

Abstract
Aims: To investigate the involvement of JARID1B histone methyltransferase in the epigenetic change of euchromatic promoter in mantle cell lymphoma (MCL) and acute leukemia. Methods: We retrospectively analyzed the protein of JARID1B and tri-methylated histone H3 lysine 4 (H3K4), histone H3 lysine 9 (H3K9), and cyclin D1 and Ki67 in 30 cases of MCL by immunohistochemistry. JARID1B was depleted by small interfering RNA (siRNA), and cell apoptosis and cell proliferation were detected by flow cytometry and MTT [3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide], histone tri-methylated H3K4 and histone acetylated H3, H4, cyclin D1, Bcl-2, procaspase-3, C-myc were studied by Western blot. Results: We demonstrated that JARID1B was upregulated and histone tri-methylated H3K4 was downregulated in MCL compared to proliferative lymphadenitis, P < 0.05. The expression of histone methylated H3K9 was similar in both. Histone methylation of H3K4 was positively correlated with Ki67 in MCL (Kappa = 0.757, P < 0.05). This study showed that depletion of JARID1B cleavage apoptotic proteins of Bcl-2, procaspase-3, C-myc and resulted in loss cell viability and inducing apoptosis in Jeko-1 and HL-60 cell lines. JARID1B siRNA improved tri-methyl H3K4 and histone acetylated H3 and inhibited cyclin D1, but did not affect histone acetylated H4. Conclusions: This study revealed hyper JARID1B expression and hypo histone H3K4 tri-methylation in MCL. We identify depletion JARID1B as a demethylase which is capable of removing three methyl groups from H3K4 and up-regulating histone acetylation of H3 in both cell lines. Interestingly, depletion of JARID1B inhibits Cyclin D1, which is one of the genes contributes to MCL pathogenesis. JARID1B might be one of therapeutic targets in acute leukemia and MCL.
Keywords: siRNA, JARID1B, histone methylation, MCL, acute leukemia
Introduction
Epigenetic markers, like DNA methylation and histone modifications, contribute to physiological and pathological states, including cancer [1]. Aberrant histone methylation has not been well characterized in human disease, especially in MCL. JARID1B, also named Plu-1, is one of the four members of the JARID1 protein family, which catalyzes the removal of three methyl groups from lysine 4 of histone H3 [2,3]. A recent discovery shows that H3K4 trimethylation is associated with active gene repression and suggests that H3K4 methylation may have a spatial and temporal effect on transcription. JARID1B has been shown to be up-regulated in breast cancer and prostate cancer, and probably involved in their development and progression [4,5]. JARID1B associates with the oncogene ZNF217 as part of a larger complex which also includes LSD1, and represses gene transcription [6] and is involved in cell cycle and signal transduction [7].
MCL is a well-defined lymphoid neoplasm characterized by a proliferation of mature B lymphocytes carrying the t(11;14)(q13;q32) translocation that leads to the overexpression of cyclin D1 [8]. It is an incurable, combining the unfavorable clinical feature of aggressive and indolent lymphomas. Conventional therapy can only make 10%-15% of patients in complete remission. Acute myeloid leukemia is the most common acute leukemia in adults. Chemotherapy induces a complete remission in 70%-80% of younger patients (age, 16 to 60 years), but many of them have a relapse and die of their disease. There is some acute leukemia with cytogenetic translocations in WHO classification, which involved in epigenetic modification change.
In this report, we investigated the state of JARID1B and histone methylation of H3K4 and H3K9 in MCL, evaluated the correlation with histone methylation of H3K4 and Ki67 that is maker of prognosis in MCL. Further we depleted JARID1B by siRNA technique and measured consequent histone modification, gene transcription, cell proliferation and apoptosis in Jeko-1 and HL-60 cell lines.
Materials and methods
Collection of patients’ samples and data
This study consisted of a total of 30 cases of MCL patients who hospitalized at the Zhangzhou Affiliated Hospital of Fujian Medical University between January 2005 and December 2010. The lymphoid tissue samples were collected from all participants after obtaining written informed consent, which was approved by the Institutional Review Board at the hospital. Diagnosis is made according to the WHO classification. A total of 30 cases in MCL was overexpression of cyclin D1. Patients’ samples with hyperplastic lymphadenitis (n = 30) were used as control.
Materials and reagents
Antibodies for rabbit against human tri-methyl-histone H3K9, rabbit against human tri-methyl-histone H3K4, rabbit against human JARID1B were purchased from Upsate Biotechnology (Lake Placid, NY, USA); Antibodies for cyclin D1 came from Dako (EP12), Ki67 from Dako (MIB1).
Tissue microarray (TMA) construction and immunohistochemistry
A representative tumor paraffin block (donor block) was collected from each case, and two tissue cores (2 mm in diameter) were obtained using a trephine apparatus. Trephinated paraffin tissue cores were then arranged in a new recipient paraffin block (tissue array block). Cores containing tumor in more than 50% of the area were considered adequate. Immunohistochemical staining was performed using commercially polyclonal rabbit antibodies to histone methyltransferase JARID1B, tri-methyl-histone H3K9 and H3K4, Ki67, cyclin D1. Tissue array blocks were section at a thickness of 4 μm and mounted on precoated glass slides. The sections were deparaffinized and hydrated prior to immunohistochemistry. The immunohistochemical S-P method was performed according to the manufacturer’s protocol. Tissues positive for all the purchased antibodies were used as positive controls, sections prepared with phosphate buffer saline instead of the primary antibody were used as negative controls. Positive control exhibited brown color in the nuclei. Chevallier’s semi-quantity system analysis was used in the calculation of immunohistochemistry results. For JARID1B, tri-methyl-histone H3K9 and H3K4, the results are presented as the sum of scores presenting color density and the percentage of stained cells. According to color density, non-stained cells were scored as 0, slightly stained were 1, intermediate-stained were 2, strongly stained were 3. When the number of positive cells was < 25%, 25%-50%, or 50%-75%, or > 75%, the immunoreactivity was scored as 1+, 2+, 3+, 4+ respectively. The two scores of presenting color density and the number of positive cells were added for each slide. The sum that was 0 was negative, 1-2 was presented as +, 3-4 as ++, 5-6 as +++, and 7 as ++++. If the sum of the two scores was less than 2, it was considered negative staining; more than two was considered positive staining. The scores of each patient group were averaged.
The Ki67 index was defined as the percentage of Ki67-positive tumor cells in representative areas of the lymphoma. To count the number of Ki67-positive cells, two representative areas were chosen. A representative area was defined not to contain residual germinal centers, hot spots of proliferation or proliferating T cells. Hot spots of proliferation were areas of tumor cells of less than two high-power fields in size (field of vision at × 400 magnification), which proliferate higher than the rest of the tumor. Counting was performed by one observer. The positive cells among 500 cells were counted using an eyepiece with a grid in a × 40 objectives. The Ki67 index was calculated as the percentage of positive cells by averaging the values obtained for the two areas (count-Ki67 index) with a semiquantitative evaluation system, consisting of four levels: < 10%, 10-30%, 30-50% and > 50% labeled cells. Negative control staining was obtained by omitting the primary antibodies.
Cell culture
Human MCL Jeko-1 and HL-60 cell line were purchased from Shanghai Institute of Cell Bank. Cells were cultured in 10% fetal bovine serum, and RPMI1640 containing 2 mM L-glutamine under 37°C, saturated humidity and 5% CO2. Cells were subcultured every 3-5 days. Cells were seeded and treated with the desired siRNA concentrations in different time points.
RNA interference
The approach by transient transfecting using LipofectamineTM 2000 was employed to deplete JARID1B expression from Jeko-1 and HL-60 cell lines. JARID1B siRNA sense: 5’-GGAGCUAUUCAAUUAACUATT-3’ Anti-sense 5’-UAGUUAAUUGAAUAGCUCCAG-3’ were synthsized by Shanghai GenePharma Co. Ltd. (China). Transfection with siRNA was performed according to the LipofectamineTM 2000 manufacturer’s protcol (Invitrogen, Carlsbad, CA). Inducible Jeko-1 and HL-60 cells (1 × 105 cells/well) were seeded onto 24-well plates, (Costar Life Science, Acton, MA) and transiently transfected with desired concentration of JARID1B siRNA. All experiments were conducted in triplicate using independent cultures. Both total RNA and protein were extracted after 24 hours transfection.
Reverse transcription polymerase chain reaction (PCR)
Total RNA was extracted from samples of 1 × 106 cells using TRIzol reagent (Invitrogen). The quantity and quality of RNA samples were measured by absorbance at 260 and 280 nm. RNA samples with an A260:A280 ratio 1.8~2.0 were stored at -80°C until use. Copy DNA synthesis was performed using an Avian Myeloblastosis Virus Reverse Transcriptase kit, according to manufacturer’s protocol (Promega, Madison, WI, USA). PCR β-actin was used as internal control. Primers used in PCR amplifications were: JARID1B forward, 5’-ATAGAATTCGGGAATCTTAAATTTG-3’; JARID1B reverse, 5’-TATCTCGAGTTCCTGTTCGGAATAGG-3’; β-actin forward: 5’-ATGTCACGCACGATTTCCCGC-3; β-actin reverse: 5’-GGCATGGGTCAGAAGGATTCC-3’. Amplicon size was 580 base pairs (bp) and 420 bp for JARID1B and β-actin respectively. PCR reaction conditions were: 95°C 30 seconds, 60°C 30 seconds, 72°C 40 seconds, and was repeated 32 cycles. PCR-amplified products were electrophoresed in 1.5% (w/v) agarose gels with 1 μg/ml ethidium bromide. Experiments were repeated twice.
Cell proliferation measured by MTT
The Jeko-1 and HL-60 cells were seeded at a density of 1 × 105/well in 96-well culture dishes (Costar Life Science). After 0, 24, 48, 72 and 96 hours transfection (n = 5) with indicated concentration of JARID1B siRNA, cell proliferation was measured using MTT assays (0.5 mg/ml; Sigma). The spectrophotometric absorbance of the samples was determined by using Ultra Multifunctional Microplate Reader (Tecan, Durham, NC) at 492 nm and 630 nm for absorbance (OD value). Suppression ratio was also calculated. The experiment was repeated in triplicated. Cell proliferation rate (%) = (A experiment-A blank)/(A control-A blank) × 100%.
Evaluation of apoptosis by flow cytometry
To detect the phosphatidylserine translocation on the surface of plasma membrane, cells were seeded in 6-well plates (1 × 106 cells/well, Costar Life Science) with sterile cover glass placed at the bottom of each well, and then transfected with negative control siRNA, 60, 120, 240 nmol/L JARID1B siRNA for 24 hours while cells at logarithmic phase. Cells were collected, resuspended in Annexin V binding buffer and then incubated with Annexin V-FITC and PI following the manufacture’s instruction(BD) by flow cytometry (Epics-XL, Beckman). 10000 events per sample were acquired on the FAC Scan. Fluorescent commissions were collected through 5.30 and 5.70 nm had pass filters for FITC and PI respectively.
Western blot analysis
Total protein lysates and Western blotting analysis were performed as previously described [9]. Briefly, Jeko-1 and HL-60 cells were plated on culture dishes and transfected by JARID1B siRNA with 60, 120, 240 nmol/L for 24 hours. Control cells were incubated in the medium with Na2CO3 using same time points. After incubation, total proteins were prepared from each culture condition with a lysis buffer containing freshly prepared protease inhibitors. Protein concentration was then measured using BCA protein assay (Pierce, Rockford, IL). Cell extracts were subjected to 12% SDS-PAGE and electrophoretically transferred to nitrocellulose membrane. Membranes were incubated with monoclonal anti-tri-methylated-histoneH3K4, anti-histone acetylation of H3, H4, JARID1B (Upstate, UAS), BCL-2, proaspase-3, C-myc, cyclin D1 (Santa Cruz USA). After being washed with TBS, membrane incubated with secondary antibody conjugated with peroxidase. The signal was then detected using the chemiluminescent detection system (Pierce) and analyzed by a color image analysis system (AlphaDigiDoc), (Alpha Innotech).
Statistical analysis
All statistical calculations were performed by SPSS version 18.0 for Windows software (SPSS Inc, IL, USA). Results are presented as means ± standard deviation. The t test was done to compare the levels of the parameters between the two groups. Different between the valus were assessed for statistical significance by Chi-square, WILCOXOM rank sum test, one-way ANOVA and repeated measure ANOVA. Agreement measure were calculated by Cohen’s kappa. P < 0.05 was considered statistically significant.
Results
Overexpression of JARID1B and lower expression of histone tri-methylation of H3K4 in MCL
We assessed the staining score of JARID1B, histone methylated H3K4 and H3K9 for 30 cases of MCL. The stained position was in cell nucleus. The expression of JARID1B in MCL was 37% (11/30), higher than that in hyperplastic lymphadenitis of 13% (4/30), P < 0.05. The expression of histone tri-methylation of H3K4 in MCL was 23% (7/30), lower than that in hyperplastic lymphadenitis of 57% (17/30), P < 0.01. The expression of histone tri-methylation of H3K9 in MCL was 87% (26/30), with similar to hyperplastic lymphadenitis of 97% (29/30), P > 0.05 (Figure 1, Table 1). The expression of cyclin D1 in MCL was 100% positive, but that in hyperplastic lymphadenitis was 100% negative (Figure 1).
Figure 1.
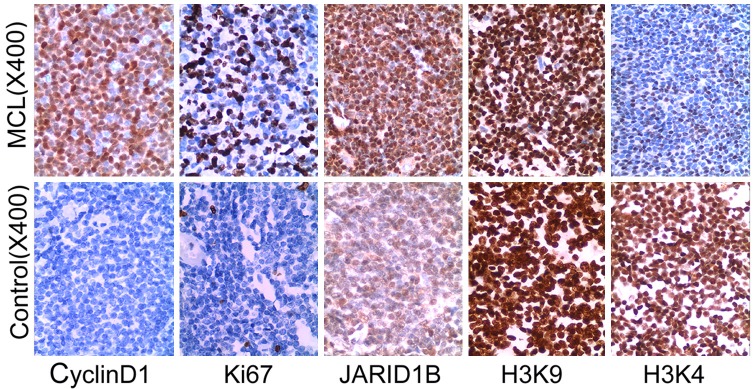
Expression of histone H3K4, H3K9 methylation, JARID1B, Ki67 and cyclin D1 in MCL. Histone H3K9, H3K4 methylation, JARID1B, Ki67 and cyclin D1 were detected by Immunohistochemistry. Tissue microarray was constructed and immunohistochemical staining was performed. Chevallier’s semi-quantity system analysis was used in the calculation of immunohistochemistry results in histone H3K4, H3K9 methylation, JARID1B and cyclin D1. Ki-67 values obtained via counting of 500 cells using an eyepiece with a grid in a × 40 objectives.
Table 1.
Expression of H3K4, H3K9 and JARID1B in MCL
| MCL | Control | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||||||
| Number | - | + | ++ | +++ | rate | - | + | ++ | +++ | rate | X2 | P | |
| H3K4 | 30 | 0 | 11 | 12 | 7 | 23% | 0 | 4 | 9 | 17 | 57% | 6.94 | < 0.01 |
| H3K9 | 30 | 0 | 3 | 1 | 26 | 87% | 0 | 0 | 1 | 29 | 97% | 0.87 | > 0.05 |
| JARID1B | 30 | 0 | 11 | 8 | 11 | 37% | 0 | 12 | 14 | 4 | 13% | 4.35 | < 0.05 |
Chi-square was done to compare the positive rate of H3K, H3K9, and JARID1B in MCL and control.
Correlation of histone methylated H3K4 with Ki67 in MCL
Ki67 labeling index was expressed 100% in MCL, including with ≤ 10% in 66.7% (20/30) in MCL, 11% to 30% in 16.7% (5/30), 31% to 50% in 6.7% (2/30), > 50% in 10.0% (3/30). The mean index was 12.53 ± 23.70%. Ki67 in hyperplastic lymphadenitis with ≤ 10% was in 93.3% (28/30). Only 6.7% was expressed in 11% to 30%. The mean index was 1.97 ± 2.70%, P < 0.05 (Figure 1, Table 2). We further analyzed the correlation between histone methylation of H3K4 and Ki67 in MCL. The data showed that the Ki67 labeling index was positive correlation with histone methylation of H3K4 (Kappa = 0.757, P < 0.05) (Table 3).
Table 2.
Expression of Ki67 in MCL
| Ki67 | ||||||
|---|---|---|---|---|---|---|
|
|
||||||
| < 10% | 11-30% | 31-50% | > 51% | mean ± SD | P | |
| MCL | 20 (66.7%) | 5 (16.7%) | 2 (6.7%) | 3 (10%) | 12.53 ± 23.70 | < 0.05 |
| Control | 28 (93.3%) | 2 (6.7%) | 0 | 0 | 1.97 ± 2.70%. | |
WILCOXOM rank sum test was done to compare the positive degree of ki67 in MCL and control (Z = -2.65 P < 0.05).
Table 3.
Positive staining of H3K4 and Ki67 in MCL
| Ki67 | |||
|---|---|---|---|
|
| |||
| + | - | ||
| H3K4 | + | 7 | 0 |
| - | 3 | 20 | |
| Ki67 > 10% and H3K4 +++ are given as positive. (Kappa = 0.757, P < 0.05) | |||
Cohen’s kappa was measured agreement between the positive staining of H3K4 and Ki67 in MCL.
Silencing efficiency of JARID1B gene by transfection with JARID1B siRNA in Jeko-1 and HL-60 cells
After 24 hours transfection with indicated concentration of JARID1B siRNA into Jeko-1 and HL-60 cells, green fluorescence in the transfected cells were counted by inverted fluorescence microscope, and transfected efficiency was calculated. Transfection efficiency was 96% ± 3.31% in Jeko-1 (n = 5), and 93% ± 2.56% in HL-60 cells (n = 5) (Figure 2A). The amplification of JARID1B mRNA attenuated with concentration dependent manner. Gray value (to β-actin) showed the amplification of JARID1B was (1.07 ± 0.15) with 60 nmol/L, (0.63 ± 0.17) with 120 nmol/L, (0.22 ± 0.7) with 240 nmol/L respectively in Jeko-1 cells, compared to control (1.25 ± 0.13), χ2 = 12.81, P < 0.05. Gray value (to β-actin) showed the amplification of JARID1B was (0.87 ± 0.15) with 30 nmol/L, (0.56 ± 0.09)with 60 nmol/L, (0.12 ± 0.03) with 120 nmol/L respectively in HL-60 cells, compared to control (1.13 ± 0.15), χ2 = 23.25, P < 0.05 (Figure 2B).
Figure 2.

A. Jeko-1 cells and HL-60 cells were transfected with JARID1BsiRNA. A. Jeko-1 cells were transfected with JARID1B siRNA after 24 hours in fluorescence microscope (left), transfection efficiency was 96% ± 3.31% (n = 5). HL-60 cells were transfected with JARID1B siRNA after 24 hours in fluorescence microscope (right), transfection efficiency was 93% ± 2.56% (n = 5). B. The expression of JARID1B mRNA by different concentrations of siRNA in Jeko-1 and HL-60 cell line. After 24 hours transfection of JARID1B, the amplification of JARID1B mRNA attenuated with concentration dependent manner. Amplification of JARID1B was (1.07 ± 0.15) with 60 nmol/L, (0.63 ± 0.17) with 120 nmol/L, (0.22 ± 0.7) with 240 nmol/L respectively in Jeko-1, compared to control (1.25 ± 0.13), χ2 = 12.81, P < 0.05 (upper). Amplification of JARID1B was (0.87 ± 0.15) with 30 nmol/L, (0.56 ± 0.09) with 60 nmol/L, (0.12 ± 0.03) with 120 nmol/L respectively in HL-60, compared to control (1.13 ± 0.15), χ2 = 23.25, P < 0.05 (lower).
JARID1B siRNA inhibited cell proliferation and promoted apoptosis in Jeko-1 and HL-60 cells
Depletion of JARID1B significantly suppressed cell proliferation in Jeko-1 and HL-60 cells. Cells were transfected with various concentrations of JARID1B siRNA for 24 hours and cell viability was measured by MTT assay. Approximately 83.53 ± 4.59% cell proliferation was seen in 15 nmol/L, 79.47 ± 5.88% in 30 nmol/L, 52.33 ± 3.76% in 60 nmol/L, 37.43 ± 4.47% in 120 nmol/L, 19.54 ± 5.18% in 240 nmol/L, 10.35 ± 4.70% in 480 nmol/L of JARID1B siRNA transfection in Jeko-1 cell line. The effect on HL-60 cells is similar to Jeko-1 cell line. JARID1B siRNA induced cell viability loss in dose -dependent manner (Figure 3A). In further experiment, 60 nmol/L of JARID1B siRNA was selected due to its moderated cytotoxic effect on both cell lines. After 60 nmol/L transfection, cell proliferation rate was 86.55 ± 5.00%, 52.33 ± 3.15%, 36.47 ± 4.50%, 28.41 ± 9.05%, 19.90 ± 5.75% in 12, 24, 36, 48, 60 hours Jeko-1 cell line, respectively. The effect on HL-60 cells was similar to Jeko-1 cell line. JARID1B siRNA induced cell viability loss in a time-dependent manner (Figure 3B).
Figure 3.
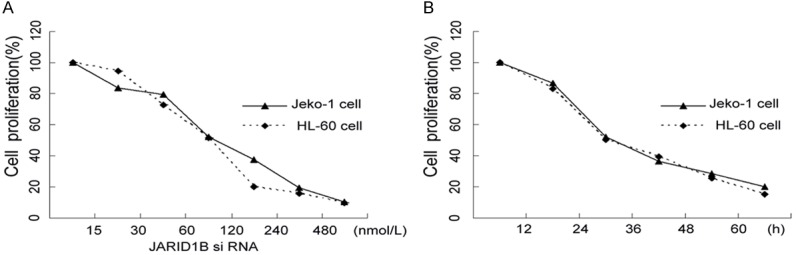
JARID1B siRNA induce cell proliferation in Jeko-1 and HL-60 cells. 1 × 105 cells were seeded into a 96-well culture dishes and treated with JARID1B siRNA in different concentration and time point. Cell proliferation was measured using MTT assays (0.5 mg/ml; Sigma). The spectrophotometric absorbance of the samples was determined by using Ultra Multifunctional Microplate Reader (Tecan, Durham, NC) at 492 nm and 630 nm for absorbance (OD value). Suppression ratio was also calculated. The experiment was repeated in triplicated. A. Cell proliferation was conducted in different concentrations in Jeko-1 and HL-60 cells. Repeated measure ANOVA: F = 109.3 P < 0.01 B. 60 nmol/L JARID1B siRNA treated to the cells in different time in Jeko-1 and HL-60 cells.Repeated measure ANOVA: F = 115.1 P < 0.01.
To verify if JARID1B siRNA induced cytotoxicity was due to apoptosis induction, Annexin V-FITC labeling was performed following 24 hours. In Jeko-1 cells, Apoptotic rate was 7.16 ± 3.45%, 25.2 ± 4.63%, 52.2 ± 3.49% after transfected with siRNA JARID1B with 30, 60, 120 nmol/L for 24 hrs respectively, compared to the untreated cells (3.57 ± 1.31%), P < 0.05. In HL-60 cells, Apoptotic rate was 35.2 ± 5.1%, 52.7 ± 3.8%, 62.0 ± 5.7% at the same time points, compared to the untreated cells (11.0 ± 3.6%), P < 0.05 (Figure 4). The effect on apoptosis was similar in both cell lines. The apoptotic rate was increased in a concentration dependent manner. We further investigated the apoptosis associated proteins, BCL-2, pro-caspase-3, and C-myc. It demonstrated that depletion of JARID1B induced breakup of BCL-2, pro-caspase-3 and C-myc significantly. Depletion of JARID1B gene with JARID1B siRNA 30, 60, 120 nmol/L for 24 hrs reduced Bcl-2 expression in 0.92 folds, 0.43 folds and 0.14 folds respectively. The expression of procaspase-3 was reduced 0.52 folds, 0.25 folds, 0.13 folds respectively. The expression of C-myc was also reduced by 0.73 folds, 0.63 folds, and 0.22 folds in Jeko-1 cells. It was the same effect in HL-60 cells (Figure 5).
Figure 4.

JARID1B siRNA produce cell apoptosis in Jeko-1 and HL-60 cells. 1 × 105 cells were seeded into a 96-well culture dishes and treated with JARID1B siRNA in 0, 30, 60, 120 nmol/L for 24 hours. Annexin V-PI staining was performed to determine apoptotic cells population. A. Apoptosis was increased gradually after transfected with siRNA JARID1B in 30, 60, 120 nmol/L for 24 hours respectively in Jeko-1, One-way ANOVA: F = 67.5 P < 0.05. B. Apoptosis was increased gradually after transfected with siRNA JARID1B in 30, 60, 120 nmol/L for 24 hours respectively in HL-60, One-way ANOVA: F = 34.8 P < 0.05. Fluorescence signals from Annenxin V-FITC and from PI are reported on the x-axis and y-axis, respectively. Numbers shown in the four quadrants represent the percentage of viable (lower left), necrotic (upper right), early apoptotic (lower right) and late apoptotic (upper right) cells.
Figure 5.
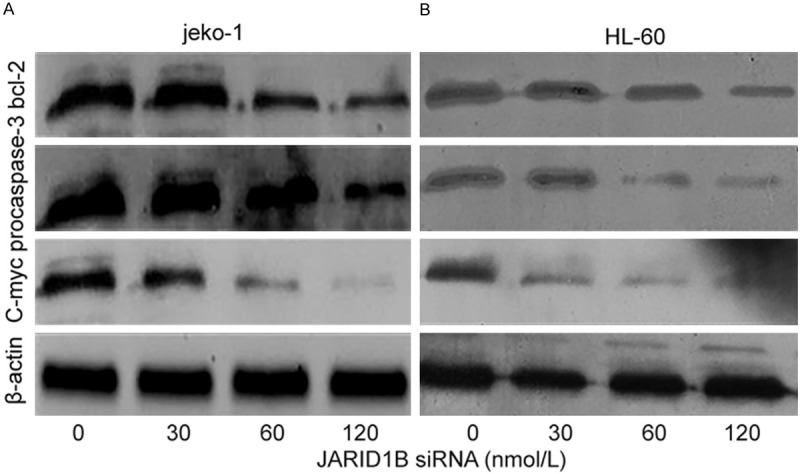
siRNA JARID1B-induced apoptosis was mitochondria-mediated and caspase dependent. Jeko-1 and HL-60 cells were treated respectively for 24 hours with siRNA JARID1B in 0, 30, 60, 120 nmol/L. After siRNA JARID1B treatment, cytosolic proteins were isolated and separated in 12% SDS gel, transferred onto PVDF membrane and blotted with BCL-2, procaspase-3, C-myc antibodys. The proteins were determined by immunoblotting using appropriate antibody. β-actin was used as a loading control. A siRNA JARID1B breakup BCL-2, pro-caspase-3 and C-myc significantly in Jeko-1 cell. B siRNA JARID1B breakup BCL-2, pro-caspase-3 and C-myc in HL-60 cell.
Depletion of JARID1B gene modulated histone methylation and acetylation, downregulated cyclin D1
To test our hypothesis, siRNA JARID1B depleted JARID1B gene expression, and upregulated histone tri-methylated H3K4 in joke-1 and HL-60 cell lines. The effect on histone lysine methylation was examined by immunocytochemistry using an antibody against H3K4me3. 30, 60, 120 nmol/L of JARID1B siRNA processing Jeko-1 cell for 24 hrs, the expression of JARID1B protein was decreased 0.89 folds, 0.54 folds, 0.21 folds, the expression of histone tri-methylated H3K4 was increased 2.12 folds, 8.12 folds, 10.24 folds. The effect of siRNA JARID1B on HL-60 cells was similar to Jeko-1 cells (Figure 6). These data indicate that JARID1B is probably H3K4me3 demethylase. JARID1B siRNA improved histone acetylation of H3. After 24 hours transfection with JARID1B siRNA in 30, 60, 120 nmol/L, the expression of histone acetylation of H3 increased 1.18 folds, 1.53 folds, 2.13 folds in HL-60 cells, which is similar to Jeko-1. But the histone acetylation of H4 was not changed.
Figure 6.

JARID1B siRNA modulated histone methylation and acetylation. The effect of JARID1B siRNA in different concentration on histone modulation in Jeko-1 and HL-60 cells was determined by immunoblotting. Following 24 hours JARID1B siRNA treatment, total proteins were isolated and separated in 12% SDS gel, transferred onto FVDF membranes and blotted with JARID1B, Tri-methy-H3K4, Act-H3, Act-H4 and Cyclin-D1. β-actin was used as a loading control. A. JARID1B siRNA while 30, 60, 120 nmol/L depleted JARID1B, upregulated histone tri-methylated H3K4, acetylated H3 in concentration-dependent manner in Jeko-1 cell for 24 hours. Alteration was not seen in acetylated H4. It declined cyclin D1 in concentration-dependent manner in Jeko-1 cell. B. JARID1B siRNA with 30, 60, 120 nmol/L depleted JARID1B, upregulated histone tri-methylated H3K4, acetylated H3 in concentration-dependent manner in HL-60 cell for 24 hours. Alteration was not seen in acetylated H4.
Because MCL is characterized by carrying the t(11;14)(q13;q32) translocation that leads to the overexpression of cyclin D1 [8]. We further study the effect of siRNA JARID1B on cyclin D1 in Jeko-1. The expression of cyclin D1 was declined after transfection of siRNA JARID1B. Cyclin D1 was reduced by 0.81 folds, 0.53 folds and 0.18 folds respectively in indicated concentration of siRNA JARID1B, as compared to control (Figure 6).
Discussion
JARID1B has been implicated as an oncogene in breast cancers and prostate [4,10,11]. In 2007, the demethylase activity of JARID1B was evidenced and it was shown to act as a transcriptional repressor in breast cancer, promoting tumor progression by repression of tumor suppressor genes, including BRCA1 [5]. JARID1B is up-regulated in prostate cancer tissues and associates with androgen receptor and regulates its transcriptional activity. On the other hand, a protein described as a JARID1B isoform, RBP2-H1, is down-regulated in melanomas where it may have tumor suppressive effects [12]. In this report, we studied 30 cases of MCL in clinical and pathological diagnosis of using immunohistochemical method with tissue microarray. We found that JARID1B is higher expression in MCL, H3K4 tri-methylation is lower expression compared to hyperplastic lymphadenitis. H3K9 methylation showed no obvious change. Overexpression of JARID1B resulted in loss of methyl H3K4. This data is coincident with our previous study [13], which showed downreguation of histone methylated H3K4, histone acetylation of H3, H4 in acute leukemia. But histone methylated H3K9 is upregulated. The evidence might prove that over expression of JARID1B and low expression of histone tri-methylated H3K4 might involve in the initiation and development of MCL.
Ki67 antigen is a cell cycle associated nuclear antigen and is present in all stages of the cell cycle (G1, S, G2, and M phases) in proliferating cells, but is absent in G0 and early G1 phases of cells re-entering the cell cycle [14]. The monoclonal antibody of Ki67 protein, MIB1, has been documented useful for the diagnosis [15,16] and prognosis [17] of some neoplasms. In this study, the average index of Ki67 in MCL is 12.53 ± 23.70%, higher than that in hyperplastic lymphadenitis (1.97 ± 2.70%), P < 0.05. The result is much similar to the report of Katzenberger et al. [18] and Gao et al. [19]. We demonstrated that in addition to prognostic factor, Ki67 might be a useful marker for distinguishing MCL and hyperplastic lymphadenitis. The expression of Ki67 has positively correlation with H3K4. It showed that histone methylation of H3K4 might be one of the prognostic factors in MCL.
Next, we investigated the effects of depletion of JARID1B on modulation of histone methylated H3K4, histone aceytlation of H3, H4, cyclin D1, improved cell apoptosis and inhibit cell proliferation in Jeko-1 and HL-60 cell lines. We identified JARID1B as a H3K4 demethylase. It supported the concept that JARDIB has affect to tri-methylated H3K4 which is a demethylase enzyme that in humans is encoded by the KDM5B gene [20,21]. It is reported that all JARID1B target genes are associated with H3K4me3 and depletion of JARID1B in ESCs leads to a global increase of H3K4me3 levels [22]. The results delineate an essential role for JARDIB-mediated transcriptional control during ESC differentiation. Our study showed that depletion of JARID1B improved histone acetylation of H3 in both cell lines. Acetylation and methylation are the two histone modification that has been clinically associated with pathological epigenetic disruption in cancer cells. It is reported that PLU-1/JARID1B has the ability to physically recruit the class I and class IIa HDACs direct. In addition, PLU-1/JARID1B can bind to the transcriptional corepressor N-CoR via an indirect interaction [23].
In this study, we found that JARID1B siRNA repressed cyclin D1, which characterized by 98% MCL. Activation of cyclin D1 in these malignancies is associated with the recruitment of RNA polymerase II to the cyclin D1 promoter and IgH regulatory regions. The cyclin D1 promoter contains a CpG island which can be potentially regulated by DNA methylation [24]. Suppression of JARID1B resulted in an accumulation of MCF-7 cells in G1 [25]. Cyclin D1 is suppressed in JARID1B knockdown cells [25]. Cyclin D1 is a therapeutics target for MCL, and JARDIB might be a new gene therapeutics target for MCL.
It of the he pathogenesis in MCL tnd in This study demonstrated that depletion of JARID1B could reduce cell proliferation and induce cell apoptosis in time and dose-dependent manner in both jeko-1 and HL-60 cell lines. Depletion of JARID1B breakup the apoptosis related proteins, procaspase-3, Bcl-2, C-myc, and induced cell apoptosis. It might contribute to upregulation of tri-methylated H3K4 which is a critical histone modification regulating transcription. High levels of H3K4-me3 and H3K4-me2 are associated with the 5’ regions of virtually all active genes and are in positive correlation with transcription rates, active polymerase II occupancy, and histone acetylation [26].
In conclusion, our studies indicated that MCL has higher JARID1B expression and lower tri-methylated H3K4 expression in Depletion of JARID1B result in suppressed cyclin D1, inhibited cell proliferation and induced cell apoptosis in jeko-1 and HL-60 cell lines. We discovered that the demethylase for H3K4 has equal effect on the transcription activity inboth cell lines. Therefore, JARID1B and H3K4 could be a therapeutic target for MCL and acute myeloid leukemia.
Acknowledgements
This work was partly supported by grant-in-aid from the Foundation of Science and Technology of Fujian Medical University, Fujian, China (No. FZS08018), Science Research Foundation of Ministry of Health, United Fujian Provincial Health, and Education Project for Tackling the Key Research, P. R. China (WKJ2008-2-55), Science Research Foundation of Fujian Province, P. R. China (2012J01420), Medical Innovations Research Foundation of Fujian Province, P. R. China (2012-CX-32) and the major project funded by R & D institutions of Fujain (2012I2004).
Disclosure of conflict of interest
None.
References
- 1.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 2.Lee MG, Norman J, Shilatifard A, Shiekhattar R. Physical and functional association of a trimethyl H3K4 demethylase and Ring6a/MBLR, a polycomb-like protein. Cell. 2007;128:877–887. doi: 10.1016/j.cell.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 3.Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH, Whetstine JR, Bonni A, Roberts TM, Shi Y. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128:1077–1088. doi: 10.1016/j.cell.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 4.Xiang Y, Zhu Z, Han G, Ye X, Xu B, Peng Z, Ma Y, Yu Y, Lin H, Chen AP, Chen CD. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc Natl Acad Sci U S A. 2007;104:19226–19231. doi: 10.1073/pnas.0700735104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamane K, Tateishi K, Klose RJ, Fang J, Fabrizio LA, Erdjument-Bromage H, Taylor-Papadimitriou J, Tempst P, Zhang Y. PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol Cell. 2007;25:801–812. doi: 10.1016/j.molcel.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Banck MS, Li S, Nishio H, Wang C, Beutler AS, Walsh MJ. The ZNF217 oncogene is a candidate organizer of repressive histone modifiers. Epigenetics. 2009;4:100–106. doi: 10.4161/epi.4.2.7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scibetta AG, Santangelo S, Coleman J, Hall D, Chaplin T, Copier J, Catchpole S, Burchell J, Taylor-Papadimitriou J. Functional analysis of the transcription repressor PLU-1/JARID1B. Mol Cell Biol. 2007;27:7220–7235. doi: 10.1128/MCB.00274-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sabattini E, Bacci F, Sagramoso C, Pileri SA. WHO classification of tumours of haematopoietic and lymphoid tissues in 2008: an overview. Pathologica. 2010;102:83–87. [PubMed] [Google Scholar]
- 9.Ma X, Fang Y, Beklemisheva A, Dai W, Feng J, Ahmed T, Liu D, Chiao JW. Phenylhexyl isothiocyanate inhibits histone deacetylases and remodels chromatins to induce growth arrest in human leukemia cells. Int J Oncol. 2006;28:1287–1293. [PubMed] [Google Scholar]
- 10.Lu PJ, Sundquist K, Baeckstrom D, Poulsom R, Hanby A, Meier-Ewert S, Jones T, Mitchell M, Pitha-Rowe P, Freemont P, Taylor-Papadimitriou J. A novel gene (PLU-1) containing highly conserved putative DNA/chromatin binding motifs is specifically up-regulated in breast cancer. J Biol Chem. 1999;274:15633–15645. doi: 10.1074/jbc.274.22.15633. [DOI] [PubMed] [Google Scholar]
- 11.Barrett A, Madsen B, Copier J, Lu PJ, Cooper L, Scibetta AG, Burchell J, Taylor-Papadimitriou J. PLU-1 nuclear protein, which is upregulated in breast cancer, shows restricted expression in normal human adult tissues: a new cancer/testis antigen? Int J Cancer. 2002;101:581–588. doi: 10.1002/ijc.10644. [DOI] [PubMed] [Google Scholar]
- 12.Roesch A, Mueller AM, Stempfl T, Moehle C, Landthaler M, Vogt T. RBP2-H1/JARID1B is a transcriptional regulator with a tumor suppressive potential in melanoma cells. Int J Cancer. 2008;122:1047–1057. doi: 10.1002/ijc.23211. [DOI] [PubMed] [Google Scholar]
- 13.Ma XD, Huang YQ, Xiao LY, Zou Y. Study on aberration in histone methylation and acetylation in acute leukemia. Zhonghua Xue Ye Xue Za Zhi. 2010;31:523–526. [PubMed] [Google Scholar]
- 14.Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol. 1984;133:1710–1715. [PubMed] [Google Scholar]
- 15.Fujimori Y, Fujimori T, Imura J, Sugai T, Yao T, Wada R, Ajioka Y, Ohkura Y. An assessment of the diagnostic criteria for sessile serrated adenoma/polyps: SSA/Ps using image processing software analysis for Ki67 immunohistochemistry. Diagn Pathol. 2012;7:59. doi: 10.1186/1746-1596-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li LX, Crotty KA, McCarthy SW, Palmer AA, Kril JJ. A zonal comparison of MIB1-Ki67 immunoreactivity in benign and malignant melanocytic lesions. Am J Dermatopathol. 2000;22:489–495. doi: 10.1097/00000372-200012000-00002. [DOI] [PubMed] [Google Scholar]
- 17.Engellau J, Persson A, Bendahl PO, Akerman M, Domanski HA, Bjerkehagen B, Lilleng P, Weide J, Rydholm A, Alvegard TA, Nilbert M. Expression profiling using tissue microarray in 211 malignant fibrous histiocytomas confirms the prognostic value of Ki-67. Virchows Arch. 2004;445:224–230. doi: 10.1007/s00428-004-1065-6. [DOI] [PubMed] [Google Scholar]
- 18.Katzenberger T, Petzoldt C, Holler S, Mader U, Kalla J, Adam P, Ott MM, Muller-Hermelink HK, Rosenwald A, Ott G. The Ki67 proliferation index is a quantitative indicator of clinical risk in mantle cell lymphoma. Blood. 2006;107:3407. doi: 10.1182/blood-2005-10-4079. [DOI] [PubMed] [Google Scholar]
- 19.Gao J, Peterson L, Nelson B, Goolsby C, Chen YH. Immunophenotypic variations in mantle cell lymphoma. Am J Clin Pathol. 2009;132:699–706. doi: 10.1309/AJCPV8LN5ENMZOVY. [DOI] [PubMed] [Google Scholar]
- 20.Lahoud MH, Ristevski S, Venter DJ, Jermiin LS, Bertoncello I, Zavarsek S, Hasthorpe S, Drago J, de Kretser D, Hertzog PJ, Kola I. Gene targeting of Desrt, a novel ARID class DNA-binding protein, causes growth retardation and abnormal development of reproductive organs. Genome Res. 2001;11:1327–1334. doi: 10.1101/gr.168801. [DOI] [PubMed] [Google Scholar]
- 21.Zhu L, Hu J, Lin D, Whitson R, Itakura K, Chen Y. Dynamics of the Mrf-2 DNA-binding domain free and in complex with DNA. Biochemistry. 2001;40:9142–9150. doi: 10.1021/bi010476a. [DOI] [PubMed] [Google Scholar]
- 22.Schmitz SU, Albert M, Malatesta M, Morey L, Johansen JV, Bak M, Tommerup N, Abarrategui I, Helin K. Jarid1b targets genes regulating development and is involved in neural differentiation. EMBO J. 2011;30:4586–4600. doi: 10.1038/emboj.2011.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barrett A, Santangelo S, Tan K, Catchpole S, Roberts K, Spencer-Dene B, Hall D, Scibetta A, Burchell J, Verdin E, Freemont P, Taylor-Papadimitriou J. Breast cancer associated transcriptional repressor PLU-1/JARID1B interacts directly with histone deacetylases. Int J Cancer. 2007;121:265–275. doi: 10.1002/ijc.22673. [DOI] [PubMed] [Google Scholar]
- 24.Kitazawa S, Kitazawa R, Maeda S. Transcriptional regulation of rat cyclin D1 gene by CpG methylation status in promoter region. J Biol Chem. 1999;274:28787–28793. doi: 10.1074/jbc.274.40.28787. [DOI] [PubMed] [Google Scholar]
- 25.Mitra D, Das PM, Huynh FC, Jones FE. Jumonji/ARID1 B (JARID1B) protein promotes breast tumor cell cycle progression through epigenetic repression of microRNA let-7e. J Biol Chem. 2011;286:40531–40535. doi: 10.1074/jbc.M111.304865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]