape.org) and the published literature. Putative CHOP DNA binding sites were identified using the Algen Promo database (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3).
Western blot analysis
Western blotting was performed as previously described [17]. Proteins were extracted from hippocampus, separated by SDS-PAGE and transferred to nitrocellulose membranes and then immunoblotted with the following primary antibodies: CHOP (Santa Cruz Biotechnology, Heidelberg, Germany), KDEL (Millipore, Cork, Ireland), Mbp (ABCAM, Cambridge, UK) and β-actin and α-tubulin (Sigma-Aldrich, Dublin, Ireland). Horseradish peroxidase-conjugated antibodies (Cell Signaling Technology, Denver, USA) were then applied and used as secondary antibodies. Protein bands were visualized using SuperSignal® West Pico Chemiluminescent Substrate (Pierce). Gel band image densities were captured using a Fuji-film LAS-3000 and analyzed using Alpha-EaseFC4.0 software.
Data analysis
Data are presented as mean ± standard error of the mean (SEM). Data were analysed using ANOVA with post hoc Fisher’s protected least significant difference test (StatView) and Student’s t-test for two-group comparison. Significance was accepted at P < 0.05.
Results
Hippocampal CHOP protein levels are increased after status epilepticus and during process of epileptogenesis
To identify novel CHOP target genes expressed after cellular stress we used our well characterized intra-amygdala status epilepticus mouse model which shows a robust CHOP induction [17,23]. Mice were microinjected with pro-convulsive KA into the amygdala and experienced first seizure bursts shortly afterwards (Figure 1A). Mice then went on and developed continuous status epilepticus. To reduce morbidity and mortality mice were injected with the anticonvulsant lorazepam 40 minutes after KA injection. Intra-hippocampal recording confirmed the recruitment of hippocampal structures during status epilepticus (Figure 1A). As reported previously [25], neuronal cell death was most prominent in the ipsi-lateral CA3 subfield (Figure 1B). Hippocampal cell death was absent in vehicle-injected mice (data not shown and [25]).
Figure 1.
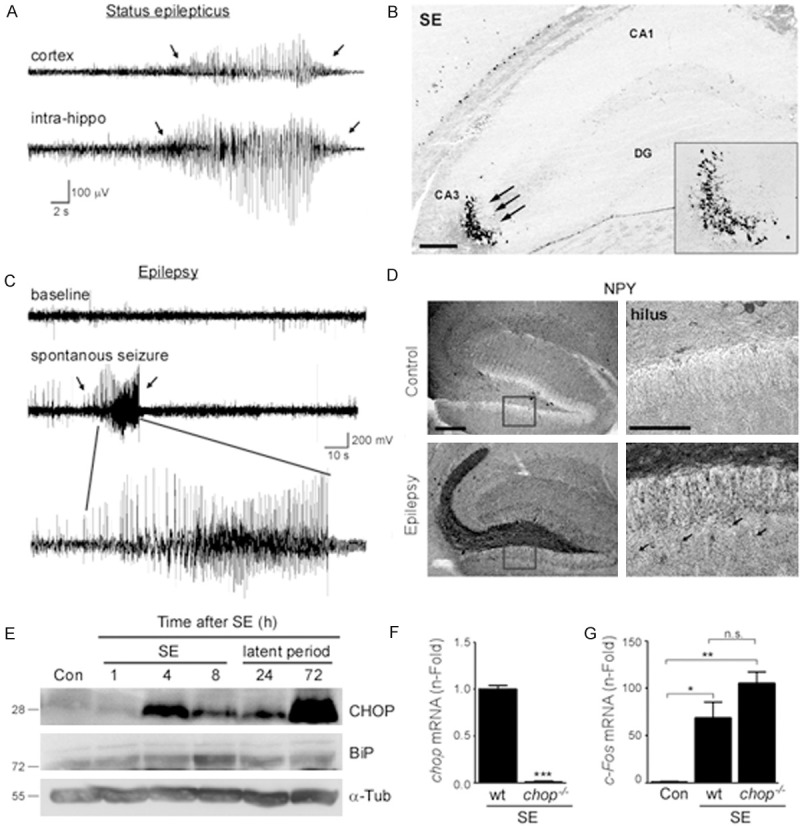
Increased hippocampal CHOP protein levels after status epilepticus and during epileptogenesis. (A)Representative EEG traces from simultaneous cortical surface electrode recording (cortex) and intra-hippocampal recording (intrahippo) during status epilepticus. (B) Representative FJB staining 24 h after status epilepticus of the ipsilateral hippocampus showing cell death (arrows) mainly restricted to the CA3 hippocampal subfield. (C) Representative EEG traces of spontaneous seizure 14 days post KA injection recorded from cortical surface electrodes. (D) Representative photomicrographs showing distribution of Neuropeptide Y (NPY) in the hippocampus of control and epileptic mice 14 days post KA injection. Note, extensive staining of mossy fibers and neuronal extensions (arrows) in epileptic mice. (E) Representative western blots (n = 1 per lane) showing increased hippocampal ER-stress marker KDEL (BiP) and CHOP protein levels after status epilepticus. α-Tubulin is shown as loading control. (F) Graph showing absence of chop mRNA expression in chop knock-out mice 6 h after status epilepticus (n = 4). (G) Increased levels of the activity-regulated gene c-fos in wt and chop-deficient mice 6 h after status epilepticus. No significant differences between wt and chop-deficient mice (p = 0.1). (n = 4 (chop-/- and 6 (wt)).*p < 0.05; **p < 0.01; Scale bar = 150 μm in (B) and 100 μm in (D). CA (cornu ammonis); DG (dentate gyrus).
We monitored a subset of mice to confirm they developed spontaneous seizures after a short latent period of approximately 2 to 3 days (Figure 1C and [25]). To analyze a possible reorganization of the hippocampus, we stained sections from mice killed 14 days after status epilepticus against Neuropeptide Y (NPY), which is a marker of synaptic rearrangement (Figure 1D). NPY staining in vehicle injected control mice was mainly present in hilar neurons and occasionally in cells surrounding the stratum pyramidale layer. In contrast, NPY staining was markedly increased in the hippocampus of epileptic wild-type mice. This was most evident along the mossy fiber pathway and in the outer molecular layer of the dentate gyrus (Figure 1D).
As published previously, ER-stress markers BiP and CHOP protein levels increase shortly after status epilepticus (Figure 1E and [17,23]). Interestingly, CHOP protein levels are not only up-regulated shortly after status epilepticus and during chronic epilepsy (Figure 1E and [17]), but also during the seizure-free latent period (Figure 1E).
Gene ontology analysis of potential CHOP-regulated target genes after status epilepticus
In the present model, chop-deficient mice display increased susceptibility to seizure-induced cell death, a shorter latent period and an aggravated epileptic phenotype, experiencing up to three times more spontaneous seizures than wild-type mice [17]. The increased cell death has been linked to higher p53 levels in chop-deficient mice, possibly due to a decreased expression of the E3 ubiquitin ligase MDM2 which targets p53 for its degradation by the proteasome [17]. However, increased p53 levels have also been linked to decreased seizure frequency in epileptic mice [26] which suggest that other mechanisms might contribute to the increased seizure susceptibility in chop-deficient mice. To test this idea we looked again at the microarray profile of wild-type (wt) and mice lacking chop (chop-/-) subjected to status epilepticus. Previous analysis revealed a strong increase of the genes down-stream of the transcription factor p53 in chop-deficient mice when compared to wt vehicle injected mice [26]. To extend these data, we performed an in depth re-analysis of the microarray data focusing on different pathways altered in chop-/- mice. In contrast to our previous analysis, where gene expression profile of chop-/- mice and wt mice subjected to status epilepticus was compared to vehicle injected seizure- free wt mice [26], this time we chose a direct comparison of chop-/- mice which underwent status epilepticus to wt mice which also experienced status epilepticus. In addition, minimum fold change was lowered from 2 to 1.5.
As previously reported [17] and expected, chop mRNA expression was absent in chop knock-out mice (Figure 1F) and impact on seizures was similar between wt and chop-/- mice, although chop-/- showed slightly increased c-fos mRNA expression (Figure 1G). Because CHOP has the ability to either decrease or increase target gene expression [5], we compared fold-changes of genes which were up- or downregulated in chop-/- mice after status epilepticus when compared to wt seizure mice. Total gene numbers (Figure 2B) and fold genes (Figure 2A, 2C) were similar between groups, suggesting that loss of CHOP has similar effects to both, increase and decrease gene expression after status epilepticus.
Figure 2.
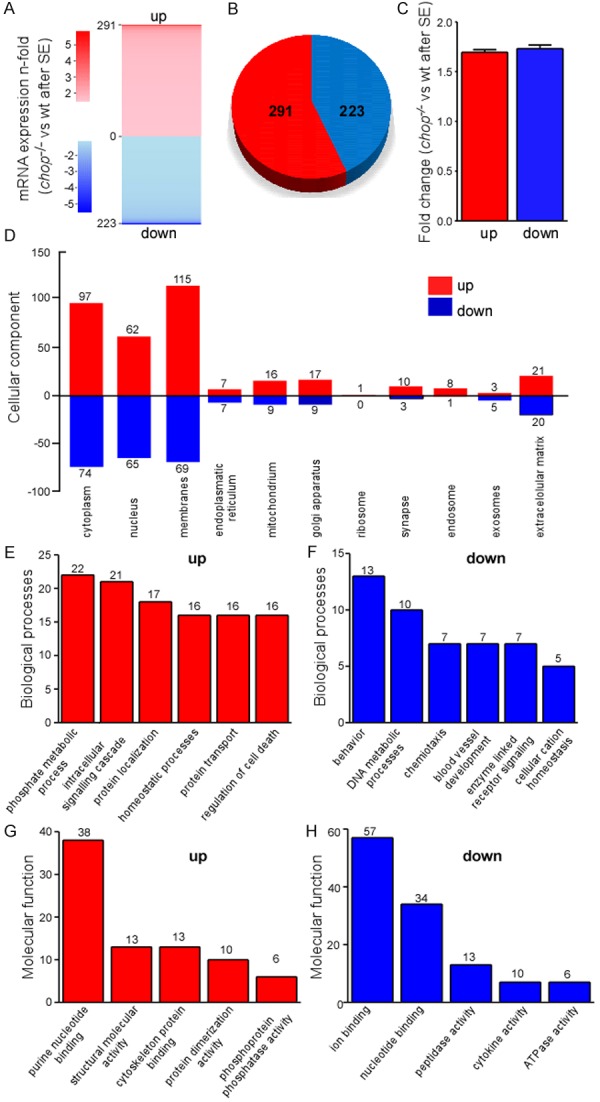
Gene expression profiling in chop-deficient mice after status epilepticus. (A) ‘Heatmap’ representing microarray analysis of the ipsilateral hippocampus 6 h after status epilepticus of up- and downregulated genes in chop-deficient mice when compared to wt mice (n = 1 per group, each sample was a pool of 4 hippocampi from different animals). (B) Pie chart showing total amount of genes up- (red) and down-regulated (blue) in chop-deficient mice when compared to wt mice 6 h after status epilepticus. (C) Graph showing average fold change of up-and down-regulated genes in chop-deficient mice when compared to wt mice. (D-H) Gene ontology analysis of genes up-and down-regulated in chop-deficient mice when compared to wt mice 6 h after status epilepticus. The DAVID bioinformatic tool was used to categorize the chop-regulated gene list according to GO processes (D) Cellular component; (E, F) Biological process and (G, H) Molecular function.
To explore the biological functions of CHOP, we used the gene ontology software DAVID and analyzed biological processes, molecular function and cellular component (Figure 2D-H). Similar numbers for up- and down-regulated genes were obtained for cellular component, although up-regulation was more common for genes associated with membrane and intracellular organelles (Figure 2D). Among the most enriched biological processes in the up-regulated genes were processes involved in phosphorylation, intracellular signalling, protein localization and regulation of cell death (Figures 2E and 4) whereas in the down-regulated genes the most prominent processes involved behaviour, DNA metabolism and chemotaxis (Figures 2F and 3). When we analyzed molecular function, nucleotide binding was highly enriched among up- and down-regulated gene sets (Figures 2G, 2H, 3 and 4) whereas ion binding was the major group among the downregulated genes (Figures 2H and 3). Interestingly, GO terms relating to ”Action potential/Ion homeostasis” and “Axon remodelling” were one of the most highly represented groups identified in enrichment maps of the up-regulated genes which were created using the bioinformatics program cytoscape [27] (Figure 4).
Figure 4.
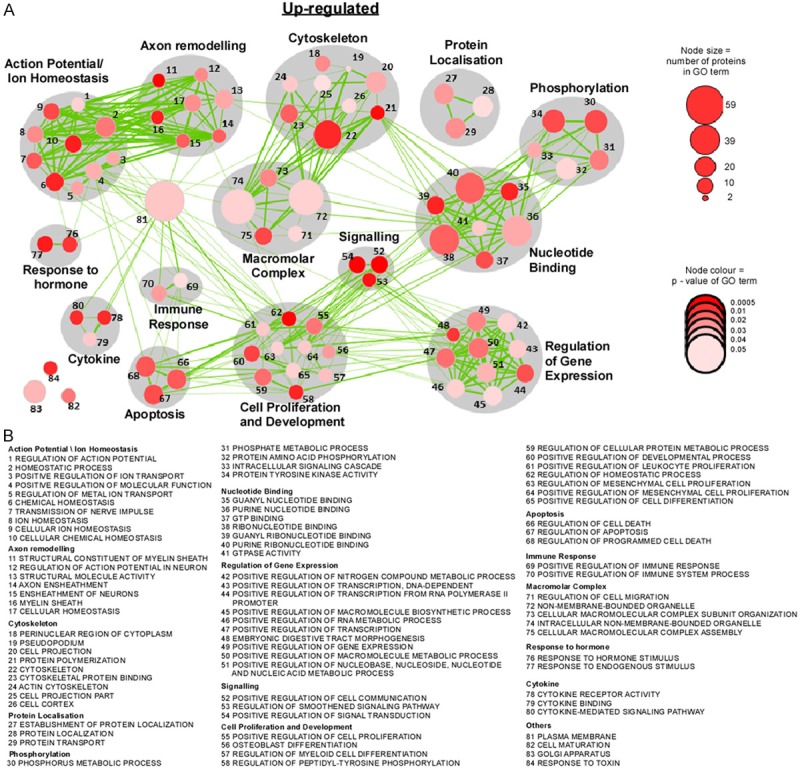
Biological functional pathways up-regulated in chop-deficient mice after status epilepticus. A. Diagram showing network of nodes related by mutual overlap where grey color indicates the class of gene sets. The size of the node corresponds to the number of genes in the GO term and colour of the node (red) relates to the P-value of GO term. B. GO term associated with each gene set.
Figure 3.
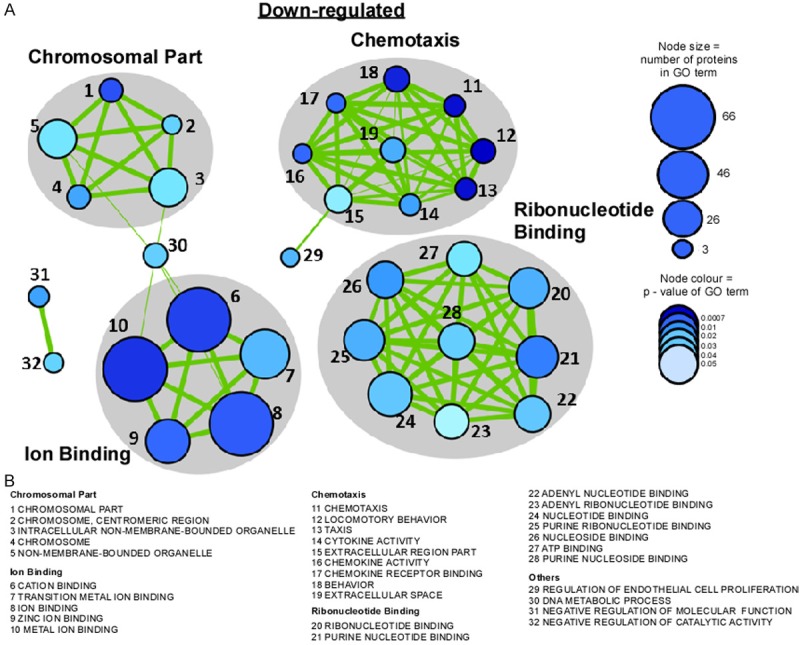
Biological functional pathways down-regulated in chop-deficient mice after status epilepticus. A. Diagram showing network of gene sets (nodes) related by mutual overlap (edges, green), where color (grey) indicates the class of gene sets. The size of the node corresponds to the number of genes in the GO term and colour of the node (blue) relates to the p-value of GO term. B. GO term associated with each gene set.
Table 1 gives a summary of the most up- and down-regulated genes of each of the categories identified under molecular functions including numbers of putative binding sites in the CHOP promoter region.
Table 1.
Potential CHOP-regulated genes after seizures identified by microarray analysis
| Upregulated | ||||||||
|
| ||||||||
| Molecular Function | Gene Symbol | Gene Name | Accession no. | Fold Change | Biological Function | Epilepsy Citations | CHOP Citations | CHOP PBS |
|
| ||||||||
| Purine Nucleotide Binding (38) | Prkcd | Protein kinase C, delta | NM_011103 | 2.62965 | Negative regulation of MAP kinase activity, apoptosis, phosphorylation, negative regulation of actin filament polymerisation | 6 | 7 | 25 |
| Gnai3 | Guanine nucleotide binding protein (G protein), alpha inhibiting 3 | NM_010306 | 1.94658 | GPCR signalling, cell division, vesicle fusion | 0 | 0 | 17 | |
| Rab37 | RAB37, member of RAS oncogene family | NM_001163753 | 1.7972 | GTP catabolic process, protein transport | 0 | 0 | 29 | |
| Structural Molecular Activity (13) | Ctse | Cathepsin E | NM_007799 | 2.32074 | Processing of antigenic peptides during MHC class II-mediated antigen presentation | 0 | 0 | 16 |
| Mbp | Myelin basic protein | NM_001025245 | 1.93847 | Membrane organisation, myelination | 29 | 4 | 29 | |
| Cytoskeleton Protein Binding (13) | Synpo2 | Synaptopodin 2 | NM_080451 | 2.00806 | Actin-binding, actin-bundling activity | 0 | 0 | 27 |
| Gsn | Gelsolin | NM_146120 | 1.68303 | Regulation of cell adhesion, cell projection, cilium morphogenesis | 3 | 2 | 29 | |
| Protein Dimerisation Activity (10) | Pm20d1 | Peptidase M20 domain containing 1 | NM_178079 | 1.79775 | Metabolic process, regulation of neuron death, proteolysis | 0 | 0 | 23 |
| Phosphoprotein Phosphotase Activity (6) | Ptpdc1 | Protein tyrosine phosphatase domain containing 1 | NM_207232 | 1.93937 | Protein dephosphorylation, cell projection | 0 | 0 | 28 |
|
| ||||||||
| Downregulated | ||||||||
|
| ||||||||
| Molecular Function | Gene Symbol | Gene Name | Accession no. | Fold Change | Biological Function | Epilepsy Citations | CHOP Citations | CHOP PBS |
|
| ||||||||
| Ion Binding (57) | Mid1 | Midline 1 | NM_010797 | 4.85018 | Negative regulation of microtubule depolymerisation | 0 | 0 | 27 |
| Cdh7 | Cadherin 7, type 2 | NM_172853 | 2.73727 | Cell adhesion | 1 | 0 | 34 | |
| Rnf41 | Ring finger protein 41 | NM_001164237 | 1.51254 | Negative regulation of cell proliferation, protein ubiquitination | 1 | 0 | 32 | |
| Zkscan6 | Zinc finger with KRAB and SCAN domains 6 | NM_026107 | 1.98704 | Regulation of transcription, transcription | 0 | 0 | 22 | |
| Nucleotide Binding (34) | Mcm6 | Minichromosome maintenance deficient 6 (MIS5 homolog, S. Pombe) (S. Cerevisiae) | NM_008567 | 2.45372 | DNA replication | 0 | 0 | 26 |
| Kif1a | Kinesin family member 1A | NM_001110315 | 2.11829 | Vesicle-mediated transport, microtubule-based movement, ATP catabolic process | 1 | 0 | 25 | |
| Stk25 | Serine/threonine kinase 25 (yeast) | NM_021537 | 2.10611 | Golgi localisation, apoptosis, establishment/maintenance of cell polarity, positive regulation of axonogenesis | 0 | 0 | 25 | |
| Peptide Activity (13) | Serpinb2 | Serine (or cysteine) peptidase inhibitor, clade B, member 2 | NM_001174170 | 1.93399 | Negative regulation of apoptosis and peptidase activity | 8 | 3 | 19 |
| Cytokine Activity (7) | Ccl21a | Chemokine (C-C motif) ligand 21A (serine) | NM_011124 | 6.26678 | Cell chemotaxis, positive regulation of T cell migration | 0 | 0 | 16 |
| ATPase Activity (7) | ATP8a1 | Atpase, aminophospholipid transporter (APLT), class I, type 8A, member 1 | NM_001038999 | 3.61592 | Phospholipid transport, cation transport | 0 | 0 | 29 |
Table 1 showing list of up- and down-regulated genes identified by microarray analysis in chop-/- mice when compared to wt mice (both genotypes underwent status epilepticus before). Top 10% of genes with highest fold change for each molecular function category are shown. Epilepsy and CHOP citations according to Pubmed. (PBS) - Potential Binding Sites.
Loss of CHOP alters expression of genes associated with the formation of the myelin sheath after seizures
A common cellular response following status epilepticus is axonal sprouting and rewiring of the brain, which may increase the brain’s susceptibility to increased seizure frequency [28,29] and mossy fibre sprouting is also common in resected human hippocampus [30]. To identify CHOP-controlled genes which might impact on axon sprouting and rewiring we focused on the enriched gene group “Axon remodelling” (Figure 4A). Here, we identified several genes which code for key proteins of the myelin sheath in the CNS (Figure 5A, 5B). The insulating multilamellar myelin sheath improves neuronal communication by increasing impulse propagation velocity along axons and demyelination has been described as a pathological characteristic in animal seizure models of status epilepticus and in some forms of human epilepsies [31,32]. Furthermore, CHOP has been proposed to be actively involved in the demyelination process, however, without any specifications of its potential targets [33]. We therefore decided to pursue further genes involved in myelination and validate Mbp (myelin basic protein), one of the major components of CNS myelin [34], as a possible CHOP target gene (Figure 5A-C). Quantification of qPCR data showed that mbp mRNA was significantly down-regulated in wt mice after status epilepticus compared to seizure-free vehicle-injected wt mice and to chop-/- mice subjected to status epilepticus (Figure 5D), suggesting CHOP acts as a negative regulator of mbd transcription. Western blotting against Mbp protein in epileptic mice confirmed higher Mbp levels in the hippocampus of chop-/- mice when compared to wt mice (Figure 5E, 5F).
Figure 5.
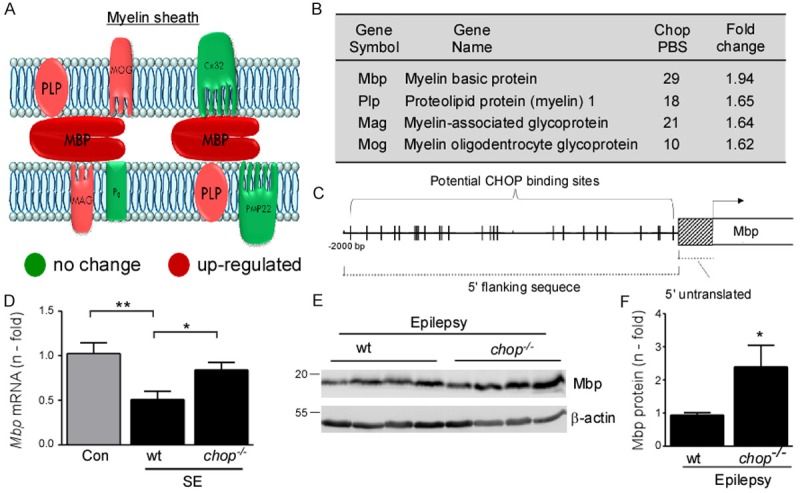
Increased induction of genes related to myelin sheath in chop-deficient mice after status epilepticus. A. Diagram showing main components of the myelin sheath in the CNS and PNS (Cx32, Connexin32; PMP22, peripheral myelin protein 22). (red = up-regulated in chop-/- mice after status epilepticus; green = no change in mRNA induction in chop-/- mice after status epilepticus). B. Table showing myelin-associated proteins up-regulated in chop-/- mice after status epilepticus detected by microarray (PBS = potential CHOP binding site). C. Cartoon showing putative binding sites for CHOP in the promoter region of the mbp gene (Mus musculus, myelin basic protein (Mbp), transcript variant 1, mRNA, NCBI Reference Sequence: NM_001025251.2). D. Graph showing decreased mbp mRNA in hippocampal tissue of wt mice when compared to vehicle injected control mice and chop-/- mice 6 h after status epilepticus. No significant change in mbp mRNA levels between vehicle wt mice and chop-/- mice after status epilepticus (n = 4 (Con wt and chop-/-) and 6 (SE wt). E. Western blot and F. graph showing increased hippocampal Mbp protein levels in epileptic chop-/- mice when compared to epileptic wt mice at day 14 post KA injection. β-actin was used as loading control (n = 5 (wt) and 4 (chop-/-)).
Discussion
In the present study we identified potential novel CHOP-regulated target genes by using mRNA microarray analysis of chop-deficient mice subjected to status epilepticus. Our study confirmed a robust increase in hippocampal CHOP protein after seizures and showed, in addition, a strong up-regulation of CHOP during the seizure free latent period [25]. Bioinformatic analysis identified cell death-promoting pathways but also cell death-related pathways to be altered in chop knockout mice. One of these pathways included several key proteins of the myelin sheath and further validation confirmed a blocked down-regulation of the myelin basic protein after seizures in the absence of CHOP. These results suggest that CHOP acts in a similar way to myelin-associated inhibitors suppressing the transcription of myelin-associated genes.
One of our first findings was the strong up-regulation of CHOP during the latent period (24 to 72 h post KA injection), during which mice normally show no seizure activity [25]. These results are in line with CHOP being upregulated by chronic stress of the endoplasmatic reticulum which has been shown to be up-regulated during epileptogenesis [35]. These results also imply CHOP as not simply an inducer of apoptosis because major cell death does not increase beyond 24 h post KA injection in our seizure model [17]. Moreover, CHOP protein has been reported to be increased in cell death-resistant brain regions after status epilepticus [17].
CHOP was initially described as a dominant negative factor, blocking the activation of other C/EBP proteins or transcription factors by forming heterodimers, but it can also bind to DNA as a heterodimer and induce transcription of target genes [1]. The fact that we found similar numbers of genes up-and down-regulated, with similar fold changes in chop-deficient mice after status epilepticus suggests that CHOP acts as both an inducer and blocker of transcription after seizures. In fact, previous results demonstrated that CHOP is able to target MDM2 and chop-deficient mice fail to induce MDM2 expression after status epilepticus in our model, thereby increasing p53 levels [17]. Our present results are consistent with CHOP being able to block the expression of many other genes, suggesting that CHOP has the ability to respond to the same stress stimulus by either up- or-down-regulating the expression of target genes. However, we do not know what percentage of up- or down-regulated target genes are direct targets of CHOP or a mere consequence of CHOP altering up-stream signalling such as altering the expression of transcription factors (e.g. p53 [17]). In depth analysis of gene pools showed that more functional pathways are altered among the up-regulated genes, which might be the consequence of indirect CHOP targeting. Interestingly, even though apoptotic pathway changes in chop-/- mice have been detected, they seem to be not the only major contributors to the overall gene changes observed. Apoptotic pathways were also mainly detected in the up-regulated gene pool, implying that CHOP mainly acts as a transcription inhibitor of cell death-related genes after seizures. Again, we cannot rule out additional effects of CHOP altering up-stream pathways such as the pro-apoptotic p53 pathway previously mentioned [17].
The most striking difference between up- and down-regulated genes in the chop-/- mice is the overrepresentation of genes involved in ion binding in the down-regulated gene pool. Interestingly, this list includes genes which regulate potassium channels (e.g. Kcnq2 (potassium voltage-gated channel, subfamily Q, member 2) and Kcnj6 (potassium inwardly-rectifying channel, subfamily J, member 6)), both of which have been implicated in epilepsy [36,37]) and the E3 ubiquitin ligase MID1 [38], which belongs to the same protein family as the E3 ubiquitin ligase MDM2, previously identified as a CHOP target gene in our model [17].
Among the up-regulated genes we identified genes involved in numerous processes with the most enriched gene sets being related to cell proliferation and development, regulation of gene expression, cell death and the generation of action potentials and axonal remodelling. Interestingly, previous mRNA microarray analysis of CHOP-overexpressing cells also identified genes involved in cell proliferation, development and apoptosis as one of the major pathways altered [2].
We focused our attention then to genes involved in axonal remodelling, as this is one of the main pathological changes during epileptogenesis [39] and identified here several genes involved in myelination. Demyelination is a pathological characteristic of animal epilepsy models and has been described in some types of epilepsy [31,32]. Here, the myelin basic protein Mbp seems to play a pivotal role. Myelin sheath damage seems to be an early pathological characteristic of epileptogenesis [31] and epileptic seizures have been associated with Mbp release from the damaged regions of the myelin sheaths [40]. In another study, Mbp expression was significantly lower in epileptic rats when compared to control rats [41]. Our results now extend these data showing a significant reduction in mbp mRNA induction after seizures. The expression of myelin-associated proteins is tightly regulated by transcription factors which induce their expression such as SP1 [42], Sox10 [43] or OLIG1 [44] and negative regulators of their expression (myelin associated inhibitors) such as Nogo-A, MAG or OMgp [45]. Critically, myelin-associated inhibitors have also been implicated in regulating axonal regeneration, plasticity and sprouting [45]. Our results suggest that CHOP can act as such a myelin-associated inhibitor, blocking the transcription of myelin-associated genes such as Mbp. Our results are in line with previous findings where chop deletion in a Charcot-Marie-Tooth 1B mouse model completely rescued motor deficit and reduced demyelination in the peripheral nerves [46] and with the observed demyelination after spinal cord injury following valproate treatment which decreased CHOP protein levels [33]. The decreased induction of myelin-associated proteins rather than a direct consequence of CHOP-regulated transcriptional repression might be a consequence of oligodentrocyte death as a response to CHOP overexpression. However, this is unlikely, as chop-deficiency has been reported to increase cell death after excitotoxicity [17] and CHOP overexpression led to increased oligodentrocyte survival [18].
An obvious caveat is that seizure-induced cell death is increased in chop-/- mice [17], therefore gene expression changes may be secondary to neurodegeneration. However, this is mitigated somewhat by the early sampling time used for the microarray analysis (6 h).
We do not know what the consequences are of a down-regulation of myelin-associated proteins on the epileptic phenotype. Mice deficient in mbd display spontaneous seizures [47] and mice deficient in mag showed increased susceptibility to kainic acid [48]. Therefore, it is tempting to speculate that the transcriptional repression of myelin-associated proteins through CHOP renders mice more susceptible to seizures and therefore CHOP over-expression has pro-epileptogenic potential. However, chop-deficient mice show an increased seizure frequency [17] and myelin-associated inhibitors have been involved in axonal plasticity and sprouting [45], processes believed to contribute to the epileptic phenotype [28,29]. Future experiments will have to examine the exact role of CHOP-induced demyelination, possibly by reducing genetically, either by cross breeding or viral infections, the gene load of mbp in chop-deficient mice.
In conclusion, the present work describes novel molecular pathways altered after seizures in the absence of chop and identified myelin-associated proteins as potential novel CHOP target genes. This study extends the potential role of CHOP beyond control of apoptosis and suggests that CHOP could be targeted to influence axonal regeneration or reorganisation.
Acknowledgements
The authors thank Prof David Henshall for critical reading of the manuscript and Prof Jose Lucas for kindly providing and Alicia Tomico for organizing shipment of chop-deficient mice. The authors would also like to thank Prof Marina Lynch for kindly providing the Mbd antibody. This work was sponsored by grants from the Health Research Board Ireland (HRA_2011/41 (TE)), BioAT (PRTU4 (Claire M)) and Science foundation Ireland (SFI 14/ADVRC2721 (Catherine M); 13/SIRG/2114 GR 14-0866 (EJ), SFI 13/SIRG/2114 (TE)).
Abbreviations
- CHOP
C/EBP homologous protein
- ER
Endoplasmatic reticulum
- Mbp
Myelin-binding protein
- SE
status epilepticus.
References
- 1.Ramji DP, Foka P. CCAAT/enhancer-binding proteins: structure, function and regulation. Biochem J. 2002;365:561–575. doi: 10.1042/BJ20020508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jauhiainen A, Thomsen C, Strombom L, Grundevik P, Andersson C, Danielsson A, Andersson MK, Nerman O, Rorkvist L, Stahlberg A, Aman P. Distinct cytoplasmic and nuclear functions of the stress induced protein DDIT3/CHOP/GADD153. PLoS One. 2012;7:e33208. doi: 10.1371/journal.pone.0033208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luethy JD, Holbrook NJ. Activation of the gadd153 promoter by genotoxic agents: a rapid and specific response to DNA damage. Cancer Res. 1992;52:5–10. [PubMed] [Google Scholar]
- 4.Wang XZ, Lawson B, Brewer JW, Zinszner H, Sanjay A, Mi LJ, Boorstein R, Kreibich G, Hendershot LM, Ron D. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153) Mol Cell Biol. 1996;16:4273–4280. doi: 10.1128/mcb.16.8.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Y, Guo Y, Tang J, Jiang J, Chen Z. New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim Biophys Sin (Shanghai) 2014;46:629–640. doi: 10.1093/abbs/gmu048. [DOI] [PubMed] [Google Scholar]
- 7.Maytin EV, Ubeda M, Lin JC, Habener JF. Stress-inducible transcription factor CHOP/gadd153 induces apoptosis in mammalian cells via p38 kinase-dependent and -independent mechanisms. Exp Cell Res. 2001;267:193–204. doi: 10.1006/excr.2001.5248. [DOI] [PubMed] [Google Scholar]
- 8.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 9.Shirakawa K, Maeda S, Gotoh T, Hayashi M, Shinomiya K, Ehata S, Nishimura R, Mori M, Onozaki K, Hayashi H, Uematsu S, Akira S, Ogata E, Miyazono K, Imamura T. CCAAT/enhancer-binding protein homologous protein (CHOP) regulates osteoblast differentiation. Mol Cell Biol. 2006;26:6105–6116. doi: 10.1128/MCB.02429-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang XZ, Kuroda M, Sok J, Batchvarova N, Kimmel R, Chung P, Zinszner H, Ron D. Identification of novel stress-induced genes downstream of chop. EMBO J. 1998;17:3619–3630. doi: 10.1093/emboj/17.13.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsukano H, Gotoh T, Endo M, Miyata K, Tazume H, Kadomatsu T, Yano M, Iwawaki T, Kohno K, Araki K, Mizuta H, Oike Y. The endoplasmic reticulum stress-C/EBP homologous protein pathway-mediated apoptosis in macrophages contributes to the instability of atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2010;30:1925–1932. doi: 10.1161/ATVBAHA.110.206094. [DOI] [PubMed] [Google Scholar]
- 12.Morse E, Schroth J, You YH, Pizzo DP, Okada S, Ramachandrarao S, Vallon V, Sharma K, Cunard R. TRB3 is stimulated in diabetic kidneys, regulated by the ER stress marker CHOP, and is a suppressor of podocyte MCP-1. Am J Physiol Renal Physiol. 2010;299:F965–972. doi: 10.1152/ajprenal.00236.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Puthalakath H, O’Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, Gotoh T, Akira S, Bouillet P, Strasser A. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 14.Galehdar Z, Swan P, Fuerth B, Callaghan SM, Park DS, Cregan SP. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J Neurosci. 2010;30:16938–16948. doi: 10.1523/JNEUROSCI.1598-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prasanthi JR, Larson T, Schommer J, Ghribi O. Silencing GADD153/CHOP gene expression protects against Alzheimer's disease-like pathology induced by 27-hydroxycholesterol in rabbit hippocampus. PLoS One. 2011;6:e26420. doi: 10.1371/journal.pone.0026420. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Tajiri S, Oyadomari S, Yano S, Morioka M, Gotoh T, Hamada JI, Ushio Y, Mori M. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 2004;11:403–415. doi: 10.1038/sj.cdd.4401365. [DOI] [PubMed] [Google Scholar]
- 17.Engel T, Sanz-Rodgriguez A, Jimenez-Mateos EM, Concannon CG, Jimenez-Pacheco A, Moran C, Mesuret G, Petit E, Delanty N, Farrell MA, O’Brien DF, Prehn JH, Lucas JJ, Henshall DC. CHOP regulates the p53-MDM2 axis and is required for neuronal survival after seizures. Brain. 2013;136:577–592. doi: 10.1093/brain/aws337. [DOI] [PubMed] [Google Scholar]
- 18.Gow A, Wrabetz L. CHOP and the endoplasmic reticulum stress response in myelinating glia. Curr Opin Neurobiol. 2009;19:505–510. doi: 10.1016/j.conb.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Southwood CM, Garbern J, Jiang W, Gow A. The unfolded protein response modulates disease severity in Pelizaeus-Merzbacher disease. Neuron. 2002;36:585–596. doi: 10.1016/s0896-6273(02)01045-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halterman MW, Gill M, DeJesus C, Ogihara M, Schor NF, Federoff HJ. The endoplasmic reticulum stress response factor CHOP-10 protects against hypoxia-induced neuronal death. J Biol Chem. 2010;285:21329–21340. doi: 10.1074/jbc.M109.095299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen CM, Wu CT, Chiang CK, Liao BW, Liu SH. C/EBP homologous protein (CHOP) deficiency aggravates hippocampal cell apoptosis and impairs memory performance. PLoS One. 2012;7:e40801. doi: 10.1371/journal.pone.0040801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murphy BM, Engel T, Paucard A, Hatazaki S, Mouri G, Tanaka K, Tuffy LP, Jimenez-Mateos EM, Woods I, Dunleavy M, Bonner HP, Meller R, Simon RP, Strasser A, Prehn JH, Henshall DC. Contrasting patterns of Bim induction and neuroprotection in Bim-deficient mice between hippocampus and neocortex after status epilepticus. Cell Death Differ. 2010;17:459–468. doi: 10.1038/cdd.2009.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hatazaki S, Bellver-Estelles C, Jimenez-Mateos EM, Meller R, Bonner C, Murphy N, Matsushima S, Taki W, Prehn JH, Simon RP, Henshall DC. Microarray profile of seizure damage-refractory hippocampal CA3 in a mouse model of epileptic preconditioning. Neuroscience. 2007;150:467–477. doi: 10.1016/j.neuroscience.2007.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mouri G, Jimenez-Mateos E, Engel T, Dunleavy M, Hatazaki S, Paucard A, Matsushima S, Taki W, Henshall DC. Unilateral hippocampal CA3-predominant damage and short latency epileptogenesis after intra-amygdala microinjection of kainic acid in mice. Brain Res. 2008;1213:140–151. doi: 10.1016/j.brainres.2008.03.061. [DOI] [PubMed] [Google Scholar]
- 26.Engel T, Tanaka K, Jimenez-Mateos EM, Caballero-Caballero A, Prehn JH, Henshall DC. Loss of p53 results in protracted electrographic seizures and development of an aggravated epileptic phenotype following status epilepticus. Cell Death Dis. 2010;1:e79. doi: 10.1038/cddis.2010.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sutula TP, Dudek FE. Unmasking recurrent excitation generated by mossy fiber sprouting in the epileptic dentate gyrus: an emergent property of a complex system. Prog Brain Res. 2007;163:541–563. doi: 10.1016/S0079-6123(07)63029-5. [DOI] [PubMed] [Google Scholar]
- 29.Buckmaster PS. Does mossy fiber sprouting give rise to the epileptic state? Adv Exp Med Biol. 2014;813:161–168. doi: 10.1007/978-94-017-8914-1_13. [DOI] [PubMed] [Google Scholar]
- 30.Pitkanen A, Nissinen J, Lukasiuk K, Jutila L, Paljarvi L, Salmenpera T, Karkola K, Vapalahti M, Ylinen A. Association between the density of mossy fiber sprouting and seizure frequency in experimental and human temporal lobe epilepsy. Epilepsia. 2000;41(Suppl 6):S24–29. doi: 10.1111/j.1528-1157.2000.tb01552.x. [DOI] [PubMed] [Google Scholar]
- 31.You Y, Bai H, Wang C, Chen LW, Liu B, Zhang H, Gao GD. Myelin damage of hippocampus and cerebral cortex in rat pentylenetetrazol model. Brain Res. 2011;1381:208–216. doi: 10.1016/j.brainres.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 32.Sherafat MA, Ronaghi A, Ahmad-Molaei L, Nejadhoseynian M, Ghasemi R, Hosseini A, Naderi N, Motamedi F. Kindling-induced learning deficiency and possible cellular and molecular involved mechanisms. Neurol Sci. 2013;34:883–890. doi: 10.1007/s10072-012-1142-6. [DOI] [PubMed] [Google Scholar]
- 33.Penas C, Verdu E, Asensio-Pinilla E, Guzman-Lenis MS, Herrando-Grabulosa M, Navarro X, Casas C. Valproate reduces CHOP levels and preserves oligodendrocytes and axons after spinal cord injury. Neuroscience. 2011;178:33–44. doi: 10.1016/j.neuroscience.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 34.Jahn O, Tenzer S, Werner HB. Myelin proteomics: molecular anatomy of an insulating sheath. Mol Neurobiol. 2009;40:55–72. doi: 10.1007/s12035-009-8071-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chihara Y, Ueda Y, Doi T, Willmore LJ. Role of endoplasmic reticulum stress in the amygdaloid kindling model of rats. Neurochem Res. 2011;36:1834–1839. doi: 10.1007/s11064-011-0501-7. [DOI] [PubMed] [Google Scholar]
- 36.Meisler MH, O’Brien JE, Sharkey LM. Sodium channel gene family: epilepsy mutations, gene interactions and modifier effects. J Physiol. 2010;588:1841–1848. doi: 10.1113/jphysiol.2010.188482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wickenden AD. Potassium channels as anti-epileptic drug targets. Neuropharmacology. 2002;43:1055–1060. doi: 10.1016/s0028-3908(02)00237-x. [DOI] [PubMed] [Google Scholar]
- 38.Trockenbacher A, Suckow V, Foerster J, Winter J, Krauss S, Ropers HH, Schneider R, Schweiger S. MID1, mutated in Opitz syndrome, encodes an ubiquitin ligase that targets phosphatase 2A for degradation. Nat Genet. 2001;29:287–294. doi: 10.1038/ng762. [DOI] [PubMed] [Google Scholar]
- 39.Sutula T. Seizure-Induced Axonal Sprouting: Assessing Connections Between Injury, Local Circuits, and Epileptogenesis. Epilepsy Curr. 2002;2:86–91. doi: 10.1046/j.1535-7597.2002.00032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen J, Wang X, Zhou S, Wang R, Zhang Y, Li C, Zhou D. [The serum MBP, myelin sheath immunohistochemistry and electromicroscope of expetrimental epilepsy rats] . Hua Xi Yi Ke Da Xue Xue Bao. 1996;27:236–239. [PubMed] [Google Scholar]
- 41.Ye Y, Xiong J, Hu J, Kong M, Cheng L, Chen H, Li T, Jiang L. Altered hippocampal myelinated fiber integrity in a lithium-pilocarpine model of temporal lobe epilepsy: a histopathological and stereological investigation. Brain Res. 2013;1522:76–87. doi: 10.1016/j.brainres.2013.05.026. [DOI] [PubMed] [Google Scholar]
- 42.Wei Q, Miskimins WK, Miskimins R. The Sp1 family of transcription factors is involved in p27(Kip1)-mediated activation of myelin basic protein gene expression. Mol Cell Biol. 2003;23:4035–4045. doi: 10.1128/MCB.23.12.4035-4045.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wei Q, Miskimins WK, Miskimins R. Sox10 acts as a tissue-specific transcription factor enhancing activation of the myelin basic protein gene promoter by p27Kip1 and Sp1. J Neurosci Res. 2004;78:796–802. doi: 10.1002/jnr.20342. [DOI] [PubMed] [Google Scholar]
- 44.Li H, Lu Y, Smith HK, Richardson WD. Olig1 and Sox10 interact synergistically to drive myelin basic protein transcription in oligodendrocytes. J Neurosci. 2007;27:14375–14382. doi: 10.1523/JNEUROSCI.4456-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Akbik F, Cafferty WB, Strittmatter SM. Myelin associated inhibitors: a link between injury-induced and experience-dependent plasticity. Exp Neurol. 2012;235:43–52. doi: 10.1016/j.expneurol.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pennuto M, Tinelli E, Malaguti M, Del Carro U, D’Antonio M, Ron D, Quattrini A, Feltri ML, Wrabetz L. Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot-Marie-Tooth 1B mice. Neuron. 2008;57:393–405. doi: 10.1016/j.neuron.2007.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Popko B, Puckett C, Lai E, Shine HD, Readhead C, Takahashi N, Hunt SW 3rd, Sidman RL, Hood L. Myelin deficient mice: expression of myelin basic protein and generation of mice with varying levels of myelin. Cell. 1987;48:713–721. doi: 10.1016/0092-8674(87)90249-2. [DOI] [PubMed] [Google Scholar]
- 48.Lopez PH, Ahmad AS, Mehta NR, Toner M, Rowland EA, Zhang J, Dore S, Schnaar RL. Myelin-associated glycoprotein protects neurons from excitotoxicity. J Neurochem. 2011;116:900–908. doi: 10.1111/j.1471-4159.2010.07069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]