

Abstract
The aim of our study was to investigate the protective effects of Paeoniflorin (PF) against injury induced by AGE-modified bovine serum albumin (AGE-BSA) in human umbilical vein endothelial cells (HUVECs), and to examine the underlying mechanisms of these effects. A 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay was used to determine cell viability. Protein expression levels were determined by western blotting. For function-blocking experiments, we used small interfering RNA molecules (siRNA) for function-blocking experiments. At 6 h, we found that 100 μg/mL AGE-BSA reduced the viability of HUVECs. However, pretreatment with PF restored cell viability in a dose-dependent manner. AGE-BSA increased the levels of microtubule-associated protein light chain 3-II (LC3-II) and the receptor for advanced glycation end products (RAGE). Expression of p62 protein was also increased, but not at a statistically significant level. Pretreatment with PF further increased levels of LC3-II and RAGE, but reduced the expression of p62. In cells transfected with Atg5 and RAGE siRNA, cell viability and expression of LC3-II decreased in both the AGE-BSA and PF + AGE-BSA treatments. PF can protect HUVECs from AGE-BSA-induced injury by upregulating autophagy and promoting the completion of autophagy flux. RAGE plays an important role in this autophagic protection effect.
Keywords: Human umbilical vein endothelial cells (HUVECs), advanced glycation end products (AGEs), receptor for advanced glycation end products (RAGE), paeoniflorin (PF), autophagy flux
Introduction
Advanced glycation end products (AGEs) are the end-stage product formed in the body by a non-enzymatic glycosylation reaction that occurs between reducing sugars and proteins or lipids. They are also a group of heterogeneous compounds accumulated in diabetes because of factors including increased reactive carbohydrate substrate availability, oxidative condition favoring glycation and injured detoxification [1]. There is growing evidence that the accumulation of AGEs can trigger oxidative stress, oxidized lipids, inflammation, metabolic stress conditions and apoptosis, which can lead to the dysfunction of endothelial cells. Such dysfunction, with vascular injury, can lead to an increase in atherosclerosis plaque formation [2,3]. Recent studies have shown that AGEs can accelerate the atherosclerotic (AS) process in type 2 diabetics with coronary heart disease [4]. In diabetic patients, some studies have shown that the accumulation of interaction between AGEs and the receptor for advanced glycation end products (RAGE) is a key factor in the development of atherosclerotic lesions of vascular smooth muscle cells (VSMCs) [5,6].
RAGE, macrophage scavenger receptor type I and type II, and other receptors, like AGE-R1 (oligosacchary-l transferase-48), AGE-R2 (80K-H phosphoprotein), and AGE-R3 (galectin-3). RAGE has recently been recognized as a member of the immunoglobulin superfamily, and is encoded in the Class III major histocompatibility complex region the area adjacent to the TNF-α and the encoding regions of several complement components [7]. Several RAGE ligands have been identified, including high-mobility group protein 1 (HMGB1), AGEs, and members of the S100 protein family [8]. The interaction of RAGE and the RAGE ligand is related to the survival and inflammatory responses of cells that express the receptor, and to increased levels of phosphorylation in ERK and NF-κB p65. The expression of RAGE is significantly increased in cells under stress, and this has been linked to a variety of inflammatory diseases (including sepsis), diabetes, atherosclerosis, and to the pathogenesis of Alzheimer’s disease [9,10]. Some studies have shown that knocking out RAGE in tumor cells can lead to weakened autophagy. In contrast, increasing the expression of RAGE can enhance autophagy, and thereby enhance cell viability [11]. RAGE can also maintain the level of autophagy, which is related to increasing phosphorylation of mTOR and the formation of the Beclin-1/VPS34 autophagosome [11].
Paeoniflorin (PF), a monoterpene glycoside, is the main active ingredient derived from the dried roots of Paeonia [12]. It has been widely used in China for more than 1000 years to treat a variety of diseases, including viral hepatitis, systemic lupus erythematosus, pain, giddiness, gynecological problems, and gynecological disease [13]. A growing number of studies show that PF is associated with mitochondrial homeostasis, and is involved in processes such as the maintenance of intracellular calcium concentration, scavenging reactive oxygen species, and inhibition of the inflammatory response [14]. There is evidence that PF could have anti-inflammatory and anti-apoptotic effects through the HMGB1-RAGE/TLR-2/TLR-4-NF-κB pathway [15], some studies suggest that PF is a critical antioxidant, decreased the number of ROS, LDH, and MDA equivalents; decrease the apoptotic ratio of cells; reduce the activity of caspase-3; and repair total SOD and GSH-Px activities. Further more, PF almost completely blocked the H2O2-induced phosphor- ylation of ERK1/2 in HUVECs [16], which suggests that PF could play a relevant role through influence the RAGE pathway.
In a preliminary experiment, we used an MTT assay to investigate the effect of AGE-BSA on HUVECs. We found that 100 µg/mL AGE-BSA applied for 6 h, reduced the viability of HUVECs compared to the control group. When we added an autophagy inducer, rapamycin hormone (Rap), cell viability increased, which indicates that increased completion of autophagy in cells has a protective role. After adding an autophagy inhibitor, 3-MA, cell viability decreased, indicating that inhibition of autophagy could increase damage to the cell. In our current study, we will investigate whether PF protects HUVECs from AGE-BSA-induced injury by upregulating autophagy, and examine whether PF increases the level of autophagy through a RAGE-mediated pathway.
Material and methods
Materials
HUVECs were obtained from the Institute of Neuroscience, Soochow University (Suzhou, China). PF (purity ≥ 98%) was purchased from the Yangling Dongke Pharmaceutical Division (Shanxi, China). AGE-BSA was purchased from Sigma-Aldrich (St. Louis, MO, USA). Dulbecco’s modified Eagle’s medium (DMEM) and fetal bovine serum (FBS) were provided by Gibco-BRL (Grand Island, NY, USA). RAGE RNAi and Atg5 RNAi were designed and purchased from Shanghai GenePharma (Shanghai, China). RAGE, Atg5, LC-3, p62, and β-actin antibodies were obtained from Abcam (Cambridge, UK). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was obtained from Beyotime Biotechnology (Jiangsu, China).
Cell culture and treatment
HUVECs were cultured in DMEM containing 10% (v/v) FBS, 100 U/mL penicillin, and 100 µg/mL streptomycin, and at 37°C in a humidified atmosphere containing 5% CO2. Cells were passaged by trypsinization and seeded at approximately 105 cells/mL. When cells reached 80% confluence, the culture medium was replaced with serum-free medium for 24 h. In the appropriate treatments, cells were pretreated with 25 µmol/L PF for 0.5 h prior to a 24 h exposure to AGE-BSA (100 µg/mL). The cells used in the experiments underwent 4 to 8 passages.
Cell viability assay
MTT was used to determine cell viability. Briefly, HUVECs were seeded at a density of 1 × 10^4 cells/well in a 96-well culture plate. Following drug treatment, the cells were washed twice with PBS to remove residual medium, 10 µL of MTT (0.5 mg/mL) was added to each well, and they were incubated for an additional 4 h at 37°C. Subsequently, we added 100 µL of dimethy l sulfoxide (DMSO) to dissolve the MTT, and the absorbance at 490 nm was read on a microplate reader. We expressed cell viability data as a percentage of the control, which was considered 100% viable.
Western blot analysis
Protein expression levels of RAGE, Atg5, LC-3, p62, and β-actin were determined by western blotting. Briefly, after drug treatment, cells were lysed with ice-cold lysis buffer (0.33 mol/L Tris/HCl, 10% SDS (w/v), 40% glycerol (v/v) and 50 mmol/L DTT containing bromophenol blue), and the protein content of the lysates was measured using the bicinchoninic acid (BCA) method. Equal amounts of protein were separated by 10% SDS-PAGE and transferred to a nitrocellulose membrane. After being blocked with TBST containing 5% bovine serum albumin, membranes were incubated overnight at 4°C with the primary antibody for RAGE, Atg5, LC-3, p62 or β-actin, and subsequently incubated at room temperature for 1 h with the corresponding horseradish peroxidase-conjugated secondary antibody. The protein bands were quantified by video densitometry and the results were normalized to β-actin expression.
RNA interference
In function-blocking experiments, we used small interfering RNA molecules (siRNA) targeted to RAGE mRNA and Atg5 mRNA, which we purchased from Shanghai GenePharma. The siRNA was designed against RAGE (sense, 5’-GCCGGAAAUUGUGAAUCCUTT-3’; antisense, 5’-AGGAUUCACAAUUUCCGGCTT-3’) and Atg5 (sense, 5’-GCAACTCTGGATGGGATTG-3’; antisense, 5’-CATCTGAGCTACCCGGATA-3’). The negative control siRNA was non-targeting (sense, 5’-UUCUCCGAACGUGUCACGUTT-3’; antisense, 5’-ACGUGACACGUUCGGAGAATT-3’). Cells were transfected with siRAGE using Lipofectamine 2000 (Invitrogen, USA) for 48 h according to the manufacturer’s instructions. The final concentration of siRNA was 100 pM. The efficacy of RNA interference was determined by Western blotting.
Statistical analysis
All data were obtained from at least 3 individual experiments. Values are expressed as the mean ± SEM. Statistical analysis between groups was performed by one-way ANOVA. Statistical significance was set at P < 0.05.
Results
PF has a protective effect against AGE-BSA-induced injury in HUVECs
Using the MTT method, we found that at 6 h, 100 g/mL AGE-BSA reduced the viability of HUVECs viability. However, we found that pretreatment for 0.5 h with concentrations of PF from 12.5-100 mol/L restored cell viability in a dose-dependent manner. At a concentration of 25 mol/L, the protective effect of PF reached a plateau (Figure 1).
Figure 1.
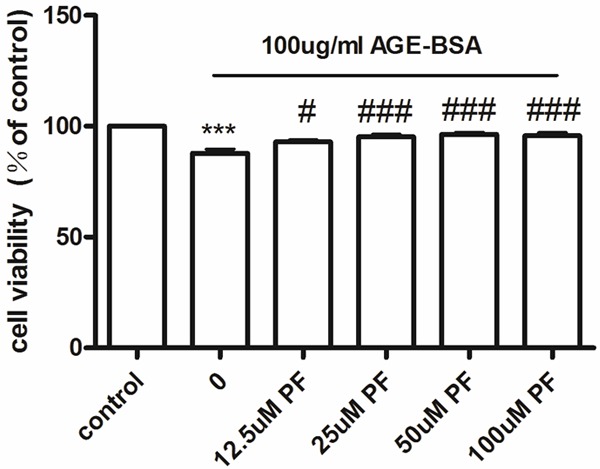
Cell viability in cultured HUVECs, measured with an MTT assay. Compared with the control, at 6 h, 100 μg/mL AGE-BSA reduces the viability of HUVECs. Pretreatment for 0.5 h with concentrations of PF ranging from 12.5-100 μmol/L increases cell viability in a dose-dependent manner. At 25 μmol/L, the protective effect of PF reaches a plateau. ***P < 0.001 vs. control; #P < 0.05 vs. AGE-BSA; ###P < 0.001 vs. AGE-BSA.
PF increases the activity of AGE-BSA-induced autophagy
Using Western blotting, we found that HUVECs treated with 100 μg/mL AGE-BSA for 6 h showed increased expression of LC3-II, which indicates the activation of autophagy, and pretreatment for 0.5 h with 25 μmol/L PF further upregulated levels of LC3-II (Figure 2A). Although p62 protein expression increased in HUVECs following treatment with 100 μg/mL AGE-BSA for 6 h, the increase was not statistically significant. Where HUVECs were pretreated with 25 μmol/L PF for 0.5 h, expression of p62 was reduced compared with AGE-BSA treatment (Figure 2B).
Figure 2.
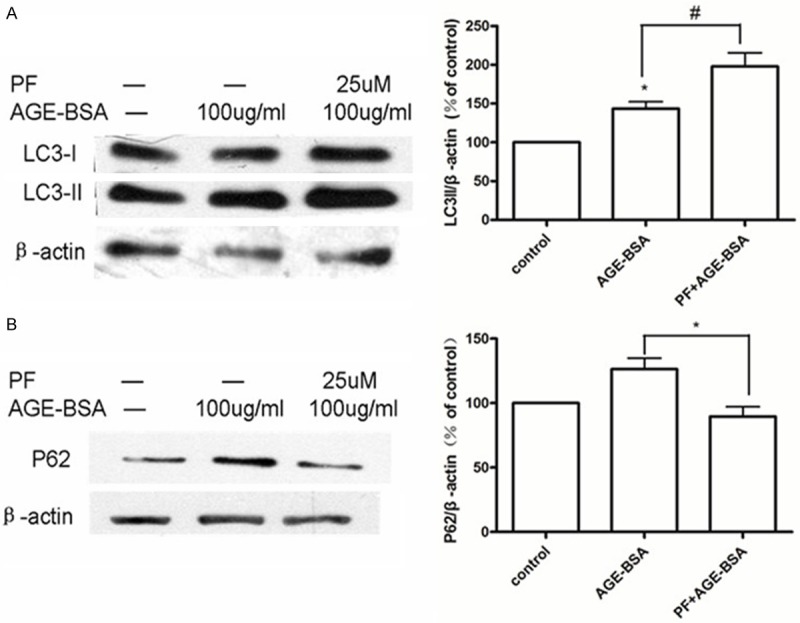
PF induces autophagy in HUVECs. A. In a western blot assay, HUVECs treated for 6 h with 100 μg/mL AGE-BSA increase expression of LC3-II. Pretreatment with 25 μmol/L PF for 0.5 h further increases expression. *P < 0.05 vs. control; #P < 0.05 vs. AGE-BSA. B. HUVECs treated with 100 μg/mL AGE-BSA for 6 h show a small, but not significant, change in expression of p62. Pretreatment with 25 μmol/L PF for 0.5 h reduces p62 protein expression. *P < 0.05 vs. AGE-BSA.
The protective role of PF is achieved via autophagy
Compared to control cells, Atg5 protein expression was reduced by almost 50% in cells transfected with Atg5 siRNA (Figure 3A). Using western blotting, we found that cells transfected with siRNA to inhibit the expression of Atg5 showed significantly reduced the expression of LC3-II (Figure 3B). Specifically, levels of LC3-II decreased in two treatments: 100 μg/mL AGE-BSA and 25 μmol/L PF + 100 μg/mL AGE-BSA (Figure 4A). Using the MTT method, we found that the viability of cells in these two treatments also decreased compared with control cells (Figure 4B).
Figure 3.
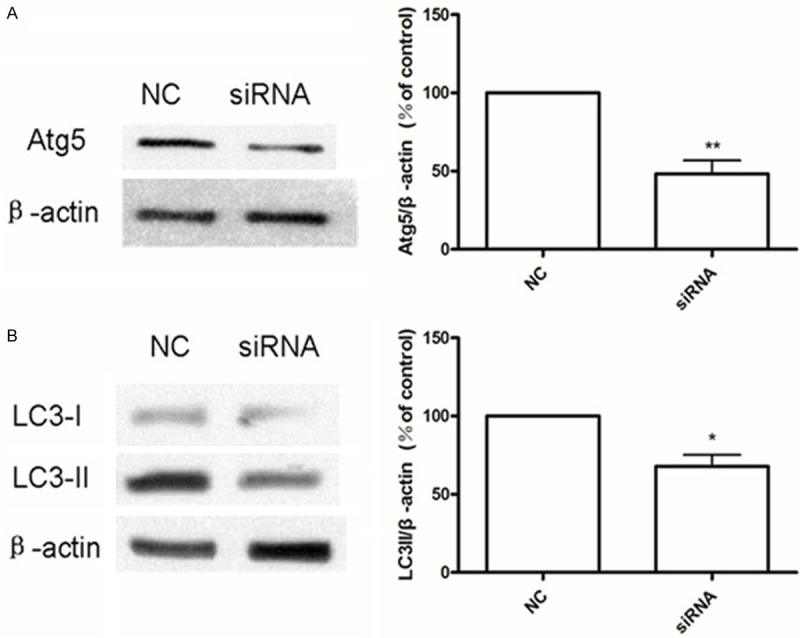
A. Atg5 siRNA reduces the expression of Atg5 protein. Compared to the negative control (NC), cells transfected with Atg5 siRNA express less Atg5 protein. **P < 0.01 vs. NC. B. Using western blotting, we observe that in cells transfected with siRNA to inhibit the expression of Atg5, the level of LC3-II expression is also reduced. *P < 0.05 vs. NC.
Figure 4.
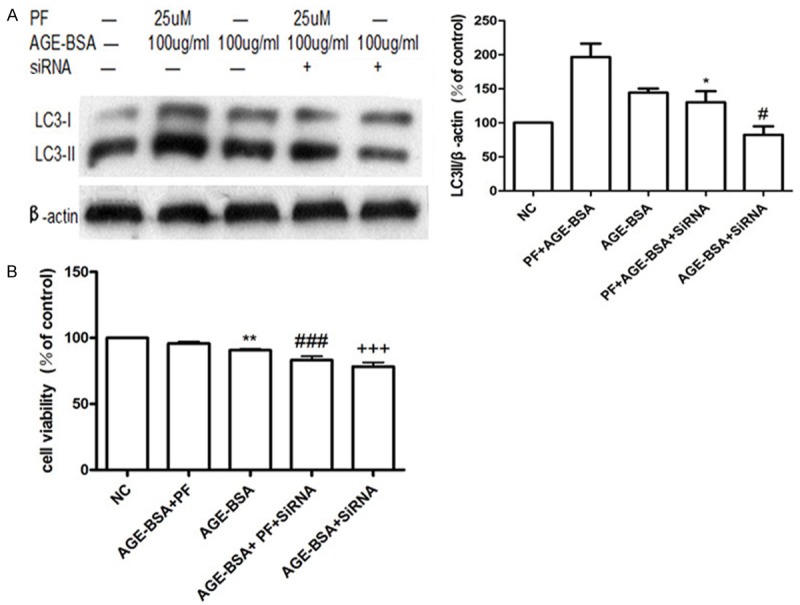
PF plays a protective role via autophagy. A. In cells transfected with siRNA to inhibit the expression of Atg5, the expression of LC3-II decreases in two treatments compared to NC: 100 μg/mL AGE-BSA and 25 μmol/L PF + 100 μg/mL AGE-BSA. *P < 0.05 vs. PF + AGE-BSA; #P < 0.05 vs. AGE-BSA. B. In cells transfected with siRNA to inhibit the expression of Atg5, cell viability decreased both in the 100 μg/mL AGE-BSA treatment and the 25 μmol/L PF + 100 μg/mL AGE-BSA treatment compared with NC. *P < 0.05 vs. PF + AGE-BSA; #P < 0.05 vs. AGE-BSA.
RAGE plays an essential role in PF-induced autophagy
The results of western blotting showed that HUVECs treated with 100 μg/mL AGE-BSA for 6 h increased their expression of RAGE. Pretreatment with 25 μmol/L PF for 0.5 h further increased the expression of RAGE (Figure 5).
Figure 5.
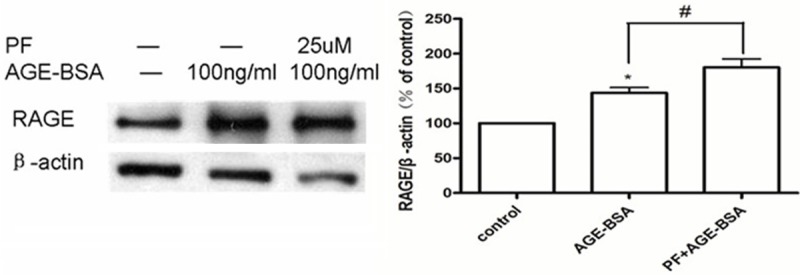
PF increases the expression of RAGE. In HUVECs treated for 6 h with 100 μg/mL AGE-BSA, either in isolation or with a pretreatment of 25 μmol/L PF for 0.5 h, expression of RAGE increases compared to that in control cells. Pretreatment with PF causes the most pronounced increase in RAGE expression. *P < 0.05 vs. control; #P < 0.05 vs. AGE-BSA.
Where we transfected cells with RAGE siRNA, their expression of RAGE protein was reduced by almost 50% relative to control cells (Figure 6).
Figure 6.
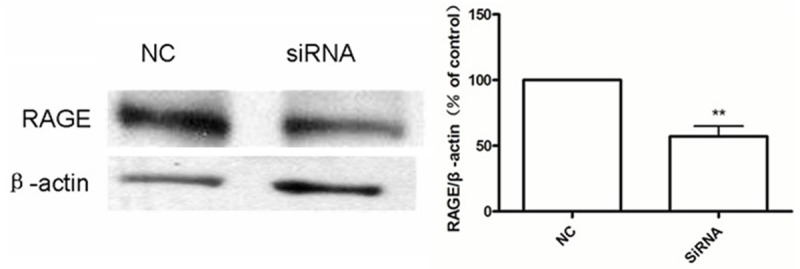
Measured by western blotting, cells transfected with RAGE siRNA show an almost 50% reduction in RAGE protein expression. **P < 0.01 vs. NC.
Compared with the control, HUVECs treated with AGE-BSA (100 µg/mL, 6 h) showed decreased viability, but pretreating cells with PF (25 μmol/L) attenuated this effect. However, in cells transfected with RAGE siRNA, cell viability was significantly decreased, by varying degrees, in both treatments (Figure 7).
Figure 7.
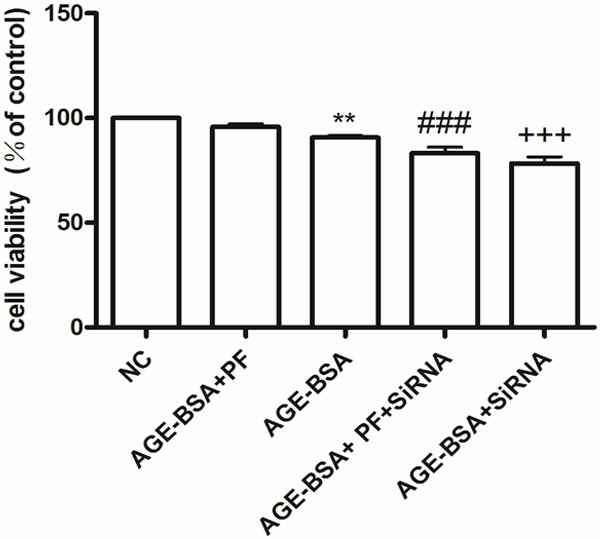
Cell viability in cultured HUVECs, measured by the MTT assay. Compared with the NC, HUVECs treated with AGE-BSA (100 μg/mL, 6 h) show decreased viability, but this effect is attenuated by pretreating cells with PF (25 μmol/L). In cells transfected with RAGE siRNA, cell viability decreases, but does so at various degrees. **P < 0.01 vs. NC; ###P < 0.001 vs. AGE-BSA + PF; +++P < 0.001 vs. AGE-BSA.
In cells transfected with RAGE siRNA, we found that treatment with AGE-BSA or PF significantly increased expression of LC3-II compared to control cells. These data illustrate that, in HUVECs, RAGE plays an essential role in PF-induced autophagy (Figure 8A) Treatment with AGE-BSA did not significantly change the expression of p62 protein compared to the control group, but after pretreatment for 0.5 h with 25 μmol/L PF expression of p62 decreased. However, in cells transfected with RAGE siRNA, expression of p62 increased at varying degrees (Figure 8B).
Figure 8.
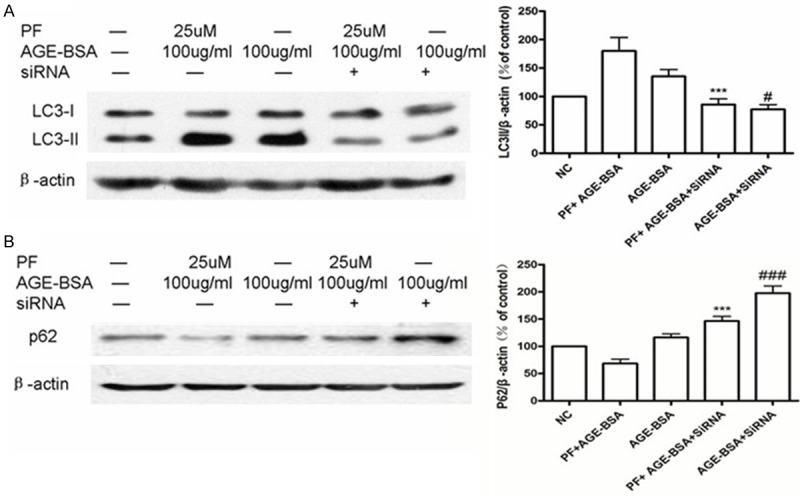
RAGE plays an essential role in PF-induced autophagy. A. Using western blotting, we found that AGE-BSA or PF + AGE-BSA induces an increase in LC3-II expression in cells, but a significant decrease in expression is observed in cells transfected with RAGE siRNA. ***P < 0.001 vs. PF + AGE-BSA; #P < 0.05 vs. AGE-BSA. B. Treatment with AGE-BSA does not significantly affect the expression of p62 protein. Pretreatment with 25 μmol/L PF for 0.5 h results in reduced p62 protein expression. However, in cells transfected with RAGE siRNA, p62 expression increases in varying degrees. ***P < 0.001 vs. PF + AGE-BSA; ###P < 0.001 vs. AGE-BSA.
Discussion
Autophagy is an evolutionarily conserved process involving the degradation of long-lived proteins [17]. It also plays a balance role in cell growth, development, and homeostasis [18]. The autophagic-lysosomal pathway (ALP) is considered the main mechanism involved in removal of misfolded proteins and cell debris. Autophagy also has a cytoprotective effect, removing the aggregation of denatured proteins and damaged organelles [19]. The autophagy process involves many genes, the critical one is microtubule-associated protein 1 light chain 3 (LC3), which is important to autophagosome formation, the best identified mammalian protein species that is specifically associated with autophagic vacuole membranes [20]. When autophagy is induced, the cytoplasmic form of LC3 (LC3-I) will transform into the membrane-associated form of LC3 (LC3-II) [21]. LC3-II is the primary protein of autophagy [21]. The role of the p62 protein is to connect the LC3 protein to an ubiquitination substrate, so that it can be incorporated into the complete phagosome, by final autophagic lysosomal to degradation, marking the completion of autophagy flux [22,23]. Atg5, a key molecule involved in autophagic vacuole formation, a protein which is necessary for autophagy, is required for autophagic cell death, stress-mediated cell death, starvation-induced apoptosis, starvation-induced cell death [24]. As autophagy research has developed, it has become clear that an increase in the number of autophagosomes alone does not necessarily correlate with increased autophagic activity or flux. Instead, the striking accumulation of autophagic vacuoles in cells likely reflects an imbalance between the rates of autophagic sequestration and completion of the degradative process. In other words, these cells can be thought of as undergoing ‘autophagic stress’ [25].
In our experiments, AGE-BSA increased the level of LC3-II and had no obviously effect on p62 protein expression in HUVECs. Meanwhile, AGE-BSA reduced cell viability in HUVECs. A cytotoxic effect may be caused by autophagy stress induced by AGE-BSA. Therefore, we propose that treating HUVECs with AGE-BSA caused autophagy dysfunction, and that autophagy flux was not completed.
Our previous experiments showed that pretreatment for 0.5 h with the autophagy inducer rapamycin (Rap) somewhat increased cell viability. Overall, our results indicate that AGE-BSA caused autophagy stress and cellular damage, while raising the level of autophagy, and promoting the completing of autophagy flux, has a protective effect in the cell. Conversely, other studies have indicated that autophagy is involved in AGE-BSA-induced proliferation of VSMCs. The interaction between AGE-BSA and RAGE significantly increased autophagy in VSMCs via the ERK and Akt pathways [20]. Recent studies have shown that PF protects PC12 cells from MPP+ and acidic damage via the autophagic pathway and enhances the autophagic degradation of α-synuclein by regulating the expression and activity of ASICs [26,27]. Researches also convey that paeoniflorin could reduce the expression of HIF-1α, p53, BNIP3, and against hypoxia-induced apoptosis of endothelial cells [28]. Thus, PF exerts protective effects against cytotoxicity. In our results, we found that pretreating cells with PF further increases levels of LC3-II and decreased expression of p62. We therefore postulate that PF promotes completion of autophagy flux and protects HUVECs from AGE-BSA damage via the autophagic pathway.
Atg5 has been characterized as a protein specifically required for autophagy. In many vivo observations, we have found higher atherosclerotic plaque formation incidence rate in macrophage-specific Atg5-knockout mice, also have an increase in both apoptosis and oxidative stress, and decreased efferocytosis [29]. In addition to its role in the formation of autophagosomes, Atg5 fragments produced by calpain cleavage have proapoptotic properties [30]. In order to verify that PF exerts a protective effect via the autophagic pathway, we silenced the expression of Atg5. We found that the protective effect of PF decreased significantly. Thus, we infer that PF can protect HUVECs from AGE-BSA damage by simultaneously upregulating autophagy and promoting the completion of autophagy flux.
RAGE is the main receptor for AGEs. Studies have shown that pretreatment with PF significantly inhibits inflammatory cytokines caused by lysophosphatidylcholine (LPC). PF downregulates the expression of RAGE, TLR-2 and TLR-4 mRNA and protein, and reduces NF-κB activity, thereby inhibiting the production of inflammatory cytokines that are induced by LPC [15]. PF can reduce the levels of proinflammatory cytokines, and has a protective action by increasing oxidative stress and by adding Methylglyoxal (MG) detoxification system, which can against MG-induced cell damage [31]. These results suggest that PF acts on RAGE and exerts anti-inflammatory effects. Studies have shown that AGE-BSA induce cardiomyocyte autophagy by inhibiting the PI3K/Akt/mTOR pathway via RAGE [32]. Investigates have indicated that use amyloid-beta peptide (Aβ) treatment SH-SY5Y cells could increase the formation of autophagosomes, which was mediated by RAGE-calcium-CaMKKβ-AMPK pathway. But inhibition the express of RAGE could attenuate autophagsome formation and AMPK signaling, to the contrary, RAGE overexpression amplified the induction of autophagy [33]. RAGE is a positive regulator of autophagy, and a negative regulator of apoptosis during oxidative stress [34]. some data convey that RAGE may prevent monocyte chemotaxis and attraction into the vessel wall where AGEs deposited form, which mean its key role in the development of vascular disease, especially in patients with diabetes [35]. Together, this suggests that RAGE is important for autophagy. We tested whether PF also increases autophagy through the RAGE pathway, and thus protects HUVECs. We found that AGE-BSA and PF could increase the level of autophagy in HUVECs. However, in HUVECs that were transfected with RAGE siRNA, the level of autophagy was significantly reduced. This illustrates the interaction between AGEs and RAGE, and their connection to the upregulation of autophagy.
In conclusion, our results demonstrate that PF can protect HUVECs from AGE-BSA damage by inducing upregulation of autophagy and promoting the completion of autophagy flux thought the RAGE pathway. But the specific mechanism and downstream signaling pathways that determine the RAGE-mediated autophagy flux completion are not yet clear. Further research is necessary to investigate and uncover these mechanisms. However, where PF exerts cytoprotection by eliminating autophagy stress and promoting the completion of autophagy flux, it may become a new therapeutic target, for example, in diabetics with atherosclerosis and other systemic diseases and complications.
Acknowledgements
The authors would like to thank Suzhou social development fund (SYS201127), program for revitalizing the youth in science and education of Suzhou (KJXW2011018), and the Jiangsu Province Natural Science Fund of China (SBK201241136).
Disclosure of conflict of interest
None.
References
- 1.Yoon MS, Jankowski V, Montag S, Zidek W, Henning L, Schluter H, Tepel M, Jankowski J. Characterisation of advanced glycation endproducts in saliva from patients with diabetes mellitus. Biochem Biophys Res Commun. 2004;323:377–381. doi: 10.1016/j.bbrc.2004.08.118. [DOI] [PubMed] [Google Scholar]
- 2.Kiffin R, Bandyopadhyay U, Cuervo AM. Oxidative stress and autophagy. Antioxid Redox Signal. 2006;8:152–162. doi: 10.1089/ars.2006.8.152. [DOI] [PubMed] [Google Scholar]
- 3.Heymann D. Autophagy: A protective mechanism in response to stress and inflammation. Curr Opin Investig Drugs. 2006;7:443–450. [PMC free article] [PubMed] [Google Scholar]
- 4.Barlovic DP, Soro-Paavonen A, Jandeleit-Dahm KA. RAGE biology, atherosclerosis and diabetes. Clin Sci (Lond) 2011;121:43–55. doi: 10.1042/CS20100501. [DOI] [PubMed] [Google Scholar]
- 5.Dzau VJ, Braun-Dullaeus RC, Sedding DG. Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat Med. 2002;8:1249–1256. doi: 10.1038/nm1102-1249. [DOI] [PubMed] [Google Scholar]
- 6.Sakata N, Meng J, Takebayashi S. Effects of advanced glycation end products on the proliferation and fibronectin production of smooth muscle cells. J Atheroscler Thromb. 2000;7:169–176. doi: 10.5551/jat1994.7.169. [DOI] [PubMed] [Google Scholar]
- 7.Yan SF, Ramasamy R, Schmidt AM. Mechanisms of disease: advanced glycation end-products and their receptor in inflammation and diabetes complications. Nat Clin Pract Endocrinol Metab. 2008;4:285–293. doi: 10.1038/ncpendmet0786. [DOI] [PubMed] [Google Scholar]
- 8.Bierhaus A, Schiekofer S, Schwaninger M, Andrassy M, Humpert PM, Chen J, Hong M, Luther T, Henle T, Kloting I, Morcos M, Hofmann M, Tritschler H, Weigle B, Kasper M, Smith M, Perry G, Schmidt AM, Stern DM, Haring HU, Schleicher E, Nawroth PP. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes. 2001;50:2792–2808. doi: 10.2337/diabetes.50.12.2792. [DOI] [PubMed] [Google Scholar]
- 9.Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–388. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- 10.Soro-Paavonen A, Watson AM, Li J, Paavonen K, Koitka A, Calkin AC, Barit D, Coughlan MT, Drew BG, Lancaster GI, Thomas M, Forbes JM, Nawroth PP, Bierhaus A, Cooper ME, Jandeleit-Dahm KA. Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes. 2008;57:2461–2469. doi: 10.2337/db07-1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kang R, Tang D, Schapiro NE, Livesey KM, Farkas A, Loughran P, Bierhaus A, Lotze MT, Zeh HJ. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 2010;17:666–676. doi: 10.1038/cdd.2009.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun LR, Cao X, Hou FQ, Zhu XH, Gao TM. [Progressive studies of paeoniflorin] . Zhongguo Zhong Yao Za Zhi. 2008;33:2028–2032. [PubMed] [Google Scholar]
- 13.Wu Y, Ren K, Liang C, Yuan L, Qi X, Dong J, Shen J, Lin S. Renoprotective effect of total glucosides of paeony (TGP) and its mechanism in experimental diabetes. J Pharmacol Sci. 2009;109:78–87. doi: 10.1254/jphs.08112fp. [DOI] [PubMed] [Google Scholar]
- 14.Su J, Zhang P, Zhang JJ, Qi XM, Wu YG, Shen JJ. Effects of total glucosides of paeony on oxidative stress in the kidney from diabetic rats. Phytomedicine. 2010;17:254–260. doi: 10.1016/j.phymed.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 15.Li JZ, Wu JH, Yu SY, Shao QR, Dong XM. Inhibitory effects of paeoniflorin on lysophosphatidylcholine-induced inflammatory factor production in human umbilical vein endothelial cells. Int J Mol Med. 2013;31:493–497. doi: 10.3892/ijmm.2012.1211. [DOI] [PubMed] [Google Scholar]
- 16.Chen T, Guo ZP, Jiao XY, Zhang YH, Li JY, Liu HJ. Protective effects of peoniflorin against hydrogen peroxide-induced oxidative stress in human umbilical vein endothelial cells. Can J Physiol Pharmacol. 2011;89:445–453. doi: 10.1139/y11-034. [DOI] [PubMed] [Google Scholar]
- 17.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–937. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 18.Morgan-Bathke M, Lin HH, Chibly AM, Zhang W, Sun X, Chen CH, Flodby P, Borok Z, Wu R, Arnett D, Klein RR, Ann DK, Limesand KH. Deletion of ATG5 shows a role of autophagy in salivary homeostatic control. J Dent Res. 2013;92:911–917. doi: 10.1177/0022034513499350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu P, Lai D, Lu P, Gao J, He H. ERK and Akt signaling pathways are involved in advanced glycation end product-induced autophagy in rat vascular smooth muscle cells. Int J Mol Med. 2012;29:613–618. doi: 10.3892/ijmm.2012.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 22.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee HM, Shin DM, Yuk JM, Shi G, Choi DK, Lee SH, Huang SM, Kim JM, Kim CD, Lee JH, Jo EK. Autophagy negatively regulates keratinocyte inflammatory responses via scaffolding protein p62/SQSTM1. J Immunol. 2011;186:1248–1258. doi: 10.4049/jimmunol.1001954. [DOI] [PubMed] [Google Scholar]
- 24.Chen B, Sun X, Zhang Y, Zhu XQ, Shen HM. Use of inducible Atg5 deletion and expression cell lines in study of the pro-survival function of autophagy under starvation. Biochem Biophys Res Commun. 2012;427:11–17. doi: 10.1016/j.bbrc.2012.08.117. [DOI] [PubMed] [Google Scholar]
- 25.Chu CT. Autophagic stress in neuronal injury and disease. J Neuropathol Exp Neurol. 2006;65:423–432. doi: 10.1097/01.jnen.0000229233.75253.be. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao BY, Yang YP, Luo WF, Mao CJ, Han R, Sun X, Cheng J, Liu CF. Paeoniflorin, a potent natural compound, protects PC12 cells from MPP+ and acidic damage via autophagic pathway. J Ethnopharmacol. 2010;131:122–129. doi: 10.1016/j.jep.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 27.Sun X, Cao YB, Hu LF, Yang YP, Li J, Wang F, Liu CF. ASICs mediate the modulatory effect by paeoniflorin on alpha-synuclein autophagic degradation. Brain Res. 2011;1396:77–87. doi: 10.1016/j.brainres.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 28.Ji Q, Yang L, Zhou J, Lin R, Zhang J, Lin Q, Wang W, Zhang K. Protective effects of paeoniflorin against cobalt chloride-induced apoptosis of endothelial cells via HIF-1alpha pathway. Toxicol In Vitro. 2012;26:455–461. doi: 10.1016/j.tiv.2012.01.016. [DOI] [PubMed] [Google Scholar]
- 29.Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, Robbins J, Martinez J, Tabas I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012;15:545–553. doi: 10.1016/j.cmet.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Codogno P, Meijer AJ. Atg5: more than an autophagy factor. Nat Cell Biol. 2006;8:1045–1047. doi: 10.1038/ncb1006-1045. [DOI] [PubMed] [Google Scholar]
- 31.Choi EM, Suh KS, Rhee SY, Kim YS. Inhibitory effect of paeoniflorin on methylglyoxal-mediated oxidative stress in osteoblastic MC3T3-E1 cells. Phytomedicine. 2014;21:1170–1177. doi: 10.1016/j.phymed.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 32.Hou X, Hu Z, Xu H, Xu J, Zhang S, Zhong Y, He X, Wang N. Advanced glycation endproducts trigger autophagy in cadiomyocyte via RAGE/PI3K/AKT/mTOR pathway. Cardiovasc Diabetol. 2014;13:78. doi: 10.1186/1475-2840-13-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Son SM, Jung ES, Shin HJ, Byun J, Mook-Jung I. Abeta-induced formation of autophagosomes is mediated by RAGE-CaMKKbeta-AMPK signaling. Neurobiol Aging. 2012;33:1006, e1011–1023. doi: 10.1016/j.neurobiolaging.2011.09.039. [DOI] [PubMed] [Google Scholar]
- 34.Kang R, Tang D, Lotze MT, Zeh HJ 3rd. RAGE regulates autophagy and apoptosis following oxidative injury. Autophagy. 2011;7:442–444. doi: 10.4161/auto.7.4.14681. [DOI] [PubMed] [Google Scholar]
- 35.Hori O, Yan SD, Ogawa S, Kuwabara K, Matsumoto M, Stern D, Schmidt AM. The receptor for advanced glycation end-products has a central role in mediating the effects of advanced glycation end-products on the development of vascular disease in diabetes mellitus. Nephrol Dial Transplant. 1996;11(Suppl 5):13–16. doi: 10.1093/ndt/11.supp5.13. [DOI] [PubMed] [Google Scholar]