

Abstract
The correlation between calcium-activated chloride channel (CaCC) and pulmonary arterial hypertension (PAH) induced by high pulmonary blood flow remains uncertain. In this study, we investigated the possible role and effects of CaCC in this disease. Sixty rats were randomly assigned to normal, sham, and shunt groups. Rats in the shunt group underwent abdominal aorta and inferior vena cava shunt surgery. The pulmonary artery pressure was measured by catheterization. Pathological changes, right ventricle hypertrophy index (RVHI), arterial wall area/vessel area (W/V), and arterial wall thickness/vessel external diameter (T/D) were analyzed by optical microscopy. Electrophysiological characteristics of pulmonary arterial smooth muscle cells (PASMCs) were investigated using patch clamp technology. After 11 weeks of shunting, PAH and pulmonary vascular structural remodeling (PVSR) developed, accompanied by increased pulmonary pressure and pathological interstitial pulmonary changes. Compared with normal and sham groups, pulmonary artery pressure, RVHI, W/V, and T/D of the shunt group rats increased significantly. Electrophysiological results showed primary CaCC characteristics. Compared with normal and sham groups, membrane capacitance and current density of PASMCs in the shunt group increased significantly, which were subsequently attenuated following chloride channel blocker niflumic acid (NFA) treatment. To conclude, CaCC contributed to PAH induced by high pulmonary blood flow and may represent a potential target for treatment of PAH.
Keywords: Calcium-activated chloride channel, pulmonary arterial hypertension, pulmonary vascular structural remodeling
Introduction
Pulmonary artery hypertension (PAH) is clinically characterized by increased pulmonary arterial pressure (PAP) in the absence of elevated left heart pressure [1]. Maladaptive changes to the pulmonary arterioles result in pulmonary vascular dysfunction, right ventricular (RV) pressure loading, and ultimately right heart failure. According to the recently updated clinical classification of pulmonary hypertension by the American College of Cardiology, both incidence and complexity of PAH due to congenital heart disease (CHD) continues to increase [2]. It is estimated that 10% of adults with CHD also have PAH [3]. Occurrence of PAH significantly contributes to a worsening prognosis of patients suffering from systemic-to-pulmonary shunting CHD.
A better understanding of the underlying mechanisms and pathophysiological processes of PAH due to high pulmonary blood flow could lead to novel therapies for a more effective management of this disease [4]. The cornerstone histopathological feature of PAH is adverse remodeling of the distal pulmonary arterioles, which is characterized by intimal fibrosis and thickening, dysregulated proliferation of apoptosis-resistant pulmonary artery endothelial cells and pulmonary artery smooth muscle cells (PASMCs), and genesis of plexogenic lesions in certain types of PAH [5]. Membrane depolarization and increased [Ca2+]i in pulmonary artery smooth muscle cells (PASMCs) are known to play a pivotal role in the induction of PASMC proliferation and increase in pulmonary arterial tone associated with PAH [6,7]. The activated voltage-dependent calcium channel is associated with the excitation-contraction coupling process and the final contraction of smooth muscles and pulmonary vessels [8]. The calcium-activated chloride channel (CaCC), one of the most vital ion channels, plays an important role in several cellular functions [9]. It plays a key role in VSM, providing a triggering mechanism during signal transduction for membrane excitability and osmotic balance. Initiation of CaCC leads to membrane depolarization and muscle contraction [10]. However, the correlation between CaCC and PAH induced by elevated pulmonary blood flow velocity remains unclear and requires further investigation. The potential electrophysiological characteristics of CaCC in PAH induced by CHD deserve further exploration. The current study was designed to explore the potential role of CaCC in high pulmonary blood flow induced PAH and the effect of niflumic acid (NFA), an inhibitor of CaCC, on the electrophysiology of PASMCs.
Materials and methods
Animal model of pulmonary hypertension
Animal experiments were conducted in accordance with the Guide to the Care and Use of Experimental Animals® issued by the Ministry of Health of the People’s Republic of China. Male Sprague Dawley® rats, weighing 180-230 g, were provided by the Animal Research Centre of Guangxi Medical University (Nanning, Guangxi, China; license No. SCXK 2009-0002). Animals were housed in plastic cages in a controlled room with 40% humidity, a temperature of 22°C, and a daily light cycle of 06:00-18:00. The systematic-to-pulmonary shunt model was established by abdominal aorta-inferior vena cava shunting surgery according to a method previously described by Garcia and Diebold [11] with some modifications [12]. Sixty rats were randomly assigned to three groups as follows: Normal Group (n = 20), Sham Group (n = 20), and Shunt Group (n = 20).
Pentobarbital sodium (0.25%, 40 mg/kg, intraperitoneal injection) was used to anesthetize Sham and Shunt Group rats. The abdominal aorta and inferior vena cava were exposed, followed by a vascular bulldog clamp placed across the aorta caudal to the left renal artery. The aorta wall was then punctured at the union of the segment one-third cephalic to the aortic bifurcation and two-thirds caudal to the renal artery using an 18-gauge disposable needle. The needle was slowly withdrawn and the wound was stitched using a 9-0 silk thread. Animals in the Sham Group underwent the same experimental procedure except for the shunt. All animals were raised for 11 weeks as previously described [13].
Measurement of pulmonary pressure parameters and right ventricle hypertrophy index (RVHI)
After the animals were anesthetized with pentobarbital sodium (40 mg/kg intraperitoneal injection), a silicone catheter (0.9 mm outer diameter) was introduced into the right jugular vein, passing the tricuspid valve and right ventricle, and into the pulmonary artery. A Biopac MP 100 transducer (BIOPAC Systems Inc., Goleta, CA, USA) was connected to the other end of the catheter. The position of the catheter was determined by the typical wave form of the pressure. The systolic pulmonary artery pressure (SPAP) and mean pulmonary artery pressure (MPAP) were recorded simultaneously. After removal of the heart, the right (RV) and left ventricles (LV) as well as the septum (SP) were dissected and weighed using an electronic scale. The right ventricle hypertrophy index (RVHI) was calculated using the ratio of the wet weight of the right ventricle to left ventricle plus septum [RV/(LV+SP)].
Sample preparation and morphological analysis
For each animal, a small section of lung tissue was dissected from the upper part of the left lung lobe, fixed in 10% paraformaldehyde, dehydrated, embedded in paraffin, and sectioned (4-6 µm). Morphological observation and analysis were carried out by hematoxylin-eosin (H&E) staining and a video-linked microscope digitizing board system (Q550CW; Leica, Wetzler, Germany). Pulmonary arterioles with a diameter range of 50-150 μm were selected (3-5 per animal) and used to estimate the arterial wall area/vessel area (W/V) and arterial wall thickness/vessel external diameter (T/D).
Cell isolation and electrophysiology
The technique described by Sun et al. [4] was used with slight modification, to isolate smooth muscle cells. In brief, lungs were quickly removed and immersed in ice-cold HEPES buffered salt solution (HBSS) containing 130 M NaCl, 5 M KCl, 1.2 MgCl2, 1.5 CaCl2, 10 HEPES, and 10 mM glucose (pH = 7.2 with NaOH). The intrapulmonary arteriole samples were prepared for enzymatic cell isolation by clearing endothelium (by rubbing the luminal surface with a cotton swab) and cutting into 1×1 mm pieces. The arteries were then digested at 37°C for 20 min in 20 μmol Ca2+ HBSS containing 1,750 U/mL type I collagenase, 9.6 U/mL papain, 2 mg/mL bovine serum albumin, and 1 mmol/L DTT. After washing with nominal Ca2+ free HBSS, the PASMCs were mechanically dispersed and kept at 4°C in the Ca2+ free HBSS for use within 6 hours after isolation.
Whole cell currents were recorded by an EPC10 amplifier (HEKA Electronik, Lambrecht/Pfalz, Germany) with pCLAMP 9 software (Axon Instruments, Inc. USA) using the patch clamp technique. Patch pipettes (6-9 MΩ when filled with pipette solution) were fabricated on a Sutter micropipette puller (Sutter Instruments, Novato, CA, USA). Step-pulse protocols and data acquisition were performed by the digital interface of the puller by computer interface. The whole-cell configuration of the patch clamp technique was performed with the objective of recording all currents; these were filtered at 3 kHz and sampled at 10 kHz unless stated otherwise. Current density (Id) versus voltage relationships was constructed by measuring the current at the end of 1 second voltage steps from -100 to +130 mV in 10 mV increments elicited every 5 seconds. Holding potential was -50 mV. All experiments were performed at room temperature (20-24°C).
Solutions and reagents for electrophysiology
A coverslip containing 0.75 mL of the cells was positioned in the recording chamber and superfused with 2-3 mL/min of the bath solution. For recording ICI, the extracellular bath solution contained 141 mM NaCl; 4.7 mM KCl; 1.8 mM CaCl2; 1.2 mM MgCl2; 10 mM N-2-hydroxyethylpiperazine-N’-2-ethanesulfonic acid (HEPES); and 10 mM glucose buffered to pH = 7.4 with 5 M NaOH. The pipette solution for whole cell ICl recording contained 135 mM CsCl, 4 mM MgCl2, 10 mM N-2-hydroxyethylpiperazine-N’-2-ethanesulfonic acid (HEPES), 5 mM Na2ATP, and 10 mM tetraethylammonium chloride (TEAC), buffered to pH 7.2 with CsOH. NFA (Sigma-Aldrich Corp., St. Louis, MO, USA) was prepared as a 300 mM stock solution in dimethyl sulfoxide. Aliquots of the stock solution were diluted into the bath solution to produce a final concentration of 100 μM. TEAC (Sigma-Aldrich) and Na2ATP were directly dissolved into the superfusate to suppress the calcium-activated potassium ion channel and ATP (adenosine triphosphate) sensitive potassium ion channel, respectively, on the day of use.
Statistical analyses
Student’s t test and analysis of variance were performed to evaluate the statistical significance of the differences between two means, multiple means, and two means at multiple test voltages as appropriate. Pearson’s correlation analysis was performed to evaluate the correlation between the variants. Pooled data were expressed as means ± standard error and statistically analyzed using IBM SPSS version 13.0 (SPSS Inc., Chicago, IL, USA). A value of P < 0.05 was considered statistically significant.
Results
General characteristics of animals and lungs
The rats in the sham groups recovered well following surgery, both normal and sham groups showing increased body weight while shunt group rats showed fatigue and diminished appetite. Except for one rat in shunt group died as a result of an intestinal obstruction, the remaining rats survived. The lungs of both normal and sham rats presented with a lighter red color and smoother surface with more elasticity compared with shunt group rats.
PSAP, MPAP, and RVHI
The waveforms of the pressure from the right atrium to the pulmonary artery were divided into three parts, as shown in Figure 1. SPAP increased significantly in the rats of the shunt group compared with that of the normal (P < 0.01) and sham groups (P < 0.01); however, the SPAP between the normal and sham groups did not significantly differ (P > 0.05). MPAP increased significantly in rats of the shunt group compared with that of the normal (P < 0.01) and sham groups (P < 0.01) while MPAP between the normal and sham groups was not significantly different (P > 0.05). RVHI was higher in rats of the shunt group compared to that of the normal (P < 0.01) and sham groups (P < 0.01). However, the normal and sham groups showed no statistically significant difference regarding RVHI (Table 1).
Figure 1.
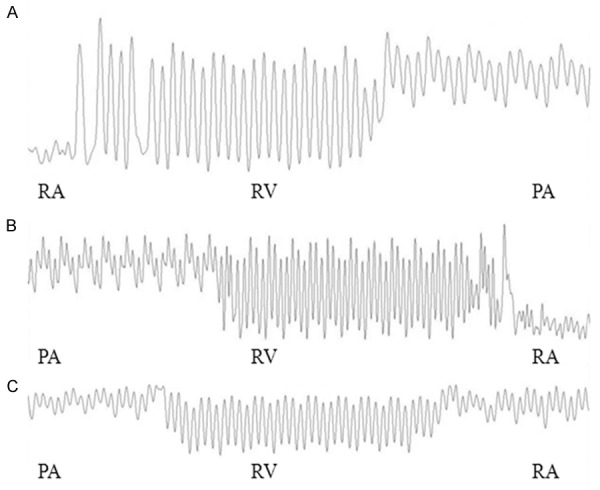
Waveforms of the RA, RV, and PA in the normal group (A), sham group (B) and shunt group (C). (Speed = 2 s/div). Abbreviations: RA, right atrium; RV, right ventricle; PA, pulmonary artery; s/div, seconds per division.
Table 1.
Changes of SPAP, MPAP and RVHI in rats of different groups
| Group | N | SPAP | MPAP | RVHI |
|---|---|---|---|---|
| Normal | 20 | 17.323 ± 2.064 | 16.722 ± 3.159 | 0.238 ± 0.019 |
| Sham | 20 | 18.787 ± 2.355 | 17.054 ± 1.755 | 0.236 ± 0.015 |
| Shunt | 19 | 32.217 ± 8.412*,# | 24.308 ± 5.239*,# | 0.276 ± 0.042*,# |
Notes: Mean ± SD.
P < 0.01 vs. normal group.
P < 0.01 vs. sham group.
Abbreviations: N, number; SPAP, systolic pulmonary artery pressure; MPAP, mean pulmonary artery pressure; RVHI, right ventricle hypertrophy index.
Pathological changes of the pulmonary mesenchyme
After 11 weeks of shunting, microscopic analysis of structure characteristic showed thickening and swelling of the pulmonary artery’s tunica media and alveolar septum, stenosis of the arterial lumen, presence of hypertrophic endothelial cells, and obvious infiltration of inflammatory cells in the pulmonary mesenchymal cells. Meanwhile, the pulmonary arterial walls in normal and sham group rats presented clearly with high integrity, without arterial tunica media thickening and pulmonary mesenchymal inflammation (Figures 2, 3).
Figure 2.
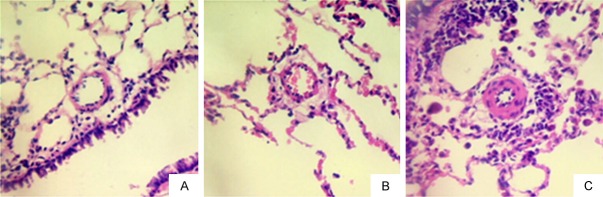
Changes in small pulmonary arteries after 11 weeks. (H&E staining, 400 ×) (A) normal group; (B) sham group; (C) shunt group.
Figure 3.
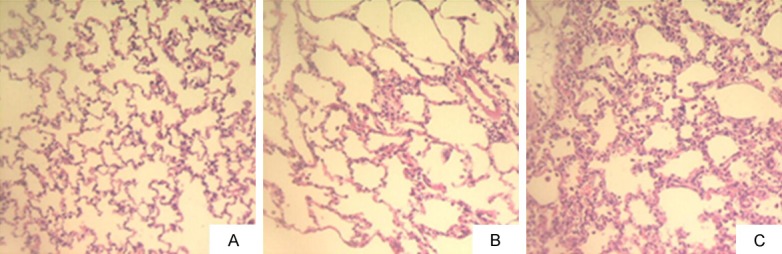
Changes in pulmonary alveoli after 11 weeks. (H&E staining, 200 ×). (A) normal group; (B) sham group; (C) shunt group.
As shown in Table 2, the proportion of arterial wall area/vessel area (W/V) and arterial wall thickness/vessel external diameter (T/D) in the shunt group increased significantly compared with that of normal and sham groups (all P < 0.01). Normal and sham groups did not differ significantly regarding W/V and T/D.
Table 2.
Changes in W/V and T/D in rats of different groups
| Group | N | W/V | T/D |
|---|---|---|---|
| Normal | 20 | 59.67 ± 4.74 | 40.00 ± 4.29 |
| Sham | 20 | 63.86 ± 4.09 | 44.00 ± 5.65 |
| Shunt | 19 | 85.83 ± 5.89*,# | 63.86 ± 10.82*,# |
Mean ± SD.
P < 0.01 vs. normal group.
P < 0.01 vs. sham group.
Abbreviations: N, number; W/V, arterial wall area/vessel area; T/D, arterial wall thickness/vessel external diameter.
Electrophysiological characteristic changes
The pulmonary artery smooth muscle cells exhibited an initial transient current (Itr) followed by a time-dependent current (Itd) that reversed to near equilibrium potential (ECl, estimated ECl = -1 mV). The currents presented the characteristics of outward rectification. The repolarizing voltage steps produced a large, long-lasting inward tail current (Itail) that also reversed potential close to that of the calculated ECl (Figure 4).
Figure 4.
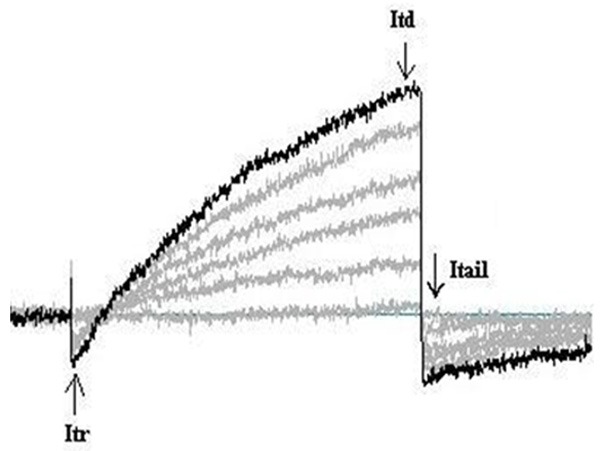
Typical types of currents elicited by depolarizing pulmonary artery smooth muscle cells.
As shown in Figure 5 and Table 3, membrane capacity (Cm) and current density (Id) in the shunt group increased significantly compared with that of normal and sham groups (all P < 0.05). The normal and sham groups did not differ significantly in Cm and Id (all P > 0.05).
Figure 5.
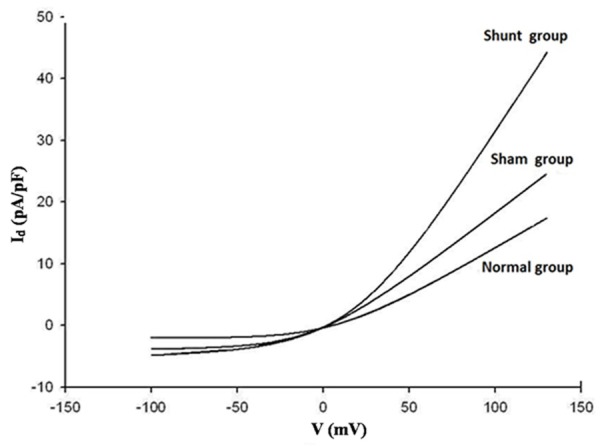
CaCC Current density-voltage curve of normal, sham and shunt groups.
Table 3.
Changes in Cm and ICl(Ca) in rats of different groups
| Group | Cm (pF) | Id (pA/pF) |
|---|---|---|
| Normal | 9.47 ± 1.51 | 11.34 ± 4.00 |
| Sham | 9.31 ± 1.41 | 14.30 ± 5.76 |
| Shunt | 13.75 ± 3.63*,# | 28.22 ± 10.75*,# |
Mean ± SD.
P < 0.05 vs. normal group.
P < 0.05 vs. sham group.
Abbreviations: Cm, membrane capacitance; Id, current density.
When treated with 100 μM NFA in a bath solution, CaCC currents significantly decreased in both normal and shunt groups. NFA almost completely eliminated the time-dependent outward and tail currents in PASMC. The current amplitude of CaCC in the shunt group decreased from 41.07 ± 2.34 pA/pF to 3.66 ± 0.24 pA/pF at 130 mV (n = 5, t = 31.788, P < 0.01). The curves of the changes are shown in Figures 6 and 7.
Figure 6.
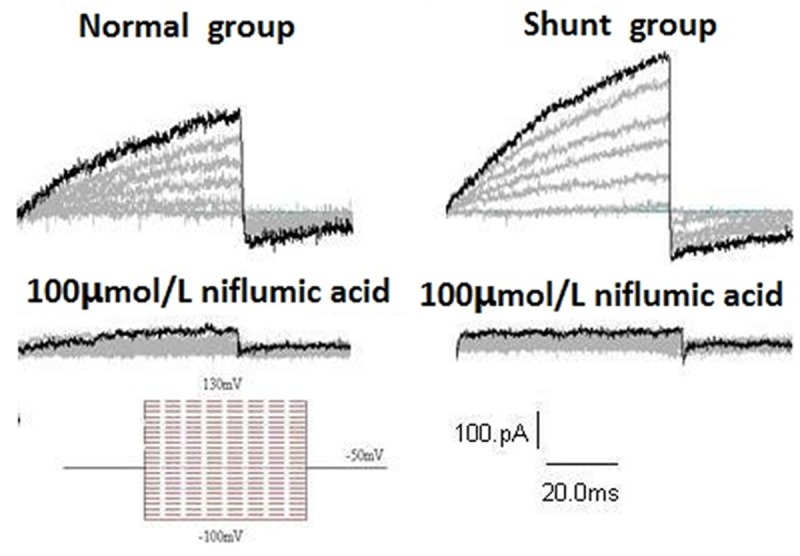
Representative recordings from normal and shunt cells in the absence or presence of 100 μM niflumic acid (NFA).
Figure 7.
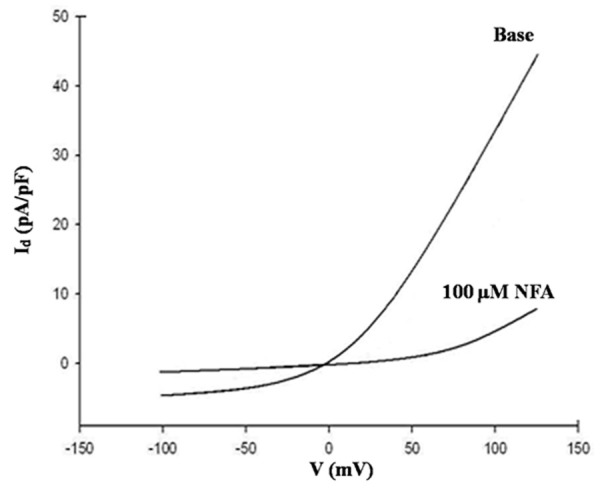
Inhibitory effect of NFA on CaCC current density in the shunt group.
Correlation analysis showed that the change in the MPAP of PASMCs was positively correlated with RVHI, Cm, and Id, as shown in Table 4.
Table 4.
Correlation between MPAP, RVHI, Cm, and Id
| Correlation | r | P |
|---|---|---|
| MPAP vs. RVHI | 0.826 | 0.00 |
| MPAP vs. Cm | 0.663 | 0.00 |
| MPAP vs. Id | 0.684 | 0.04 |
Abbreviations: Cm, membrane capacitance; Id, current density; r, correlation coefficient; P, probability.
Discussion
In the current study, SPAP and MPAP increased significantly after 11 weeks of shunting, suggesting that the rat model of high pulmonary blood flow induced PAH had been successfully established, as shown by other similar studies [11,12]. In addition, the RVHI, W/V and T/D were found to have increased significantly. The microstructure showed thickening and swelling of the pulmonary artery’s tunica media and alveolar septum, stenosis of the arterial lumen, presence of hypertrophic endothelial cells, and inflammation of the pulmonary mesenchymal cells. Furthermore, Cm and Id in the shunt group increased significantly. MPAP was closely correlated to RVHI, Cm, and Id. When treated with NFA, the current amplitude depressed significantly.
PVSR is the pathological basis of PAH [14]. The mechanisms responsible for PVSR and PAH induced by high pulmonary blood flow have not been fully elucidated. The vascular wall is formed by endothelial and smooth muscle cells, as well as fibroblasts that interact to form an autocrine-paracrine complex. Remodeling of the vascular wall is the result of changes in cellular and noncellular components depending on the disease process, during which changes in the growth and migration of vascular smooth muscle cells, endothelial dysfunction, inflammatory processes, and the synthesis or degradation of extracellular matrix components may be present [15]. Studies have shown that during the high pulmonary blood flow induced PAH that fluid mechanical forces, especially shear stress, result in endothelial and smooth muscle dysfunction as a vascular structure remodeling process, resulting in PAH [16,17]. In the current study, results identified the main characteristics of PVSR of small pulmonary arteries as well as the development process of PAH.
The hypothesis of inflammatory and endothelial dysfunction is involved during the pathophysiology of PAH; the concept of vascular inflammatory disease allows us to develop a new approach for risk stratification and treatment of PAH. Clinical treatment modalities and lifestyle modifications such as diet and physical activity can result in important benefits, which in turn can result in reductions in inflammatory processes [18]. In an experimental model of metabolic syndrome in hypertensive rats receiving a fructose-rich diet, researchers showed the presence of endothelial cells and products of NF-κB signaling and the vascular cell adhesion molecule (VCAM)-1 [19]. In the current study, we also showed the occurrence of an inflammatory pathological morphology during the PAH process as a result of high pulmonary blood flow. However, the potential mechanisms responsible for the inflammatory process of PAH require further investigation.
Ion channels in cell membranes play a significant role in regulating VSMC tone, and CaCC is one such ion channel. It is activated by Ca2+ released from ryanodine receptors located in the sarcoplasmic reticulum and is responsible for the passive flow of Cl- in and out of the cell subsequent to depolarizing of the membrane and muscle contraction [8]. CaCC possesses the following properties: anion selectivity, submicromolar sensitivity to intracellular Ca2+ concentration, voltage activation at low Ca2+ concentrations, and inhibition by the pharmacological agent NFA [20]. In the current study, the currents exhibited the main characteristics of Cl- currents. Similarly to other studies [4,10], the reversal potential of Ca2+-activated currents (~-1 mV) in our ionic conditions was close to the equilibrium potential for Cl-. PAH animal models have shown that [Ca2+]i increases compared to normal pulmonary pressure, resulting in membrane depolarization, combined with elevated levels of vasoconstrictors such as 5-HT and endothelial-1 [21]. The current study found that Cm and Id increased significantly in the shunt group, demonstrating that PASMCs under high pulmonary blood flow conditions resulted in increased amplitude of Cl- outwards currents, thereby leading to membrane depolarization and final PASMCs contraction. In addition, correlation analysis showed that the change in MPAP of PASMCs was positively correlated with RVHI, Cm, and Id. These results suggest that increased CaCC activity in PASMCs may contribute to elevated pulmonary pressure and the formation of PAH induced by high pulmonary blood flow.
We also treated PASMCs with NFA, a chloride channel blocker, and showed that the current amplitude and density of CaCC significantly decreased without change in the equilibrium potential; similar results have been reported in a rabbit model [22]. NFA showed an enhanced chloride channel effect in noradrenaline-induced contractions and basal vessel tone in PAH rats, resulting in an increased influx of extracellular Ca2+ via L-type Ca2+ channels and membrane depolarization [21]. We speculate that the mechanisms of action of NFA could be due to reduced calcium-activated chloride current (ICl(Ca)) and Cl- efflux, resulting in decreased Ca2+ influx and final vessel contraction. Moreover, being a non-steroidal anti-inflammatory drug, NFA could potentially alleviate inflammatory reaction, which in turn could attenuate the complete pathological process. NFA has also been shown to cause inhibition as well as paradoxical stimulation of a sustained ICl(Ca) in the myocytes of rabbit pulmonary arteries [23]. Additional research concerning the potential mechanisms of NFA involving the PAH process are required.
However, there remain some limitations to the current study. First, due to technological difficulties during cell sealing, we only tested the effects of one concentration of NFA; additional gradients will be required in our future studies. Second, the effect of Itail still requires consideration during electrophysiology. Finally, NFA is not specific to Cl- conductance; the molecular nature of this channel during pulmonary arterial hyperreactivity in PAH remains only partially understood and as a result, requires further study [24-26].
In conclusion and based on our demonstration and results, CaCC contributed to elevated pulmonary artery pressure and final formation of PAH induced by high pulmonary blood flow and may represent a potential target for treatment of PAH.
Acknowledgements
This work was supported by National Natural Science Foundation of China (81160040) and Guangxi Natural Science Foundation (2013GXNSFAA019177). All the authors have no interest or conflict of interest in association with this work.
Disclosure of conflict of interest
None.
References
- 1.Chan SY, Loscalzo J. Pathogenic mechanisms of pulmonary arterial hypertension. J Mol Cell Cardiol. 2008;44:14–30. doi: 10.1016/j.yjmcc.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Krishna Kumar R, Landzberg M, Machado RF, Olschewski H, Robbins IM, Souza R. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D34–41. doi: 10.1016/j.jacc.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 3.Engelfriet PM, Duffels MG, Moller T, Boersma E, Tijssen JG, Thaulow E, Gatzoulis MA, Mulder BJ. Pulmonary arterial hypertension in adults born with a heart septal defect: the Euro Heart Survey on adult congenital heart disease. Heart. 2007;93:682–687. doi: 10.1136/hrt.2006.098848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun H, Xia Y, Paudel O, Yang XR, Sham JS. Chronic hypoxia-induced upregulation of Ca2+-activated Cl- channel in pulmonary arterial myocytes: a mechanism contributing to enhanced vasoreactivity. J Physiol. 2012;590:3507–3521. doi: 10.1113/jphysiol.2012.232520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 6.Shimoda LA, Sham JS, Shimoda TH, Sylvester JT. L-type Ca(2+) channels, resting [Ca(2+)] (i), and ET-1-induced responses in chronically hypoxic pulmonary myocytes. Am J Physiol Lung Cell Mol Physiol. 2000;279:L884–894. doi: 10.1152/ajplung.2000.279.5.L884. [DOI] [PubMed] [Google Scholar]
- 7.Rodat L, Savineau JP, Marthan R, Guibert C. Effect of chronic hypoxia on voltage-independent calcium influx activated by 5-HT in rat intrapulmonary arteries. Pflugers Arch. 2007;454:41–51. doi: 10.1007/s00424-006-0178-y. [DOI] [PubMed] [Google Scholar]
- 8.Leblanc N, Ledoux J, Saleh S, Sanguinetti A, Angermann J, O’Driscoll K, Britton F, Perrino BA, Greenwood IA. Regulation of calcium-activated chloride channels in smooth muscle cells: a complex picture is emerging. Can J Physiol Pharmacol. 2005;83:541–556. doi: 10.1139/y05-040. [DOI] [PubMed] [Google Scholar]
- 9.Ni YL, Kuan AS, Chen TY. Activation and Inhibition of TMEM16A Calcium-Activated Chloride Channels. PLoS One. 2014;9:e86734. doi: 10.1371/journal.pone.0086734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manoury B, Tamuleviciute A, Tammaro P. TMEM16A/Anoctamin 1 protein mediates calcium-activated chloride currents in pulmonary arterial smooth muscle cells. J Physiol. 2010;588:2305–2314. doi: 10.1113/jphysiol.2010.189506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia R, Diebold S. Simple, rapid, and effective method of producing aortocaval shunts in the rat. Cardiovasc Res. 1990;24:430–432. doi: 10.1093/cvr/24.5.430. [DOI] [PubMed] [Google Scholar]
- 12.Ocampo C, Ingram P, Ilbawi M, Arcilla R, Gupta M. Revisiting the surgical creation of volume load by aorto-caval shunt in rats. Mol Cell Biochem. 2003;251:139–143. [PubMed] [Google Scholar]
- 13.Yan H, Du J, Tang C. The possible role of hydrogen sulfide on the pathogenesis of spontaneous hypertension in rats. Biochem Biophys Res Commun. 2004;313:22–27. doi: 10.1016/j.bbrc.2003.11.081. [DOI] [PubMed] [Google Scholar]
- 14.Li XH, Du JB, Bu DF, Tang XY, Tang CS. Sodium hydrosulfide alleviated pulmonary vascular structural remodeling induced by high pulmonary blood flow in rats. Acta Pharmacol Sin. 2006;27:971–980. doi: 10.1111/j.1745-7254.2006.00353.x. [DOI] [PubMed] [Google Scholar]
- 15.Renna NF, de Las Heras N, Miatello RM. Pathophysiology of vascular remodeling in hypertension. Int J Hypertens. 2013;2013:808353. doi: 10.1155/2013/808353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bongrazio M, Baumann C, Zakrzewicz A, Pries AR, Gaehtgens P. Evidence for modulation of genes involved in vascular adaptation by prolonged exposure of endothelial cells to shear stress. Cardiovasc Res. 2000;47:384–393. doi: 10.1016/s0008-6363(00)00111-5. [DOI] [PubMed] [Google Scholar]
- 17.Li X, Du J, Jin H, Tang X, Bu D, Tang C. The regulatory effect of endogenous hydrogen sulfide on pulmonary vascular structure and gasotransmitters in rats with high pulmonary blood flow. Life Sci. 2007;81:841–849. doi: 10.1016/j.lfs.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 18.Libby P. Inflammation and cardiovascular disease mechanisms. Am J Clin Nutr. 2006;83:456S–460S. doi: 10.1093/ajcn/83.2.456S. [DOI] [PubMed] [Google Scholar]
- 19.Viedt C, Vogel J, Athanasiou T, Shen W, Orth SR, Kübler W, Kreuzer J. Monocyte chemoattractant protein-1 induces proliferation and interleukin-6 production in human smooth muscle cells by differential activation of nuclear factor-κB and activator protein-1. Arterioscler Thromb Vasc Biol. 2002;22:914–920. doi: 10.1161/01.atv.0000019009.73586.7f. [DOI] [PubMed] [Google Scholar]
- 20.Berg J, Yang H, Jan LY. Ca2+-activated Cl- channels at a glance. J Cell Sci. 2012;125:1367–1371. doi: 10.1242/jcs.093260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guibert C, Marthan R, Savineau JP. Modulation of ion channels in pulmonary arterial hypertension. Curr Pharm Des. 2007;13:2443–2455. doi: 10.2174/138161207781368585. [DOI] [PubMed] [Google Scholar]
- 22.Wiwchar M, Ayon R, Greenwood IA, Leblanc N. Phosphorylation alters the pharmacology of Ca(2+)-activated Cl channels in rabbit pulmonary arterial smooth muscle cells. Br J Pharmacol. 2009;158:1356–1365. doi: 10.1111/j.1476-5381.2009.00405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ledoux J, Greenwood IA, Leblanc N. Dynamics of Ca2+-dependent Cl- channel modulation by niflumic acid in rabbit coronary arterial myocytes. Mol Pharmacol. 2005;67:163–173. doi: 10.1124/mol.104.004168. [DOI] [PubMed] [Google Scholar]
- 24.Oriowo MA. Chloride channels and alpha1-adrenoceptor-mediated pulmonary artery smooth muscle contraction: effect of pulmonary hypertension. Eur J Pharmacol. 2004;506:157–163. doi: 10.1016/j.ejphar.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 25.Nakazawa H, Hori M, Murata T, Ozaki H, Karaki H. Contribution of chloride channel activation to the elevated muscular tone of the pulmonary artery in monocrotaline-induced pulmonary hypertensive rats. Jpn J Pharmacol. 2001;86:310–315. doi: 10.1254/jjp.86.310. [DOI] [PubMed] [Google Scholar]
- 26.Duran C, Hartzell HC. Physiological roles and diseases of Tmem16/Anoctamin proteins: are they all chloride channels? Acta Pharmacol Sin. 2011;32:685–692. doi: 10.1038/aps.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]