

Abstract
Diabetes is a frequent and increasing public health problem with a large economic burden in modern society. Endothelial cells dysfunction was involved in the development of diabetes-associated diseases. Sirtuins are a conserved family of NAD-dependent deacetylases. However, the role of sirtuins in diabetes-associated endothelial cell dysfunction was relatively unknown. In this study, we focus on the intrinsic link between SIRT3, a mitochondrial sirtuin, and high glucose-induced endothelial cells dysfunction. We showed that loss of SIRT3 expression was associated with decreased viability in endothelial cells from diabetes patients. Knockdown of SIRT3 decreased viability of endothelia cells exposed to high glucose condition. Further, mechanistic study showed that SIRT3 repression results in SOD2 acetylation, leading to SOD2 inactivation, which enhanced high glucose-induced oxidative stress in endothelial cells. Our data suggested that SIRT3 protects endothelial cells from high glucose-induced cytotoxicity. Our findings are considered a significant step toward a better understanding of diabetes-associated vascular diseases.
Keywords: Diabetes, SIRT3, endothelial cell dysfunction, high glucose
Introduction
Diabetes is a frequent and increasing public health problem with a large economic burden in modern society. Hyperglycaemia, β-cell dysfunction and insulin resistance are the hallmark clinical manifestation of diabetes [1]. Diabetes is associated with an increased risk of multiple complications, including microvascular disorder, obesity and hypertension which may finally lead to cardiovascular disease [2]. Although cardiovascular diseases result in approximately 80% of diabetes-associated death [3], the mechanisms responsible for the development of diabetes-associated cardiovascular disease are still unclear.
Accumulating evidences had shed a light on the involvement of endothelial cells dysfunction in the development of diabetes-associated diseases. It was demonstrated the bioavailability of NO, a crucial short-lived molecule, is reduced in endothelial cells from diabetes patients, which is associated with disorder in vascular tone [4]. Further, using an Nrf2/antioxidant response element (ARE)-driven luciferase reporter gene assay, it was shown that the transcription activity of Nrf2 was enhanced under diabetic conditions. Additionally, high-fat diet-induced endothelial dysfunction was more se-vere in Nrf2-/- mice, as shown by the significantly diminished acetylcholine-induced relaxation of aorta of these animals compared with HFD-fed Nrf2+/+ mice [5]. It is also reported that expression of miR-503 in endothelial cells is upregulated under high glucose treatment and ischemia-associated starvation. Delivery of miR-503 inhibitor to the ischemic adductor of diabetic mice rescued diabetes mellitus-induced impairment of postischemic angiogenesis and blood flow recovery [6]. However, detailed illustration of the key factors in high glucose-induced cytotoxicity in endothelial cells was still needed.
Sirtuins are a conserved family of NAD-dependent ADP-ribosyltransferases and/or protein deacetylases involved in metabolism, stress response. The mammalian sirtuin family consists of seven sirtuins that are localized to the nucleus (SIRT1, SIRT6, and SIRT7), mitochondria (SIRT3, SIRT4, and SIRT5), and cytoplasm (SIRT2), respectively, and that regulate a wide range of intracellular process [7]. Among them, SIRT3 is mainly localized in mitochondria and regulates several key oxidative pathways by targeting many enzymes involved in central metabolism. For instance, SIRT3 mediates deacetylation of complex I and complex II to activate electron transport [8]. SIRT3 also induces fatty acid oxidation during fasting in hepatocytes via deacetylation of LCAD [9]. Since morphological and functional changes in mitochondrial disrupt the endothelial physiological function [10], we proposed that SIRT3 may have a role in endothelial cell dysfunction in diabetes. In this study, we showed that SIRT3 expression was associated with decreased viability in endothelial cells from diabetes patients. Loss of SIRT3 expression enhanced high glucose-induced cytotoxicity. Further, mechanistic study showed that ROS production and SOD2 deacetylation was involved in SIRT3-mediated protection of endothelial cells.
Materials and methods
Clinical samples
The blood samples were obtained from diabetes patients and healthy donors at First Affiliated Hospital of Harbin Medical University. The study was approved by the Institutional Ethics Committee of First Affiliated Hospital of Harbin Medical University. All participants gave written informed consent prior to sampling. A total of 12 ml blood sample was obtained from each diabetic patient or healthy donor. About 2 ml blood was used to determine the glucose concentration, and 10 ml blood sample was used for isolation of circulating endothelial cells.
Isolation of circulating endothelial cells
Human circulating endothelial cells (CD34+) were isolated from blood samples by using the MACS system (Miltenyi Biotec GmbH, Germany), as described previously [11,12].
Cells culture
Human umbilical vein endothelial (HUVEC) cells were maintained in low glucose Dulbecco’s modified Eagle’s medium (DMEM; Gibco, USA) containing 5 mM glucose, 10% fetal calf serum (Gibco, USA), penicillin (100 U/L) and streptomycin (10 mg/L).
Immunoblot
Cell samples were harvested and lysed in lysis buffer (Beyotime, China). Samples were separated by SDS-PAGE and transferred to PVDF membranes (Amersham Biosciences). The membranes were blocked with PBS containing 0.1% Tween 20 and 5% skimmed milk at 37°C for 1 h and then probed by primary antibodies. The blots were probed with secondary antibody (diluted 1:10000; Santa Cruz Biotechnology) conjugated to horseradish peroxidase for 2 h at room temperature. Blots were visualized by enhanced chemiluminescence reagents (Cell signaling technology).
DCF staining
Endogenous ROS production was examined by staining cells with 2’,7’-dichloro-fluorescein diacetate (DCFH-DA) (GENMED, GMS10016.2) according to the manufacturer’s instructions. The DCFH-DA signal was measured by Molecular Devices SPECTRAMAX M5 fluorimeter at 490 nm excitation and 530 nm emission.
Proliferation assay
MTT assay was performed using 96-well plate according to the manufacturer’s instructions. Cells were incubated with MTT (Sigma, St. Louis, MO) for 2 h, and then the insoluble substance dissolved in DMSO. The absorbance was read by multi-well spectrophotometer (MDC, Sunnyrale, CA) at 595 nm.
For BrdU labeling assay, cells were incubated with BrdU (Roche Applied Science, Indianapolis, IN) at final concentration at 10 mM for 4 h. BrdU signal was detected by using 5-Bromo-2’-deoxy-uridine Labeling and Detection Kit III (Roche) and read by multi-well spectrophotometer (MDC, Sunnyrale, CA) at 405 nm.
TUNEL assay
TUNEL staining was performed by using Dead End Fluorometric TUNEL system (Promega) following the manufacture’s instruction. The TUNEL-positive cells were visualized and counted via fluorescence microscope (Zeiss, Germany).
Metabolite assay
The level of G16DP, citrate and succinate was measured by using PicoProbe™ Fructose-6-Phosphate Fluorometric Assay Kit, Succinate (Succinic Acid) Colorimetric Assay Kit and Citrate Colorimetric/Fluorometric Assay Kit, respectively, all of which were purchased from Biovision.
Statistics analysis
Differences between two groups were assessed by Student’s t test. Leaner regressions were tested by using the Spearman rank correlation. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Results
Loss of SIRT3 expression was associated with decreased viability in endothelial cells from diabetes patients
As an initial test, we determined the impact of high glucose condition on SIRT3 expression in endothelial cells. Circulating endothelial cells from diabetes patients and normal healthy donors were isolated, and the expression level of SIRT3 was examined by real time RT-PCR. As shown in Figure 1A, no apparent changes were found in SIRT3 expression in circulating endothelial cells from diabetes patients versus normal donors (P = 0.97).
Figure 1.
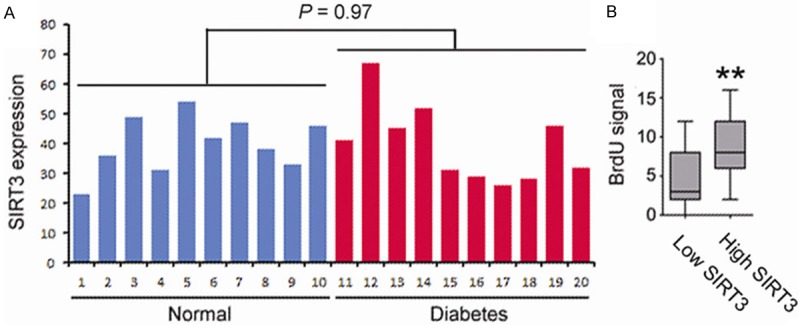
A. Circulating endothelial cells (CD34+) were isolated from blood samples from diabetes patients and normal healthy donors. Expression of SIRT3 was examined and compared by real time PCR. B. The culture of circulating endothelial cells were divided into two groups based on SIRT3 expression. The cells were then treated with 30 mM glucose for 48 h, and cell proliferation was examined by BrdU assay.
Interestingly, we found that, among the diabetic patients, the circulating endothelial cells harboring a low SIRT3 expression showed a decreased cell proliferation rate compared to those cells with high SIRT3 expression (Figure 1B). These results suggested that loss of SIRT3 expression was associated with decreased viability in endothelial cells from diabetic patients.
SIRT3 protected endothelial cells from high glucose-induced cytotoxicity
It was previously observed that exposure to diabetic mellitus causes endothelial cells dysfunction, even leading to cell death [13]. Thus we sought to determine if SIRT3 possessed a role in high glucose-induced cytotoxicity in endothelial cells. To this end, primary culture of human umbilical vein endothelial (HUVEC) cells was established. HUVEC cells were transfected with specific siRNA targeting SIRT3 (siSIRT3) or non-targeting siRNA (siNC), and then treated with 30 mM glucose.
To unveil whether SIRT3 was involved in high glucose-induced cytotoxicity in endothelial cells, proliferation and apoptosis in endothelial cells was examined. As shown, the proliferating cells were notably reduced upon SIRT3 knockdown, revealed by both MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (Figure 2A) and BrdU incorporation assay (Figure 2B). In MTT assay, the proliferation ratio in siSIRT3-treated cells was decreased by 55.6%, compared to siNC-treated cells (Figure 2A). Similarly, the BrdU signal decreased by 48.20% upon SIRT3 knockdown in BrdU incorporation assay (Figure 2B, P < 0.01). Further, loss of SIRT3 expression also increased apoptosis in endothelial cells under high glucose condition. As shown in Figure 2C, a significantly increased number of TUNLE-positive cells were found in siSIRT3-treated HUVEC cells (51.25±4.41), compared to siNC-treated cells (32.75±3.50, P < 0.01). Collectively, this data suggested that SIRT3 protected endothelial cells from high glucose-induced cytotoxicity.
Figure 2.
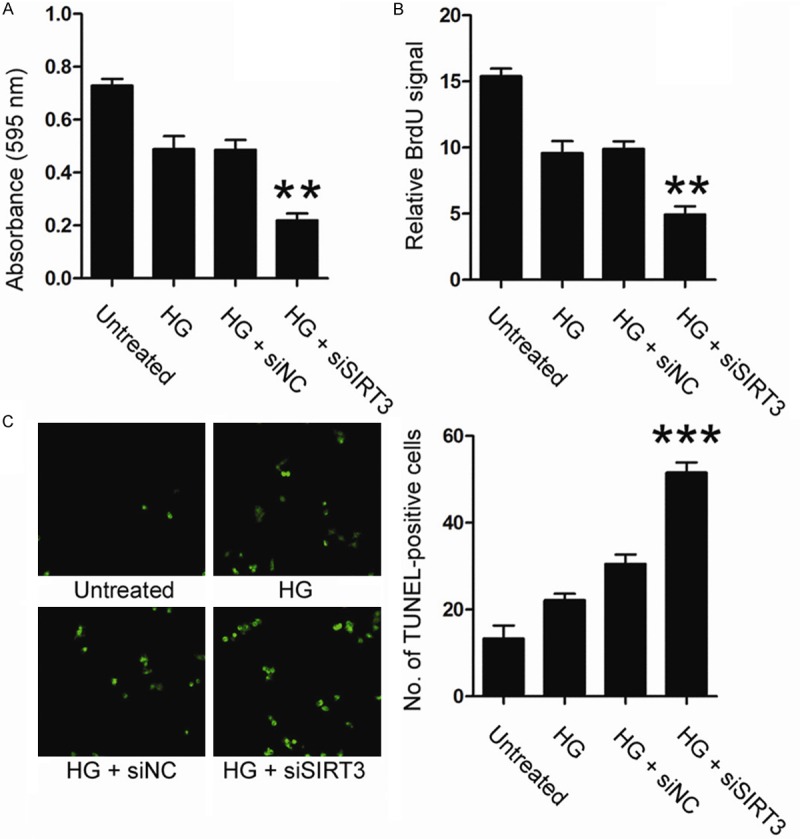
HUVEC cells were transfected with siSIRT3 or siNC, and then treated with 30 mM glucose for 48 h. Cell proliferation was examined by MTT assay (A) and BrdU assay (B). Cell apoptosis was examined by TUNEL assay (C). HG, high glucose.
SIRT3 protected endothelial cells by modulating high glucose-induced ROS production
It was reported that diabetic mellitus could cause oxidative stress, which was implicated in high glucose-mediated cytotoxicity [14]. We thus asked whether SIRT3 had an impact on ROS production in high glucose-treated endothelial cell. To this goal, endogenous ROS production in HUVEC cells was examined by using 2’,7’-dichloro-fluoresce-in diacetate (DCF) fluorescent probe. As shown in Figure 3A, loss of SIRT3 expression increased intracellular ROS level by approximately 2.5 folds, compared to control cells.
Figure 3.
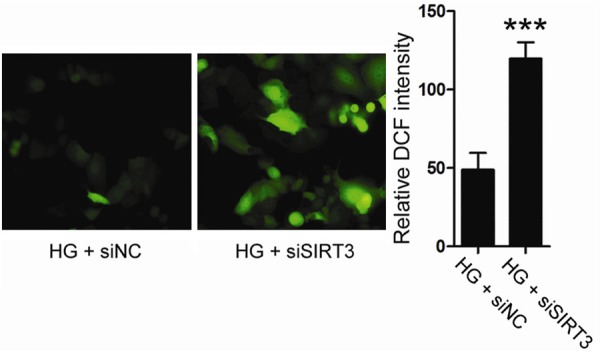
HUVEC cells were transfected with siSIRT3 or siNC, and then treated with 30 mM glucose for 48 h. Intracellular ROS level was examined by DCF staining. HG, high glucose.
To determine whether SIRT3 rescued endothelial cell viability by regulating oxidative stress, a ROS inhibitor, NAC was used. As shown in Figure 4A-C, though knockdown of SIRT3 enhanced the high glucose-induced cytotoxicity, no apparent differences were found in either proliferation or apoptosis between siSIRT3- or siNC-treated cells when these cells were pre-treated with NAC. Therefore, these data suggested that SIRT3 protected endothelial cells by modulating high glucose-induced ROS production.
Figure 4.
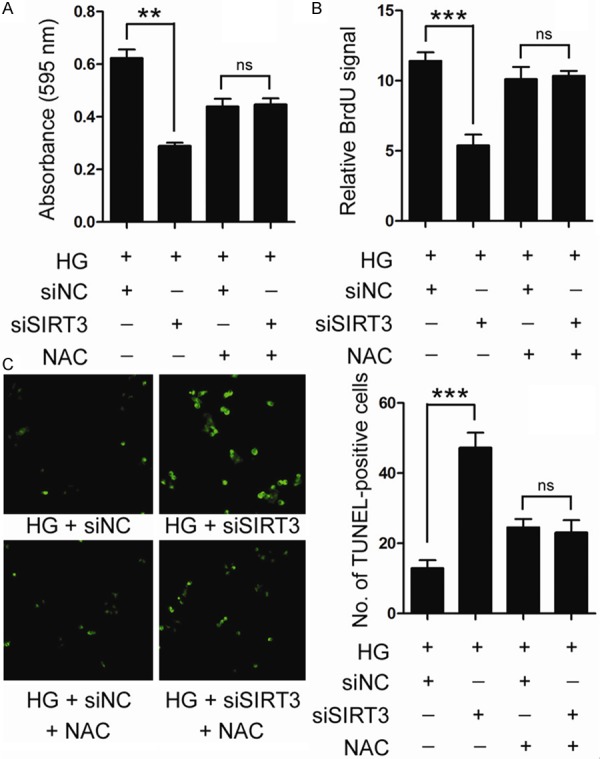
HUVEC cells were pre-treated with 10 mM NAC for 4 h. HUVEC cells were then transfected with siSIRT3 or siNC, and treated with 30 mM glucose for 48 h. Cell proliferation was examined by MTT assay (A) and BrdU assay (B). Cell apoptosis was examined by TUNEL assay (C). HG, high glucose.
SIRT3 regulates high glucose-induced oxidative stress by mediating SOD2 deacetylation
It is widely accepted that glycolysis and TAC cycle is the major source for cellular ROS production [15]. Further, SIRT3 plays a crucial role in regulating glycolysis and TAC cycle through modulating the key intermediates of these two metabolic pathways, including G16PD, citrate and succinate [8,9]. To explore the potential mechanisms underlying SIRT3-mediated ROS inhibition, the level of G16PD, citrate and succinate were examined. As results, loss of SIRT3 expression had no further impact on the level of these three intermediates (Figure 5A).
Figure 5.
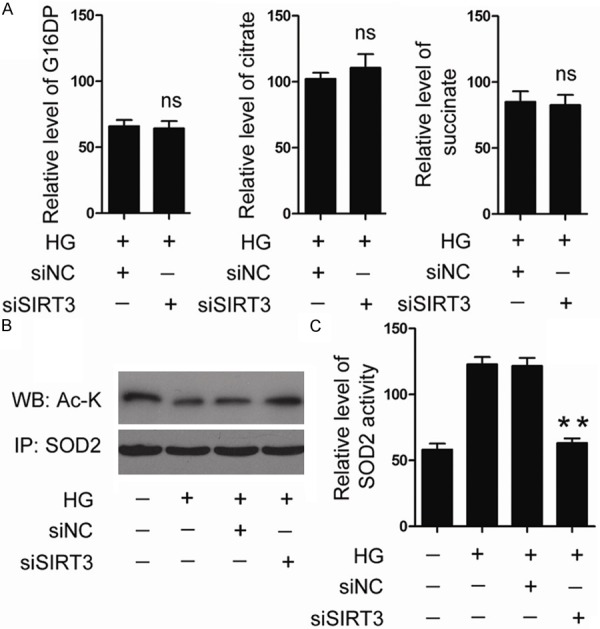
HUVEC cells were transfected with siSIRT3 or siNC, and then treated with 30 mM glucose for 48 h. Intracellular level of G16DP, citrate and succinate was examined by HPLC (A). Acetylation status of SOD2 was examined by immunoprecipitation followed by immunoblot; (B) SOD2 activity was determined by SOD2 activity assay kit. (C) HG, high glucose.
It is reported that SOD2 is an important ROS scavenger, and SIRT3 mediated SOD2 deacetylation at Lys68 which is an essential step of SOD2 activation [16]. Thus we have particular interests to examine the impact of SIRT3 on the acetylation status of SOD2. As shown in Figure 5B, 5C, treatment with 30 mM glucose reduced SOD2 acetylation and elevated SOD2 activity. However, high glucose-induced activation of SOD2 was markedly abolished by SIRT3 knockdown. These data suggested that SIRT3 regulates high glucose-induced oxidative stress by mediating SOD2 deacetylation.
Discussion
Endothelial cells dysfunction which was resulted from chronic exposure to various stressors, such as high glucose and oxidative stress was common in diabetes. Mitochondria are central to cellular metabolism, and dysregulation has been implicated in numerous diseases, including diabetes [17]. Understanding the regulation of mitochondrial enzymes and their associated pathways is essential to grasp how mitochondria function and adapt numerous metabolic processes and maintain cellular viability under the demands of diverse physical or pathological conditions [18].
Sirtuins are NAD-dependent deactylases that share homology to the yeast Sir2 protein. Their enzymatic activity is regulated by the ratio of NAD to NADH; high NAD levels activate sirtuins and conversely high NADH levels inhibit activity [19]. Of the three mitochondrial sirtuins, SIRT3 is the most studied to date and has been long been considered as a tumor suppressor. It is reported that siRNA-mediated knockdown of SIRT3 in colorectal cancer cell line augmented HIF-1α protein stabilization and transcriptional activity in hypoxic conditions, which results in a significant increase in cell proliferation and tumorigenic capability in xenograft models [20]. Further, knockout of SIRT3 in MEF induced chromosomal instability and decreased mitochondrial integrity. Though SIRT3 knockout MEFs could not immortalize, expression of a single oncogene (Myc or Ras) in SIRT3-deleted MEFs results in vitro transformation and altered intracellular metabolism [21]. Recently, accumulating evidences have linked SIRT3 to diabetes. It is shown that expression of SIRT3 was suppressed in pancreatic islets isolated from human type 2 diabetic patients, and knockdown of SIRT3 in mouse pancreatic beta cell resulted in lowered insulin secretion and increased beta cell apoptosis [22]. Moreover, apelin gene therapy increases myocardial vascular density and ameliorates diabetic cardiomyopathy via upregulation of SIRT3 [23]. In present data, we investigated the intrinsic link between high glucose-associated endothelial dysfunction and SIRT3. We demonstrated that though the expression level of SIRT3 was almost unchanged in the circulating endothelial cells from diabetes patients, the endothelial cells with a low level of SIRT3 expression exhibit reduced cell viability in response to high glucose treatment, compared to those cells with a high SIRT3 expression. Further, knockdown of SIRT3 enhanced high glucose-induced proliferation inhibition and apoptosis in endothelial cells. Therefore, our data suggested a protective role of SIRT3 in endothelial cells against high glucose-induced cytotoxicity.
Oxidative stress has long been considered as important reasons for diabetic vascular complications [24]. In the represent data, loss of SIRT3 expression enhanced high glucose-induced oxidative stress in endothelial cells. Further treatment with NAC reduced the intracellular ROS level, which in turn abolished SIRT3 knockdown-induced proliferation inhibition and apoptosis under high glucose treatment, suggesting that SIRT3 protects endothelial cell from high glucose stimulation probably through reducing intracellular ROS level.
Abnormal changes in glycolysis and mitochondrial metabolism were the major source for ROS production [14]. Aberrant accumulation of intracellular ROS could activate anti-oxidant proteins, including SOD2, which functions as ROS scavengers [25]. It is reported that, as a major mitochondrial deacetylase, SIRT3 reduced intracellular ROS level mainly by targeting either key metabolic enzymes or anti-oxidant proteins [9,16]. In this study, we demonstrated that loss of SIRT3 expression markedly increased acetylation form of SOD2 in endothelial cells exposed to high glucose condition, which led to SOD2 inactivation, while repression of SIRT3 expression had no impact on the level of several metabolic intermediate products. Therefore, it is reasonable to infer that SIRT3 regulates high glucose-induced oxidative stress mainly by mediating SOD2 deacetylation, but not modulating the activity of metabolic enzymes.
Disclosure of conflict of interest
None.
References
- 1.Quagliaro L, Piconi L, Assaloni R, Martinelli L, Motz E, Ceriello A. Intermittent high glucose enhances apoptosis related to oxidative stress in human umbilical vein endothelial cells: the role of protein kinase C and NAD(P)H-oxidase activation. Diabetes. 2003;52:2795–2804. doi: 10.2337/diabetes.52.11.2795. [DOI] [PubMed] [Google Scholar]
- 2.Morgan CL, Currie CJ, Stott NC, Smithers M, Butler CC, Peters JR. The prevalence of multiple diabetes-related complications. Diabet Med. 2000;17:146–151. doi: 10.1046/j.1464-5491.2000.00222.x. [DOI] [PubMed] [Google Scholar]
- 3.Ford ES. Risks for all-cause mortality, cardiovascular disease, and diabetes associated with the metabolic syndrome: a summary of the evidence. Diabetes Care. 2005;28:1769–1778. doi: 10.2337/diacare.28.7.1769. [DOI] [PubMed] [Google Scholar]
- 4.Kojda G, Harrison D. Interactions between NO and reactive oxygen species: pathophysiological importance in atherosclerosis, hypertension, diabetes and heart failure. Cardiovasc Res. 1999;43:562–571. doi: 10.1016/s0008-6363(99)00169-8. [DOI] [PubMed] [Google Scholar]
- 5.Ungvari Z, Bailey-Downs L, Gautam T, Jimenez R, Losonczy G, Zhang C, Ballabh P, Recchia FA, Wilkerson DC, Sonntag WE, Pearson K, de Cabo R, Csiszar A. Adaptive induction of NF-E2-related factor-2-driven antioxidant genes in endothelial cells in response to hyperglycemia. Am J Physiol Heart Circ Physiol. 2011;300:H1133–1140. doi: 10.1152/ajpheart.00402.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caporali A, Meloni M, Völlenkle C, Bonci D, Sala-Newby GB, Addis R, Spinetti G, Losa S, Masson R, Baker AH, Agami R, le Sage C, Condorelli G, Madeddu P, Martelli F, Emanueli C. Deregulation of microRNA-503 contributes to diabetes mellitus-induced impairment of endothelial function and reparative angiogenesis after limb ischemia. Circulation. 2011;123:282–291. doi: 10.1161/CIRCULATIONAHA.110.952325. [DOI] [PubMed] [Google Scholar]
- 7.Inoue T, Hiratsuka M, Osaki M, Oshimura M. The molecular biology of mammalian SIRT proteins: SIRT2 in cell cycle regulation. Cell Cycle. 2007;6:1011–1018. doi: 10.4161/cc.6.9.4219. [DOI] [PubMed] [Google Scholar]
- 8.Bell EL, Guarente L. The SirT3 divining rod points to oxidative stress. Mol Cell. 2011;42:561–568. doi: 10.1016/j.molcel.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV Jr, Alt FW, Kahn CR, Verdin E. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie Z, Zhang J, Wu J, Viollet B, Zou MH. Upregulation of mitochondrial uncoupling protein-2 by the AMP-activated protein kinase in endothelial cells attenuates oxidative stress in diabetes. Diabetes. 2008;57:3222–3230. doi: 10.2337/db08-0610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Bonanno G, Mariotti A, Procoli A, Corallo M, Scambia G, Pierelli L, Rutella S. Interleukin-21 induces the differentiation of human umbilical cord blood CD34-lineage-cells into pseudomature lytic NK cells. BMC immunology. 2009;10:46. doi: 10.1186/1471-2172-10-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peichev M, Naiyer AJ, Pereira D, Zhu Z, Lane WJ, Williams M, Oz MC, Hicklin DJ, Witte L, Moore MAS. Expression of VEGFR-2 and AC133 by circulating human CD34+ cells identifies a population of functional endothelial precursors. Blood. 2000;95:952–958. [PubMed] [Google Scholar]
- 13.Cosentino F, Hishikawa K, Katusic ZS, Lüscher TF. High glucose increases nitric oxide synthase expression and superoxide anion generation in human aortic endothelial cells. Circulation. 1997;96:25–28. doi: 10.1161/01.cir.96.1.25. [DOI] [PubMed] [Google Scholar]
- 14.Wolff SP, Jiang ZY, Hunt JV. Protein glycation and oxidative stress in diabetes mellitus and ageing. Free Radic Biol Med. 1991;10:339–352. doi: 10.1016/0891-5849(91)90040-a. [DOI] [PubMed] [Google Scholar]
- 15.Fernie AR, Carrari F, Sweetlove LJ. Respiratory metabolism: glycolysis, the TCA cycle and mitochondrial electron transport. Curr Opin Plant Biol. 2004;7:254–261. doi: 10.1016/j.pbi.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 16.Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010;12:662–667. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 17.Loomans CJ, de Koning EJ, Staal FJ, Rookmaaker MB, Verseyden C, de Boer HC, Verhaar MC, Braam B, Rabelink TJ, van Zonneveld AJ. Endothelial progenitor cell dysfunction: a novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes. 2004;53:195–199. doi: 10.2337/diabetes.53.1.195. [DOI] [PubMed] [Google Scholar]
- 18.Geary K, Knaub LA, Schauer IE, Keller AC, Watson PA, Miller MW, Garat CV, Nadeau KJ, Cree-Green M, Pugazhenthi S, Regensteiner JG, Klemm DJ, Reusch JE. Targeting mitochondria to restore failed adaptation to exercise in diabetes. Biochem Soc Trans. 2014;42:231–238. doi: 10.1042/BST20130283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haigis MC, Guarente LP. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913–2921. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- 20.Schumacker PT. SIRT3 controls cancer metabolic reprogramming by regulating ROS and HIF. Cancer Cell. 2011;19:299–300. doi: 10.1016/j.ccr.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, van der Meer R, Nguyen P, Savage J, Owens KM, Vassilopoulos A, Ozden O, Park SH, Singh KK, Abdulkadir SA, Spitz DR, Deng CX, Gius D. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell. 2010;17:41–52. doi: 10.1016/j.ccr.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caton PW, Richardson SJ, Kieswich J, Bugliani M, Holland ML, Marchetti P, Morgan NG, Yaqoob MM, Holness MJ, Sugden MC. Sirtuin 3 regulates mouse pancreatic beta cell function and is suppressed in pancreatic islets isolated from human type 2 diabetic patients. Diabetologia. 2013;56:1068–1077. doi: 10.1007/s00125-013-2851-y. [DOI] [PubMed] [Google Scholar]
- 23.Zeng H, He X, Hou X, Li L, Chen JX. Apelin gene therapy increases myocardial vascular density and ameliorates diabetic cardiomyopathy via upregulation of sirtuin 3. Am J Physiol Heart Circ Physiol. 2014;306:H585–597. doi: 10.1152/ajpheart.00821.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johansen JS, Harris AK, Rychly DJ, Ergul A. Oxidative stress and the use of antioxidants in diabetes: linking basic science to clinical practice. Cardiovasc Diabetol. 2005;4:5. doi: 10.1186/1475-2840-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med. 2002;33:337–49. doi: 10.1016/s0891-5849(02)00905-x. [DOI] [PubMed] [Google Scholar]