

Abstract
Purpose: This study was designed to investigate the effect of losartan on the myocardial interstitial fibrosis in diabetic cardiomyopathy (DCM) rats. Methods: In this study, a total of 48 male Wister rats (3 groups of 16 animals each) were examined, including the control group, DCM group and losartan-treated (DCM + L) group. Control group was fed with standard diet (14 KJ/g); DCM group and losartan-treated (DCM + L) group were both fed with high glucose and fat diet (20 KJ/g). Diabetes was induced by streptozotocin (STZ) intraperitoneal injuction (IP, 30 mg/kg body weight). Rats of DCM + L group were treated with losartan (30 mg/kg body weight) daily by oral gavage for 16 weeks. Biochemical, hemodynamic, histological and western blotting analyses were performed. Results: Compared with DCM rats, the quantity of p-JAK2 and p-STAT3 in myocardium of rats treated with losartan was lower, the expression of TGF-β1 was down-regulate, the content of collagen in myocardium decreased, LVSP and ± dp/dt increased, LVEDP decreased, the level of myocardial fibrosis reduced, and heart function improved evidently. Conclusion: Losartan has a protective effect on heart function against myocardial interstitial fibrosis of DCM by inhibiting JAK/STAT signaling pathway and lowering the expression of TGF-β1.
Keywords: Diabetes mellitus, diabetic cardiomyopathy, myocardial interstitial fibrosis, losartan, JAK, STAT
Introduction
With the improvement of people’s living standard, type 2 diabetes caused by excess nutrients which is a worldwide public health problem has become a global disease and it’s the top 5 reason causing death in the world [1]. 1972, Rubler first put the concept of diabetic cardiomyopathy (DCM) forward, and thought it was an independent cardiomyopathy from the large vessels and coronary atherosclerosis [2]. With further researches, DCM has been recognized as a unique diabetes complication, and it is closely associated with the high incidence and mortality of cardiovascular disease in diabetic patients [3-5].
The early-onset diastolic heart failure is a characteristic of DCM, with a decreased myocardial relaxation and an increased myocardial stiffness. And one important cause of the development of diastolic heart failure is myocardial interstitial fibrosis. The development of DCM is shown by the myocardial structural changes and it will lead to the myocardial interstitial fibrosis [6]. Transforming growth factor β1 (TGF-β1) is a kind of polypeptide regulates cell growth and differentiation [7]. As one of the most important factors to promote myocardial fibrosis, it promotes the expression of college, also the synthesis and deposition in extracellular matrix. Cytokine signal transduction pathway JAK/STAT is closely related to cardiac hypertrophy caused by pressure overload, heart failure, cardiac dysfunction induced by ischemia-reperfusion, etc. [8].
Studies indicate that JAK/STAT signaling pathway of vascular endothelial cells can be activated by both high glucose and angiotensin II in vitro, and JAK2 will be phosphorylated followed by the phosphorylation of STAT1 and STAT3, leading to the proliferation of endothelial cells [9]. Inhibiting JAK/STAT signaling pathway of mesangial cells cultured in high glucose can decrease the synthesis of TGF-β1 and fibrin [10].
Clinical trials have confirmed that angiotensin 1 (AT1) receptor antagonist can delay the development of diabetic nephropathy. But whether the antagonist of AT1 receptor has the protective effect on DCM is unknown. It is necessary to research the relationship between JAK/STAT signaling pathway and TGF-β1 in DCM and whether the AT1 receptor antagonist can reduce myocardial interstitial fibrosis in DCM by JAK/STAT signaling pathway. Therefore, an animal model of diabetes was established, and DCM rats are treated with losartan in this study, to investigate the effect of losartan on myocardial interstitial fibrosis in DCM.
Materials and methods
Animals
The experimental study was conducted on 60 male Wistar rats, aged 60-90 days and weighing approximately 200 ± 20 g, provided by Hebei Provincial Laboratory Animal Center. Experimental procedures were performed in accordance with the Guidance Suggestions for the Care and Use of Laboratory Animals, formulated by the Ministry of Science and Technology of the People’s Republic of China in 1998, and was approved by the Animal Ethics Committee of Hebei Medical University.
Animal models
Rats are randomly divided into three groups: control group, fed with standard diet (14 KJ/g); DCM group and losartan-treated (DCM + L) group, both fed with high glucose and fat diet (20 KJ/g). After 5 weeks, the rats of DCM and DCM + L groups were injected intraperitoneally with STZ (Sigma, USA, 30 mg/kg body weight) in citrate buffer (pH 4.2) to establish diabetic models, and the control rats were only injected with an equal volume of citrate buffer. The rats with a fasting blood glucose ≥ 16.7 mmol/L, a lower ISI (insulin sensitivity index) than control rats, polydipsia, polyphagia and polyuria were regarded as diabetic. And then losartan (30 mg/kg body weight) was administered daily by oral gavage to rats in DCM + L group for 16 weeks. After the experiments all the rats were sacrificed by carotid artery exsanguination.
Biochemical analysis
Rats were weighted weekly, and the fasting blood glucose was determined every 4 weeks. One week before and after STZ injection, and at the end of experiments, the blood was drawn to determine the fasting blood glucose (FBG), fasting insulin (FINS), total cholesterol (T-CHO) and triglycerides (TG). Insulin sensitivity index [12] [ISI = ln (FINS × FBG)-1] and insulin resistance index [13] (HOMA-IR = FBG × FINs/22.5) were calculated. Blood glucose was measured by a blood glucose meter (Johnson & Johnson, USA). TG was measured by glycerol phosphate oxidase method (Triglycerides assay kit, Biosino, China). T-CHO was measured by enzymatic colorimetric method (Total cholesterol assay kit, Biosino, China). Insulin was measured by radioimmunoassay (Rat insulin RIA kit, BNIBT).
Hemodynamics parameters
At the end of experiments, rats were connected to pressure transducer via right common carotid artery intubation, recording heart rate (HR) and mean arterial pressure (MAP) by a Powerlab/8 s polygraph (AD Instrument, Australia), and the catheter was inserted into the left ventricle to determine the left ventricular systolic pressure (LVSP), left ventricular end-diastolic pressure (LVEDP), left ventricular pressure change rate (± dp/dtmax) and other hemodynamic parameters.
Histological examinations
After hemodynamic analysis, rat hearts were harvested and weighted. Left ventricular anterior walls from each experimental group (n = 6) were fixed in 4% paraformaldehyde, dehydrated in graded ethanol, deparaffinized in xylene, routine paraffin-embedded, sectioned, HE stained and examined for pathological changes of myocardial cells under light microscope.
Apical myocardium from each experimental group (n = 8) were cut into blocks of 2 mm × 2 mm × 2 mm on ice, fixed in 2.5% glutaraldehyde, washed twice in 0.1 mol·L-1 cacodylate buffer, post-fixed in 1% osmium tetroxide, dehydrated in acetone series, impregnated in epoxy resin, embedded, ultrathin sectioned, uranyl acetate-lead citrate stained, and examined the ultrastructure changes of myocardium cells with transmission electron microscope (TEM).
Determination of collagen content in myocardium
About 100 mg left ventricular anterior walls from each experimental group (n = 6) were used to measure myocardial hydroxyproline content with a hydroxyproline detection kit (Nanjing Jiancheng Biotechnique Institute) and normalized to total protein. Because hydroxyproline is incorporated only into collagen, and it is assumed that collagen contains 13.4% of total hydroxyproline, myocardial collagen content (mg/g heart weight) = hydroxyproline content × 7.46 [14].
Llevel of phospho-JAK2 (p-JAK2) and phospho-STAT3 (p-STAT3) in myocardial tissue
Extraction of myocardial protein: about 100mg myocardium tissue was cut into pieces, and total protein was extracted with RIPA lysate (1% NP-40, 1% SDS, 150 mmol/L NaCl, 50 mmol/L Tris-Cl, pH 7.5, 10% glycerol, 1 mmol/L sodium vanadate, 1 mmol/L PMSF). The protein concentration was determined by the CBB (Coomassie brilliant blue) method.
Western blotting
50 µg sample protein per lane was separated by 8% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene-difluoride (PVDF) membrane. The membranes were incubated in 5% skimmed milk for 2 h at 37°C, and overnight at 4°C with primary antibodies (mice anti-p-JAK2, 1:200 dilution, Santa Cruz; p-STAT3, 1:200 dilutions, Santa Cruz). β-actin was used as a loading control. The membranes were exposed to the negative films to develop target bands after incubated with enhanced chemiluminescence (Santa Cruz, USA). The intensities of bands were quantitated by LabWorks 4.5 software (UVP, USA).
Expression of TGF-β1 in myocardial tissue
The expression of TGF-β1 in myocardial tissue was determined by Western blotting. TGF-β1 was separated by 12% SDS-PAGE.
Statistics analysis
SPSS 13.0 was used for statistical analysis. All data were expressed as mean ± SD (Standard deviations), and the statistical differences among different groups were assessed by one-way ANOVA. The two groups were compared using paired t test, and Spearman correlation was used to analyze the relationship between two variables. P < 0.05 indicated a significant difference; P < 0.01 indicated that there was a very significant difference.
Results
Experimental rats had clinical features compatible with type II diabetes
Since the blood glucose of 9 rats didn’t meet 16.7 mmol/L and 3 rats died during experiments, a total of 48 rats completed the experiment, and control, DCM and DCM + L groups each had 16 rats.
At the end of experiment, the heart weights of rats were normalized against their tibial length (HW/TL). HW/TL of DCM group rats was significantly lower than the control rats (P < 0.05) (Table 1). There were no significant differences between DCM and DCM + L group (P < 0.05).
Table 1.
The heart weight in each group at the end of experiment
| Group | N | Tibial length (mm, TL) | Heart weight (g, HW) | HW/TL |
|---|---|---|---|---|
| Control | 8 | 45.21 ± 1.13 | 1.45 ± 0.062 | 0.0321 ± 0.0236 |
| DCM | 8 | 44.23 ± 1.07 | 1.37 ± 0.063** | 0.0310 ± 0.0352* |
| DCM + L | 8 | 45.53 ± 1.21 | 1.39 ± 0.054** | 0.0305 ± 0.0295 |
P < 0.05 vs. control group;
P < 0.01 vs. control group.
The FBG, T-CHO and TG of DCM and DCM + L rats were significantly higher than the control (P < 0.05 or P < 0.01). Compared to the control rats, DCM and DCM + L rats had significantly lower FINS and ISI (P < 0.01), while had a higher HOMA-IR (P < 0.05) (Table 2).
Table 2.
The biochemical indexes in each group at the end of experiment (x̅ ± s, n = 8)
| Group | FBG (mmol/L) | Insulin (U/L) | ISI | HOMO-IRI | TG (mmol/L) | T-CHO (mmol/L) |
|---|---|---|---|---|---|---|
| Control | 5.96 ± 0.62 | 64.28 ± 21.06 | -5.81 ± 0.38 | 15.76 ± 5.84 | 0.32 ± 0.11 | 1.14 ± 0.22 |
| DCM | 28.68 ± 5.46** | 18.37 ± 5.95** | -6.41 ± 0.38** | 28.86 ± 12.05* | 0.68 ± 0.17* | 1.94 ± 0.19** |
| DCM + L | 28.39 ± 6.18** | 17.29 ± 6.41** | -6.32 ± 0.26** | 26.35 ± 9.47* | 0.73 ± 0.15* | 1.84 ± 0.23** |
P < 0.05 vs. control group;
P < 0.01 vs. control group.
The biochemical characteristics of experimental rats were compatible with type II diabetes, suggesting the rat model of type II diabetic was successfully established in this study.
Hemodynamics parameters changed significantly in experimental rats
Compared to the control rats, LVSP and ± dp/dt of DCM and DCM + L rats were significantly lower (P < 0.05 or P < 0.01), LVEDP was higher (P < 0.05 or P < 0.01), however HR and MAP were not significantly different (P > 0.05). Compared to DCM group, rats of DCM + L group had significantly higher LVSP and ± dp/dt (P < 0.05), a lower LVEDP (P < 0.05), and no significant differences for HR and MAP (P > 0.05) (Table 3).
Table 3.
Changes of cardiac function in each group at the end of experiment
| Group | N | MAP (mmHg) | LVSP (mmHg) | LVEDP (mmHg) | + dp/dtmax (mmHg/s) | -dp/dtmax (mmHg/s) |
|---|---|---|---|---|---|---|
| Control | 8 | 106.83 ± 13.72 | 146.00 ± 6.45 | 2.71 ± 1.11 | 6234 ± 465 | 4517 ± 819 |
| DCM | 8 | 95.00 ± 9.56* | 118.00 ± 8.56* | 8.75 ± 2.05** | 4960 ± 750* | 3430 ± 749** |
| DCM + L | 8 | 98.00 ± 10.12* | 129.00 ± 7.43*,# | 5.68 ± 1.89*,# | 5483 ± 585*,# | 3947 ± 743*,# |
P < 0.05 vs. control group;
P < 0.01 vs. control group;
P < 0.05 vs. DCM group.
Myocardial injuries were induced in experimental rats
Observed by light microscopy, myocardial cell of DCM rats were predominantly of swelling, loss or disappearance of cross striations within myocardial fibers, accumulation of myocardial extracellular matrix and increasing collagen (Figure 1). DCM + L group had decreased swelling and less extracellular matrix and collagen than DCM group.
Figure 1.
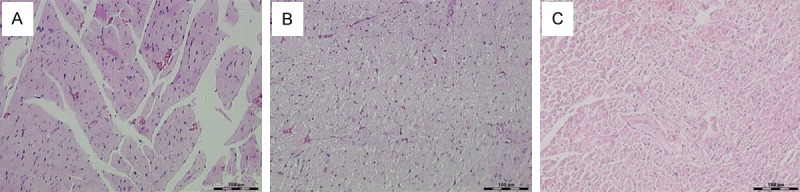
Pathological changes of myocardium by light microscope. A: Control group HE staining × 200; B: DCM group HE staining × 200; C: DCM + L group HE staining × 200.
As the ultrastructural observation by electron microscopy, DCM rats had myocardial fiber disorganization, myocardial cells and nucleus swelling, severe mitochondria swelling and cristae disorganization, breaking, lysis and disappearance (Figure 2). However DCM + L group rats were observed with normal myocardial nucleus, organized myofilament and no extracellular collagen accumulation.
Figure 2.

Pathological changes of ultrastructure of myocardium by electron microscope. A: Control group × 4000; B: DCM group × 25000; C: DCM group × 4000; D: DCM + L group × 4000.
The collagen content in myocardium was significantly higher in experimental rats
As the analysis, the collagen content in myocardium of DCM (n = 6, 12.48 ± 3.51 mg/g) and DCM + L (n = 6, 27.50 ± 6.64 mg/g) group rats was significantly higher than control group (n = 6, 20.20 ± 5.33 mg/g) (P < 0.01), and DCM + L group had a lower content of myocardial collagen than DCM group (P < 0.05) (Figure 3). It suggested that a myocardial fibrosis was carried out in experimental rats.
Figure 3.

Change of cardium collagen in each group. *P < 0.05, **P < 0.01 vs. control group; #P < 0.05, ##P < 0.01 vs. DCM group.
The expression of p-JAK2 and p-STAT3 was up-regulated in myocardium of rats treated with lorsartan
The expression of TGF-β1 in myocardium of DCM group rats was higher than control group (P < 0.05), and the level of p-JAK2 and p-STAT3 in myocardium was also higher (P < 0.05, P < 0.01) (Figure 4). As the correlation analysis, the relative content of p-JAK2 and the expression of TGF-β1 in myocardium of DCM rats were positively correlated (r = 0.643, P < 0.05), and the relative content of p-JAK2 and the collagen content in myocardium of DCM rats were also positively correlated (r = 0.67, P < 0.05). The expression of TGF-β1 and the level of p-JAK2 and p-STAT3 in myocardium of DCM + L group rats was significantly lower than DCM group (P < 0.05), but higher than control group (P < 0.05 or P < 0.01) (Figure 4).
Figure 4.
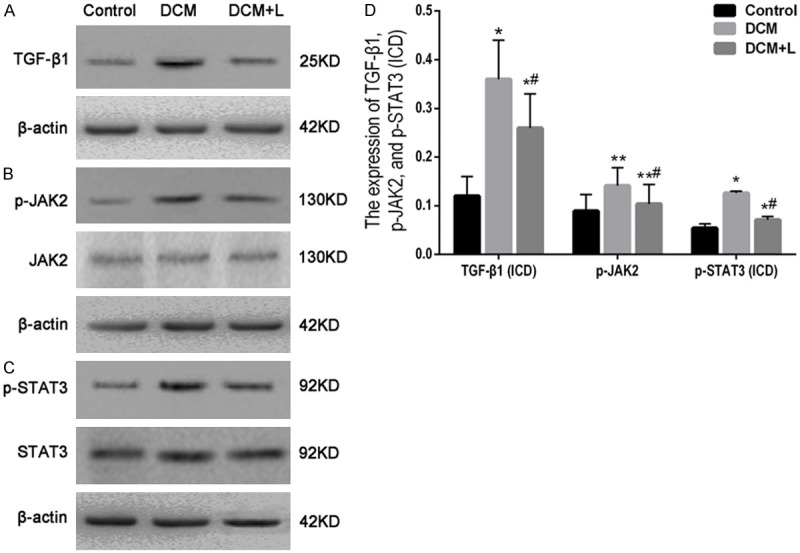
Western blotting and quantification. A: Western blotting of TGF-β1 in myocardium; B: Western blotting of p-JAK2 in myocardium; C: Western blotting of p-STAT3 in myocardium; D: Bar graph for the quantitative comparison among TGF-β1, p-JAK2 and p-STAT3 expression quantity of the three groups. *P < 0.05, **P < 0.01 vs. control group; #P < 0.05 vs. DCM group.
Discussion
Cardiovascular complication of diabetes is the main cause of hospitalization and death [15]. DCM is an independent complication of diabetes, characterized by early-onset diastolic dysfunction followed by systolic dysfunction and a diastolic heart failure of which myocardial interstitial fibrosis takes an important part in the occurrence and development at the end [16,17]. Diabetic patients are more possible to have a heart failure than hypertension or ischemic heart disease patients and have a worse prognosis [18].
Studies indicate that activation of renin-angiotensin system (RAS) is important to the occurrence and development of myocardial interstitial fibrosis in DCM [19]. Studies shown that diabetic rats have cardiomyocyte hypertrophy and myocardial interstitial collagen deposition at 10-12 weeks after the onset of diabetes while the level of Ang II in myocardium increased significantly and Ang II in serum increased 3 months after diabetes [20]. Ang II is a multifunctional hormone and it regulates the heart function by activating Angiotensin II receptor type 1 [21]. Ang II can cause cell hypertrophy and stimulate collagen production and transforming growth factor-β1 (TGF-β1) released from cardiac fibroblasts simulated by Ang II can promote the development of myocardial interstitial fibrosis. TGF-β1 is currently considered one of the most important factors to promote myocardial fibrosis. Studies have shown that TGF-β1 can regulate the growth and differentiation of many cell types and increase the collagen and extracellular matrix production [22]. Hyperglycemia, hyperinsulinemia and insulin resistance and other metabolic disorders can increase the secretion of TGF-β1 [23], and it will promote the collagen production by combining with TGF-β1 receptors high expressed in left ventricles of type II diabetic rats [24]. Therefore, in diabetes RAS is activated, and Ang II will cause heart disease and vascular fibrosis while the increasing expression of TGF-β1 in cardiac cells and fibroblasts can enhance the fibrosis induced by Ang II [25].
As the results, the TGF-β1 expression in myocardium of DCM group rats was significantly higher than control and positively correlated with the content of collagen. It suggested that TGF-β1 was very important to the myocardial interstitial fibrosis in DCM. Compared to DCM group, rats treated with losartan had less TGF-β1 expression in myocardium, less collagen, increased LVSP and ± dp/dt, decreased LVEDP, improved heart function, reduced myocardial fibrosis and lighter histopathological damages. It showed losartan intervention reduced myocardial fibrosis and improved heart function for DCM rats. As a specific AT1 receptor antagonist, losartan prevent Ang II binding to the AT1 receptor to protect myocardium from effects of Ang II on the myocardial fibrosis. But further studies are still needed to know which pathways losartan delay myocardial interstitial fibrosis in DCM through.
JAK kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway is an important cytokine signal transduction pathway, regulating diverse physiological and pathological processes including cellular immunity, proliferation, differentiation, apoptosis and inflammation.
Ang II is one of the factors can activate JAK/STAT signaling pathway. Studies have shown that Ang II can quickly induce the phosphorylation of JAK2 and Tyk2 in cultured cardiomyocytes, to phosphorylate STAT1 and STAT2 and induce the phosphorylation of STAT3 in 2 hours [26]. High glucose or glycosylated products in diabetes can stimulate RAS to produce Ang II followed by the activation of JAK/STAT signaling pathway.
In our study, the quantity of p-JAK2 and p-STAT3 was significant up-regulate in rats of DCM group contrast with control rats. And the relative content of p-JAK2 was positively correlated to the expression of TGF-β1, also the content of collagen in myocardium. Compared with DCM rats, the quantity of p-JAK2 and p-STAT3 in myocardium of rats treated with losartan was lower, the expression of TGF-β1 was down-regulate, the content of collagen in myocardium decreased, LVSP and ± dp/dt increased, LVEDP decreased, the level of myocardial fibrosis reduced, and heart function improved evidently. This declares that losartan has a protective effect on myocardial interstitial fibrosis in DCM by inhibiting JAK/STAT signaling pathway.
As a specific AT1 receptor antagonist, losartan blocks the effect of Ang II followed by inhibiting JAK/STAT signaling pathway and the down-regulated expression of TGF-β1, and delays the development of myocardial interstitial fibrosis in DCM to improve heart function. In conclusion, losartan had a protective effect on myocardial interstitial fibrosis in DCM by a down-regulated JAK/STAT signaling pathway.
Disclosure of conflict of interest
None.
References
- 1.R S, J S, P Z, R T. Diabetes atlas 2003. 2nd edition. Brussels, Belgium: International Diabetes Federation; 2003. [Google Scholar]
- 2.Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol. 1972;30:595–602. doi: 10.1016/0002-9149(72)90595-4. [DOI] [PubMed] [Google Scholar]
- 3.Hayat SA, Patel B, Khattar RS, Malik RA. Diabetic cardiomyopathy: mechanisms, diagnosis and treatment. Clin Sci (Lond) 2004;107:539–557. doi: 10.1042/CS20040057. [DOI] [PubMed] [Google Scholar]
- 4.Bell DS. Diabetic cardiomyopathy. A unique entity or a complication of coronary artery disease? Diabetes Care. 1995;18:708–714. doi: 10.2337/diacare.18.5.708. [DOI] [PubMed] [Google Scholar]
- 5.Spector KS. Diabetic cardiomyopathy. Clin Cardiol. 1998;21:885–887. doi: 10.1002/clc.4960211205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watanabe K, Thandavarayan RA, Harima M, Sari FR, Gurusamy N, Veeraveedu PT, Mito S, Arozal W, Sukumaran V, Laksmanan AP, Soetikno V, Kodama M, Aizawa Y. Role of differential signaling pathways and oxidative stress in diabetic cardiomyopathy. Curr Cardiol Rev. 2010;6:280–290. doi: 10.2174/157340310793566145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Massague J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 8.Kiu H, Nicholson SE. Biology and significance of the JAK/STAT signalling pathways. Growth Factors. 2012;30:88–106. doi: 10.3109/08977194.2012.660936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marrero MB, Schieffer B, Paxton WG, Heerdt L, Berk BC, Delafontaine P, Bernstein KE. Direct stimulation of Jak/STAT pathway by the angiotensin II AT1 receptor. Nature. 1995;375:247–250. doi: 10.1038/375247a0. [DOI] [PubMed] [Google Scholar]
- 10.Boengler K, Hilfiker-Kleiner D, Drexler H, Heusch G, Schulz R. The myocardial JAK/STAT pathway: from protection to failure. Pharmacol Ther. 2008;120:172–185. doi: 10.1016/j.pharmthera.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 11.Srinivasan K, Viswanad B, Asrat L, Kaul CL, Ramarao P. Combination of high-fat diet-fed and low-dose streptozotocin-treated rat: a model for type 2 diabetes and pharmacological screening. Pharmacol Res. 2005;52:313–320. doi: 10.1016/j.phrs.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 12.G L. The answers to some quetions of the applications of insulin sensitivity index 1/(FPG × FINS) Chinese Journal of Diabetes. 1998;22:44–48. [Google Scholar]
- 13.X C. Determination and evaluation of β-cell function and insulin resistance in type 2 diabetes. Chinese Journal of Microcirculation. 2004;26:24–26. [Google Scholar]
- 14.Neuman RE, Logan MA. The determination of collagen and elastin in tissues. J Biol Chem. 1950;186:549–556. [PubMed] [Google Scholar]
- 15.Bauters C, Lamblin N, Mc Fadden EP, Van Belle E, Millaire A, de Groote P. Influence of diabetes mellitus on heart failure risk and outcome. Cardiovasc Diabetol. 2003;2:1. doi: 10.1186/1475-2840-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodrigues B, McNeill JH. The diabetic heart: metabolic causes for the development of a cardiomyopathy. Cardiovasc Res. 1992;26:913–922. doi: 10.1093/cvr/26.10.913. [DOI] [PubMed] [Google Scholar]
- 17.Conrad CH, Brooks WW, Hayes JA, Sen S, Robinson KG, Bing OH. Myocardial fibrosis and stiffness with hypertrophy and heart failure in the spontaneously hypertensive rat. Circulation. 1995;91:161–170. doi: 10.1161/01.cir.91.1.161. [DOI] [PubMed] [Google Scholar]
- 18.Solang L, Malmberg K, Ryden L. Diabetes mellitus and congestive heart failure. Further knowledge needed. Eur Heart J. 1999;20:789–795. doi: 10.1053/euhj.1998.1472. [DOI] [PubMed] [Google Scholar]
- 19.Fiordaliso F, Li B, Latini R, Sonnenblick EH, Anversa P, Leri A, Kajstura J. Myocyte death in streptozotocin-induced diabetes in rats in angiotensin II- dependent. Lab Invest. 2000;80:513–527. doi: 10.1038/labinvest.3780057. [DOI] [PubMed] [Google Scholar]
- 20.Lassila M, Davis BJ, Allen TJ, Burrell LM, Cooper ME, Cao Z. Cardiovascular hypertrophy in diabetic spontaneously hypertensive rats: optimizing blockade of the renin-angiotensin system. Clin Sci (Lond) 2003;104:341–347. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 21.Touyz RM, Schiffrin EL. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev. 2000;52:639–672. [PubMed] [Google Scholar]
- 22.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 23.Li JH, Huang XR, Zhu HJ, Johnson R, Lan HY. Role of TGF-beta signaling in extracellular matrix production under high glucose conditions. Kidney Int. 2003;63:2010–2019. doi: 10.1046/j.1523-1755.2003.00016.x. [DOI] [PubMed] [Google Scholar]
- 24.Campbell SE, Katwa LC. Angiotensin II stimulated expression of transforming growth factor-beta1 in cardiac fibroblasts and myofibroblasts. J Mol Cell Cardiol. 1997;29:1947–1958. doi: 10.1006/jmcc.1997.0435. [DOI] [PubMed] [Google Scholar]
- 25.Yang F, Chung AC, Huang XR, Lan HY. Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-beta-dependent and -independent Smad pathways: the role of Smad3. Hypertension. 2009;54:877–884. doi: 10.1161/HYPERTENSIONAHA.109.136531. [DOI] [PubMed] [Google Scholar]
- 26.Mascareno E, Siddiqui MA. The role of Jak/STAT signaling in heart tissue renin-angiotensin system. Mol Cell Biochem. 2000;212:171–175. [PubMed] [Google Scholar]