

Abstract
Objectives: To develop an “overlap syndrome (OS)” rat model by intermittent hypoxia (IH) exposure on the base of pre-existing emphysema, and to explore whether “OS” exposure results in more severe systemic inflammation, and whether the inflammation changes levels of coagulant/anticoagulant factors and oxidative stress status. Methods: Sixty Wistar rats were put into 4 groups: Control group; IH group, IH exposure; Emphysema group, smoke exposure; Overlap group, smoke exposure and IH exposure. We obtained peripheral blood for apoptosis of CD3+CD4+, CD3+CD8+ T lymphocytes and neutrophils, and for endothelial progenitor cell (EPC) counts. Tumor necrosis factor (TNF)-α, interleukin (IL)-6 and coagulant/anticoagulant factors [antithrombin (AT), fibrinogen (FIB), Factor VIII (FVIII) and von Willebrand factor (vWF)] were evaluated. We also obtained tissue blocks of lung, liver, pancreas, and right carotid artery for pathologic scoring and measurements of liver oxidative stress [superoxide dismutase (SOD) activity, catalase (CAT) activity and malondialdehyde (MDA) concentration]. Results: The levels of TNF-α and IL-6, CD3+CD4+ T lymphocyte apoptosis, EPC counts, coagulant factors and MDA are the highest in Overlap group, the lowest in Control group, when the levels of neutrophil apoptosis, CD3+CD8+ T lymphocyte apoptosis, AT, SOD and CAT are the lowest in Overlap group, the highest in Control group (all P values < 0.05). Conclusion: In model animals, when IH is combined with emphysema, there will be a more severe or an “overlapped” systemic/multiple organic inflammation, oxidative stress and hyper-coagulability. And the pro-inflammatory and pro-thrombotic status resulted from “OS” exposure may elicit a robust EPC mobilization, which needs further investigation.
Keywords: Overlap syndrome, intermittent hypoxia, emphysema, inflammation, coagulant factors
Introduction
Chronic obstructive pulmonary disease (COPD) is a common disease in respiratory system whose prevalence has been estimated to be 1% in general population and up to 5%~10% in older adult population. It is a condition of progressive deterioration of the respiratory system characterized by an obstruction of the pulmonary airways and decreasing expiratory airflow [1]. Obstructive sleep apnea (OSA), a common condition affecting 3%~7% of adult men, 2%~5% of adult women [2], and up to 4% of children [3], is characterized by repeated episodes of complete or partial obstruction of the upper airway during sleep, resulting in interruptions of breathing during sleep, recurring episodes of hypoxemia, sleep fragmentation, and daytime sleepiness [4]. Their coexistence, which is denominated as “overlap syndrome (OS)”, indicates a prevalence of 0.5%~1% in the population over 40 years of age [5], and is known to have greater degree of hypoxemia and hypercapnia than either OSA or COPD alone [6], thus increase the risk of systemic inflammation and cardiovascular diseases [7].
Both diseases are characterized by local and systemic inflammations. For COPD, systemic inflammation has been implicated in the pathogenesis of majority of its systemic effects [8], therefore, COPD is suggested as a chronic systemic inflammatory syndrome [9]. On the other hand, there is increased inflammatory cell infiltration within upper airway tissue in OSA patient, which may be related to mechanical trauma and/or increased tissue oxidative stress.
The potential damaging mechanism in OSA may be that the intermittent hypoxia (IH), may cause delayed lymphocyte and neutrophil apoptosis through a range of factors including pro-inflammatory cytokines and hypoxia [10], so as to extend their functional lifespan, leading to exacerbate their functions of producing reactive oxygen species (ROS) and releasing active pro-inflammatory mediators, which may contribute to the systemic inflammation, endothelial dysfunction and subsequent cardiovascular diseases from OSA [11]. Patients with COPD show signs of atherosclerosis which is associated with endothelial dysfunction, hypoxemia, and increased levels of oxidative stress and inflammation biomarkers [12]. These pro-inflammatory processes may stimulate and increase the mobilization of endothelial progenitor cell (EPC) [13], bone-marrow-derived circulating cells that act as an endothelial reservoir for replacing damaged or dysfunctional endothelium and counteracting ongoing endothelial injury [14].
Systemic inflammation and endothelial damages may be particularly worsened in patients with OS, where, systemic inflammation associated with IH, might have overlapped adverse impact on vasculature and even cardiovascular diseases. In this animal study, we exposed emphysematous rats to IH during sleep in a custom-made chamber therefore developing a novo rat model of “OS” exposure. The systemic inflammatory status, including apoptosis of CD3+CD4+, CD3+CD8+ T lymphocyte and neutrophil, EPC counts, serum TNF-α and IL-6 levels, were measured. Liver coagulant/anticoagulant factors [antithrombin (AT)-3, fibrinogen (FIB), Factor VIII (F VIII) and von Willebrand factor (vWF)] and liver oxidative stress status [superoxide dismutase (SOD) activity, catalase (CAT) activity and malondialdehyde (MDA) concentration], were studied. We also obtained tissue blocks of lung, liver, pancreas, and right carotid artery for pathologic scoring.
Methods
The overall rat model experimental protocol (overlap exposure) is IH exposure on the base of pre-existing emphysema. Institutional Review Board of Tianjin Medical University General Hospital approved the ethical and methodological aspects of the investigation (TMU IRB Approving Number: EA-20120002.). Sixty male Wistar rats weighing 120~150 g at age of 4 weeks were divided into 4 groups according to exposure conditions as follows: Control group; IH group, IH exposure; Emphysema group, smoke exposure; and Overlap group, smoke exposure and IH exposure.
Animal cigarette smoke exposure
We used a cigarette smoke exposure device similar with that seen in one of our previous studies [15]. Briefly, rats were passively exposed to the smoke of 15 commercial unfiltered cigarettes for 30 minutes twice daily, in the morning (before 9 am) and in the evening (after 5 pm), for 16 weeks continuously in a custom-made plexiglas chamber.
Animal IH exposure
Model rats were exposed to IH for 4 weeks, 9 AM to 5 PM in every day, from the start of the 13th week of cigarette smoke exposure. We used an IH exposure device similar with that seen in one of our previous studies [15]. Briefly, a gas control delivery system was designed to regulate the flow of nitrogen (IH phase) or clean air (ROX phase) alternatively into a customized IH housing chamber to maintain a designated IH (30 s) or normoxia (90 s) environment.
PMN and PBMC isolation
Polymorphonuclear (PMN) and peripheral blood mononuclear cell (PBMC) were isolated from rat peripheral blood by light density separation using Histopaque 1083 (Histopaque-1083: 1.083 g/ml at 25°C) and Histopaque 1119 (Histopaque-1.119: 1.119 g/ml at 25°C) according to the instructions supplied by the manufacturer.
Neutrophil/lymphocyte apoptosis and quantification of EPC
Neutrophil and lymphocyte Apoptosis were measured using the PE-Annexin V apoptosis Detection Kit I (BD Pharmingen, San Diego, CA) according to the manufacturer’s instructions and analyzed by flow cytometry using a Becton Dickinson FACS Calibur. EPC numbers were analyzed immediately by flow cytometry after isolation of PBMC, and were defined as positive events to PE-conjugated mouse anti-rat CD 133 and FITC-conjugated mouse anti-rat vascular endothelial growth factor receptor (VEGFR)-2 (Becton Dickinson, San Jose, CA, USA).
Measurements for TNF-α and IL-6
The serum levels of TNF-α and IL-6 in peripheral blood of these rats were determined by enzyme-linked immunosorbent assay (ELISA) using commercially available Quantakine® kits.
Measurements for AT, FIB, F VIII and vWF
Plasma was prepared by centrifugation at 1500 g for 10 min at room temperature and was performed for AT, FIB, F VIII and vWF tests through an ACL 9000 automated coagulation analyzer (Beckman Coulter, CA) with proper reagent kits (Dade Behring, IL).
Bronchoalveolar lavage fluid (BALF)
The percentage of macrophages, neutrophils, and lymphocytes in rat BALF was determined using light microscopy. Details are provided as Supporting File.
Tissue preparation for lung, liver, pancreas, and right carotid artery
After BALF, tissue blocks of lung, liver, pancreas, and right carotid artery were got and fixed, and were transversely cut into four μm-thick tissue slices and stained for routine histopathological diagnosis with haematoxylin and eosin (HE).
Histological measurements of lung, liver, pancreas, and right carotid artery
Stratified random pathologic slices were used for making the following measurements, which will also be referred to as “objective measurements” [16]. Assessing criteria of the lung includes mean linear intercept (MLI), mean alveolar number (MAN), and the pathological score of lung inflammation. We also measured pathological score of liver (near hepatic hilar region) inflammation, pathological score of pancreas inflammation and the ratio of carotid intima-media thickness of whole thickness of vascular wall expressed in percent (C-IMT) (%).
Measurements of SOD activity, CAT activity and MDA concentration in liver tissue
Liver tissue blocks were homogenized and then centrifuged, and the resultant supernatant were used for the determinations of SOD, CAT and MDA using commercial analysis kits (Jiancheng Institute of Bioengineering, Nanjing, China) as manufacturer’s instructions.
Statistical analysis
SPSS 11.5 (SPSS Inc., Chicago, IL) software package was used for statistical analysis and illustration. One way analysis of variance (ANOVA) was performed for whole difference and Bonferroni post hoc multiple comparisons were used to evaluate differences between internal groups. Unless otherwise stated, values were reported as mean ± standard deviation (SD), and P < 0.05 is considered statistically significant.
Results
MLI is significantly increased while MAN is decreased in Emphysema or Overlap group compared with IH or Control group (Table 1). In addition, pathological scores show higher levels in Emphysema or Overlap group compared with IH group or Control group (Table 3), suggesting the emphysematous rat model has been established, see Figure 4.
Table 1.
Assessment of the emphysematous lung
| Groups | MAN (/mm3) | MLI (μm) |
|---|---|---|
| Control | 169.9790 ± 5.09127a | 41.5310 ± 1.61759a |
| IH | 164.3760 ± 8.55632a | 43.1280 ± 2.08544a |
| Emphysema | 103.4310 ± 4.24439* | 91.1470 ± 2.50658* |
| Overlap | 83.6860 ± 4.39768* | 98.4160 ± 3.93629* |
| F | 270.505 | 1289.084 |
| P | 0.000 | 0.000 |
MAN: mean alveolar number; MLI: mean linear intercept;
There is statistical difference significantly compared with other groups.
There is no statistical difference between IH and Control group.
Table 3.
Pathological score in lung, liver, pancreas, carotid artery
| Groups | Lung (%) | Liver (%) | Pancreas (%) | C-IMT (%) |
|---|---|---|---|---|
| Control group | 10.30 ± 1.947# | 8.00 ± 1.491# | 7.00 ± 2.582# | 48.8370 ± 1.28601# |
| IH group | 19.20 ± 1.1814# | 13.60 ± 2.951# | 18.50 ± 4.743# | 53.9950 ± 1.80079# |
| Emphysema group | 55.00 ± 4.522# | 58.00 ± 4.619# | 51.50 ± 5.297# | 60.7240 ± 3.24484# |
| Overlap group | 74.60 ± 4.169# | 80.00 ± 6.325# | 77.50 ± 4.859# | 72.0620 ± 3.65512# |
| F | 812.676 | 672.494 | 508.402 | 139.833 |
| P | 0.000 | 0.000 | 0.000 | 0.000 |
Unless otherwise stated, there is significant in Bonferroni post hoc multiple comparisons compared with groups.
Different with others.
Figure 4.
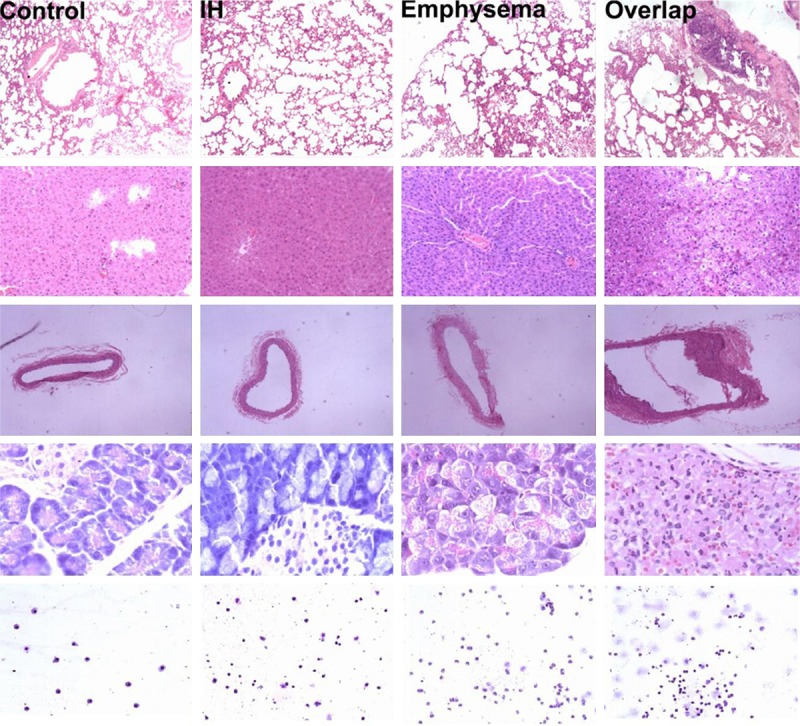
Row 1~5 from the top down are histological photographs of lung, liver, right carotid artery, pancreas and cytological photographs of BALF. Histological photographs are stained with hematoxylin/eosin (H/E) and cytological photographs of BALF are stained with modified Wright-Giemsa. Photographs from lung are captured with 40 × light microscopy; liver, 100 ×; right carotid artery, 40 ×; pancreas, 400 ×; BALF, 100 ×.
From Table 2, neutrophil apoptosis (Figure 1) and CD3+CD8+ T lymphocyte apoptosis (Figure 2) are the highest in Control group, lowest in Overlap group. CD3+CD4+ T lymphocyte apoptosis (Figure 2) and EPC level (Figure 3) are the least in Control group, the most in Overlap group.
Table 2.
Neutrophil/Lymphocyte apoptosis and quantification of EPCs
| Groups | PMN apoptosis (%) | CD3+CD4+ apoptosis (%) | CD3+CD8+ apoptosis (%) | EPCs (cells/μl) |
|---|---|---|---|---|
| Control | 30.87 ± 1.6506228* | 13.02 ± 1.1820885* | 31.75 ± 1.5342389* | 4.502745 ± 0.4343691* |
| IH | 22.71 ± 1.4715449* | 20.65 ± 1.3066071* | 23.65 ± 1.2929295* | 7.21344 ± 0.5855154* |
| Emphysema | 19.45 ± 1.6119347* | 18.49 ± 1.6736188* | 28.42 ± 1.4482173* | 5.84597 ± 0.4310491* |
| Overlap | 8.54 ± 2.5526457* | 22.62 ± 1.5512003* | 18.42 ± 1.1583513* | 8.88927 ± 0.5854478* |
| F | 244.228 | 87.183 | 166.354 | 133.118 |
| P | 0.000 | 0.000 | 0.000 | 0.000 |
PMNs: Polymorphonuclears; EPCs: endothelial progenitor cells.
There is statistical difference significantly compared with other groups.
Figure 1.
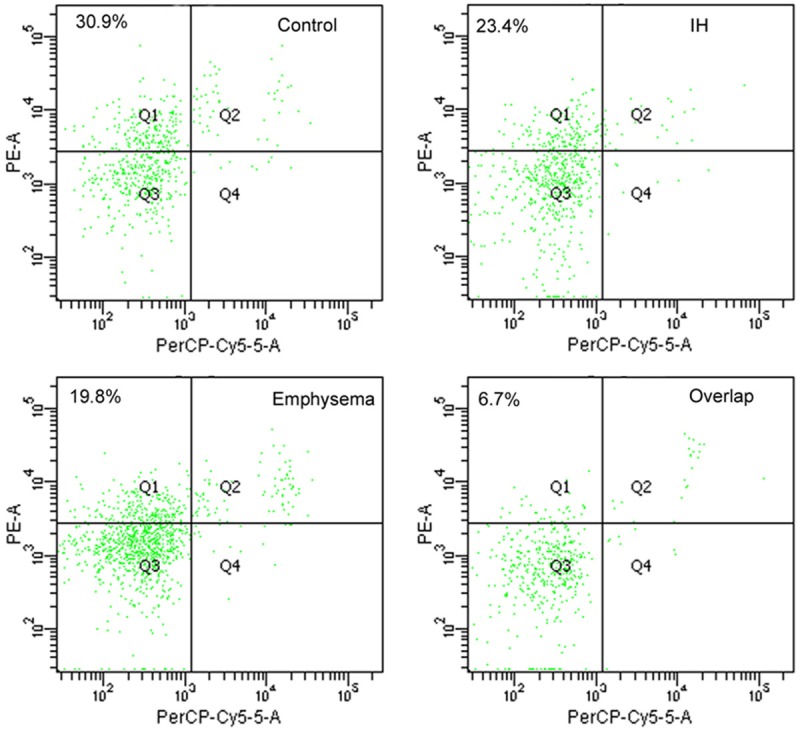
Flow cytometric analysis for early-stage apoptotic neutrophils from rat PMNs, where early-stage apoptotic neutrophils are defined as positive at PE-Annexin V and negative to PerCP-Cy5-5-7-AAD and shown as cells in left upper quadrant.
Figure 2.
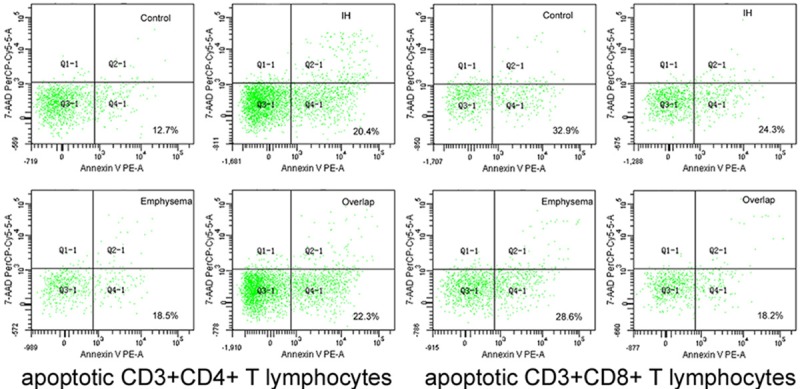
Flow cytometric analysis for early-stage apoptotic CD3+CD4+, CD3+CD8+ T lymphocytes from rat PBMCs, where early-stage apoptotic lymphocytes are defined as positive at PE-Annexin V and negative to PerCP-Cy5-5-7-AAD and shown as cells in right lower quadrant.
Figure 3.
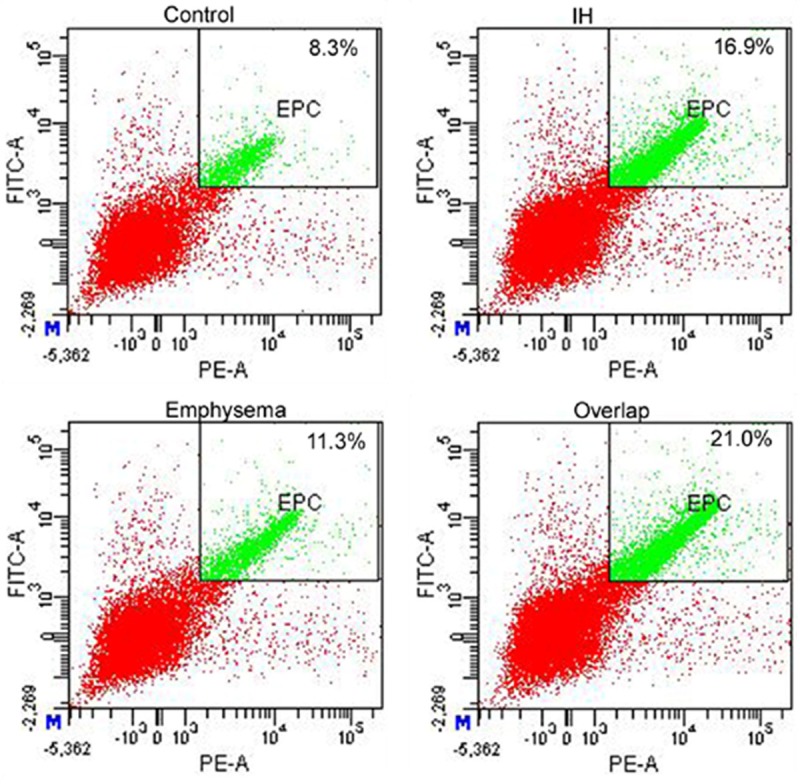
Flow cytometric analysis for EPCs from rat PBMCs, where EPCs are defined as positive events both to CD133 and VEGFR-2 and were shown in green.
The serum levels of TNF-α and IL-6 and histological inflammatory scores of lung, liver, pancreas, and right carotid artery are the lowest in Control group, highest in Overlap group when those in Emphysema group are higher than IH group, see Table S1 (details are provided as Supporting File) and Table 3.
It is shown that the percentage of neutrophils is the most in Emphysema group when there is no statistical difference among other groups. And the percentage of lymphocytes is the least in Emphysema group, see Table S2. Details are provided as Supporting File.
FIB, FVIII, vWF and MDA are the highest in Overlap group, the lowest in Control group, when those values are higher in Emphysema group than IH group, see Table 4 and Table S3. AT, SOD and CAT are the least in Overlap group, the most in Control group, when those are less in Emphysema group than IH group, see Table 4 and Table S3. Details and Table S3 are provided as Supporting File.
Table 4.
Measurements of AT, FIB, F VIII and vWF
| Groups | AT: A (%) | FIB (mg/dl) | vWF: Ag (%) | F VIII: C (%) |
|---|---|---|---|---|
| Control | 123.10 ± 3.281* | 134.730 ± 6.4790* | 48.890 ± 2.4978* | 116.350 ± 3.8254* |
| IH | 117.10 ± 2.885* | 153.120 ± 6.8579* | 57.520 ± 2.3569* | 198.680 ± 7.9140* |
| Emphysema | 105.37 ± 5.779* | 175.790 ± 7.8053* | 65.310 ± 2.3695* | 231.030 ± 13.5187* |
| Overlap | 96.12 ± 5.351* | 201.330 ± 6.6291* | 72.570 ± 3.8268* | 315.920 ± 26.1305* |
| F | 71.570 | 171.073 | 129.446 | 289.047 |
| P | 0.000 | 0.000 | 0.000 | 0.000 |
AT: antithrombin; FIB: fibrinogen; FVIII: Factor VIII; vWF: von Willebrand factor.
There is statistical difference significantly compared with other groups.
Discussion
Our rat model of emphysema, caused by smoke exposure and identified with pathologic criteria, reflects partly the pathophysiologic features of emphysematous COPD which is induced commonly by inhalation of noxious gases and particulate matter resulting in a chronic inflammation in the lung [17].
The apoptosis mechanisms of neutrophils are essential for the resolution of progressive inflammatory responses and for the prevention of related organ or tissue damages [18]. Apoptosis may be delayed by a range of factors such as pro-inflammatory cytokines or hypoxia [10], ostensibly to extend their functional lifespan, resulting in producing larger quantities of ROS. Oxidative stress/inflammation pathways in PMNs mediated by various mechanisms including delayed apoptosis could possibly be activated in patients subject to repeated nightly episodes of IH, leading to cardiovascular morbidities similar to those caused by ischemia/reperfusion injury [19]. Dyugovskaya etc. had the same idea that neutrophil survival increases in response to IH in-vitro as well as in-vivo [20]. Accordingly, reduced levels of neutrophil apoptosis are also well documented in COPD. Delayed cellular apoptosis can prolong the life of neutrophils and lead to their accumulation, resulting in a persistent inflammation in the lung and airway, which is considered to be critical step in COPD pathogenesis [21]. Our data show that neutrophil apoptosis is the highest in Control group, lowest in Overlap group. These results can make a sense that, prolonged neutrophil survival could be further elevated, and this may be particularly true in patients with the OS where, systemic inflammation overlapped with IH, could have additional effects on prolonged neutrophil survival.
CD4+ and CD8+ T lymphocytes are critical against inappropriate immune responses, and the disruption of their differentiation or function can result in autoimmunity diseases and inflammation. More recent studies, defining their phenotypes, show that levels of apoptotic CD3+ and CD4+ T cells are increased when apoptotic CD8+ T cells are reduced [22], and these increased levels of CD8+ cells, are correlated inversely with the degree of airflow limitation [23]. These findings are suggestive of the possible roles of CD8+ T cells in the pathogenesis of airway abnormalities in COPD. It has been established that OSA is associated with increased oxidative stress and inflammatory cell activation, thus propagating inflammatory or immune responses by adhesion molecule releasing, free radical formation and cytotoxicity towards endothelial cells [24]. It has been reported that increased proliferative activity of CD8+ T cells in tonsils may be predominant response in OSA being associated with higher expression and release of the pro-inflammatory cytokines, promoting the development of local inflammatory [25]. Our data show that CD3+CD4+ T lymphocyte apoptosis is the least in Control group, the most in Overlap group, while CD3+CD8+ T lymphocyte apoptosis is the most in Control group, the least in Overlap group. So, when OSA and COPD, with different mechanisms but the same basis of the increased airway inflammation, are presented simultaneously, like in the OS, a more severe inflammatory status maybe expected.
Circulating EPCs could mediates endothelial repair at sites of vascular injury directly by providing a circulating pool of cells to counteract ongoing endothelial cell injury, replace dysfunctional endothelium [13], or indirectly by secreting vasoactive substances to promote repair and angiogenesis [26]. In order to facilitate repair sequences and new vessel formation [27], the increase in EPC correlating with the severity of nocturnal IH is consistent with mobilization of EPC in response to acute hypoxia-reoxygention injury in subjects relatively free of conventional cardiovascular risk factors or disease. Whereas, baseline EPC is reduced in the number or becomes dysfunctional in chronic conditions as later time points of the natural course of OSA. Besides that, several studies demonstrate that both pulmonary and systematic vascular alterations in patients with COPD closely relate to endothelial dysfunction and inflammation [28]. Levels of circulating CD34+ cells appear to increase during exacerbations of COPD, which has been correlated with an elevation in the plasma levels of VEGF from these patients [29]. According to the data in our study, we may hypothesize that a variety of mechanisms are associated between increasing EPCs and IH or emphysema, when both IH and emphysema are exist like those in OS, a further more EPCs should be observed and greater contribution is expected to the development of cardiovascular events.
A systemic inflammatory status can be seen in IH or Emphysema group, not only in blood, but also in lung, liver, pancreas and right carotid artery. Meanwhile, a prothrombotic tendency is also seen in IH or Emphysema group, indicated by AT, FIB and FVIII results. There is an increased risk of thrombotic events occurs in COPD and our concerned coagulant factors, AT, FIB and F VIII, can all be influenced by systemic inflammation comes from emphysema [30]. The underlying mechanism may be that exposure to IH typically shifts the endothelial phenotype toward where anticoagulant characteristics are reduced and pro-inflammatory features dominate the endovascular milieu, resulting in elevated pro-coagulant activities. Another explanation may be IH triggers the release of inflammatory factors and ROS that alter the blood coagulability [31]. The potential influence of inflammation on coagulation may contribute further to thrombogenesis, while abnormal blood coagulation contributes directly to the pathophysiology and mortality of COPD, which means a more severe inflammatory state and a vicious cycle may be created [32].
In conclusion, in model animals, when IH exposure is combined with emphysema, there will be a more severe or an “overlapped” systemic/multiple organic inflammation, oxidative stress and hyper-coagulability which may lead to further health problems as its clinical implication. And the systemic proinflammatory and prothrombotic status resulted from “OS” exposure may elicit a robust EPC mobilization, the consequence of which needs further investigation.
Acknowledgements
We are grateful to the information and correction from Danielle Speer and Ambrose Chiang of Division of Pulmonary and Critical Care Medicine in Duke University Medical Center, Durham, North Carolina, USA. This study was supported by grants from the National Natural Science Foundation of China (81270144, 30800507, and 81170071). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Wysocka E, Cofta S, Cymerys M, Gozdzik J, Torlinski L, Batura-Gabryel H. The impact of the sleep apnea syndrome on oxidant-antioxidant balance in the blood of overweight and obese patients. J Physiol Pharmacol. 2008;59(Suppl 6):761–9. [PubMed] [Google Scholar]
- 2.Young T, Peppard PE, Gottlieb DJ. Epidemiology of obstructive sleep apnea: a population health perspective. Am J Respir Crit Care Med. 2002;165:1217–39. doi: 10.1164/rccm.2109080. [DOI] [PubMed] [Google Scholar]
- 3.Lumeng JC, Chervin RD. Epidemiology of pediatric obstructive sleep apnea. Proc Am Thorac Soc. 2008;5:242–52. doi: 10.1513/pats.200708-135MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carotenuto M, Gimigliano F, Fiordelisi G, Ruberto M, Esposito M. Positional abnormalities during sleep in children affected by obstructive sleep apnea syndrome: the putative role of kinetic muscular chains. Med Hypotheses. 2013;81:306–8. doi: 10.1016/j.mehy.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 5.Weitzenblum E, Chaouat A, Kessler R, Canuet M. Overlap syndrome: obstructive sleep apnea in patients with chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2008;5:237–41. doi: 10.1513/pats.200706-077MG. [DOI] [PubMed] [Google Scholar]
- 6.Marin JM, Soriano JB, Carrizo SJ, Boldova A, Celli BR. Outcomes in patients with chronic obstructive pulmonary disease and obstructive sleep apnea: the overlap syndrome. Am J Respir Crit Care Med. 2010;182:325–31. doi: 10.1164/rccm.200912-1869OC. [DOI] [PubMed] [Google Scholar]
- 7.McNicholas WT. Chronic obstructive pulmonary disease and obstructive sleep apnea: overlaps in pathophysiology, systemic inflammation, and cardiovascular disease. Am J Respir Crit Care Med. 2009;180:692–700. doi: 10.1164/rccm.200903-0347PP. [DOI] [PubMed] [Google Scholar]
- 8.Agustí A. Systemic effects of chronic obstructive pulmonary disease: what we know and what we don’t know (but should) Proc Am Thorac Soc. 2007;4:522–5. doi: 10.1513/pats.200701-004FM. [DOI] [PubMed] [Google Scholar]
- 9.Fabbri LM, Rabe KF. From COPD to chronic systemic inflammatory syndrome? Lancet. 2007;370:797–9. doi: 10.1016/S0140-6736(07)61383-X. [DOI] [PubMed] [Google Scholar]
- 10.Leuenroth SJ, Grutkoski PS, Ayala A, Simms HH. Suppression of PMN apoptosis by hypoxia is dependent on Mcl-1 and MAPK activity. Surgery. 2000;128:171–7. doi: 10.1067/msy.2000.107609. [DOI] [PubMed] [Google Scholar]
- 11.Li C, Jackson RM. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am J Physiol Cell Physiol. 2002;282:C227–41. doi: 10.1152/ajpcell.00112.2001. [DOI] [PubMed] [Google Scholar]
- 12.Kluchová Z, Petrásová D, Joppa P, Dorková Z, Tkácová R. The association between oxidative stress and obstructive lung impairment in patients with COPD. Physiol Res. 2007;56:51–6. doi: 10.33549/physiolres.930884. [DOI] [PubMed] [Google Scholar]
- 13.Fan Y, Ye J, Shen F, Zhu Y, Yeghiazarians Y, Zhu W, Chen Y, Lawton MT, Young WL, Yang GY. Interleukin-6 stimulates circulating blood-derived endothelial progenitor cell angiogenesis in vitro. J Cereb Blood Flow Metab. 2008;28:90–8. doi: 10.1038/sj.jcbfm.9600509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yeh ET, Zhang S, Wu HD, Körbling M, Willerson JT, Estrov Z. Transdifferentiation of human peripheral blood CD34+ -enriched cell population into cardiomyocytes, endothelial cells, and smooth muscle cells in vivo. Circulation. 2003;108:2070–3. doi: 10.1161/01.CIR.0000099501.52718.70. [DOI] [PubMed] [Google Scholar]
- 15.Feng J, Wang QS, Chiang A, Chen BY. The effects of sleep hypoxia on coagulant factors and hepatic inflammation in emphysematous rats. PLoS One. 2010;5:e13201. doi: 10.1371/journal.pone.0013201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thurlbeck WM. Internal surface area and other measurements in emphysema. Thorax. 1967;22:483–96. doi: 10.1136/thx.22.6.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barnes PJ, Celli BR. Systemic manifestations and comorbidities of COPD. Eur Respir J. 2009;33:1165–85. doi: 10.1183/09031936.00128008. [DOI] [PubMed] [Google Scholar]
- 18.Kobayashi SD, Voyich JM, Somerville GA, Braughton KR, Malech HL, Musser JM, DeLeo FR. An apoptosis-differentiation program in human polymorphonuclear leukocytes facilitates resolution of inflammation. J Leukoc Biol. 2003;73:315–22. doi: 10.1189/jlb.1002481. [DOI] [PubMed] [Google Scholar]
- 19.Kin H, Wang NP, Halkos ME, Kerendi F, Guyton RA, Zhao ZQ. Neutrophil depletion reduces myocardial apoptosis and attenuates NFkappaB activation/TNFalpha release after ischemia and reperfusion. J Surg Res. 2006;135:170–8. doi: 10.1016/j.jss.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 20.Dyugovskaya L, Polyakov A, Ginsberg D, Lavie P, Lavie L. Molecular pathways of spontaneous and TNF-{alpha}-mediated neutrophil apoptosis under intermittent hypoxia. Am J Respir Cell Mol Biol. 2011;45:154–62. doi: 10.1165/rcmb.2010-0025OC. [DOI] [PubMed] [Google Scholar]
- 21.Plataki M, Tzortzaki E, Rytila P, Demosthenes M, Kousopoulos A, Siafakas NM. Apoptosis mechanism in the pathogenesis of COPD. Int J COPD. 2006;1:161–171. doi: 10.2147/copd.2006.1.2.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baraldo S, Lokar Oliani K, Turato G, Zuin R, Saetta M. The Role of Lymphocytes in the Pathogenesis of Asthma and COPD. Curr Med Chem. 2007;14:2250–6. doi: 10.2174/092986707781696573. [DOI] [PubMed] [Google Scholar]
- 23.Chrysofakis G, Tzanakis N, Kyriakoy D, Tsoumakidou M, Tsiligianni I, Klimathianaki M, Siafakas NM. Perforin expression and cytotoxic activity of sputum CD8+ lymphocytes in patients with COPD. Chest. 2004;125:71–6. doi: 10.1378/chest.125.1.71. [DOI] [PubMed] [Google Scholar]
- 24.Lavie L. Obstructive sleep apnoea syndrome--an oxidative stress disorder. Sleep Med Rev. 2003;7:35–51. doi: 10.1053/smrv.2002.0261. [DOI] [PubMed] [Google Scholar]
- 25.Dyugovskaya L, Lavie P, Lavie L. Phenotypic and functional characterization of blood gammadelta T cells in sleep apnea. Am J Respir Crit Care Med. 2003;168:242–9. doi: 10.1164/rccm.200210-1226OC. [DOI] [PubMed] [Google Scholar]
- 26.Hacievliyagil SS, Mutlu LC, Temel İ. Airway inflammatory markers in chronic obstructive pulmonary disease patients and healthy smokers. Niger J Clin Pract. 2013;16:76–81. doi: 10.4103/1119-3077.106771. [DOI] [PubMed] [Google Scholar]
- 27.Shantsila E, Watson T, Tse HF, Lip GY. New insights on endothelial progenitor cell subpopulations and their angiogenic properties. J Am Coll Cardiol. 2008;51:669–71. doi: 10.1016/j.jacc.2007.09.057. [DOI] [PubMed] [Google Scholar]
- 28.Kamota T, Li TS, Morikage N, Murakami M, Ohshima M, Kubo M, Kobayashi T, Mikamo A, Ikeda Y, Matsuzaki M, Hamano K. Ischemic pre-conditioning enhances the mobilization and recruitment of bone marrow stem cells to protect against ischemia/reperfusion injury in the late phase. J Am Coll Cardiol. 2009;53:1814–22. doi: 10.1016/j.jacc.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 29.Matsuoka S, Yamashiro T, Diaz A, Estépar RS, Ross JC, Silverman EK, Kobayashi Y, Dransfield MT, Bartholmai BJ, Hatabu H, Washko GR. The relationship between small pulmonary vascular alteration and aortic atherosclerosis in chronic obstructive pulmonary disease: quantitative CT analysis. Acad Radiol. 2011;18:40–46. doi: 10.1016/j.acra.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vaidyula VR, Criner GJ, Grabianowski C, Rao AK. Circulating tissue factor procoagulant activity is elevated in stable moderate to severe chronic obstructive pulmonary disease. Thromb Res. 2009;124:259–61. doi: 10.1016/j.thromres.2008.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ryan S, Taylor CT, McNicholas WT. Selective activation of inflammatory pathways by intermittent hypoxia in obstructive sleep apnea syndrome. Circulation. 2005;112:2660–7. doi: 10.1161/CIRCULATIONAHA.105.556746. [DOI] [PubMed] [Google Scholar]
- 32.Feng J, Chen BY, Guo MN, Cao J, Zhao HY, Liang DC, Zuo AJ. Interleukin-6 and tumor necrosis factor-alpha levels of endothelial cells in different hypoxiamodes: in vitro experiment. Zhonghua Yi Xue Za Zhi. 2007;87:774–7. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.