

Abstract
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is an uncommon extranodal non-Hodgkin lymphoma, with an aggressive course with no well-defined treatment. This article describes a 56-year-old man, treated surgically 7 months earlier for a subcutaneous nodosity near the left axilla, presenting with a progressive inflamed wound, pain, and high fever (39°C). Treatment with systemic antibiotics and topical anti-inflammatory dressings failed. After 7 months, the patient was diagnosed with SPTCL based on biopsy results and a multidisciplinary consultation. While undergoing systemic chemotherapy with corticosteroid therapy, his wound become more painful, larger, and covered with necrotic tissue. Fifty days after chemotherapy with corticosteroid therapy, his wound became seriously painful and increasingly necrotic. He developed a serious stomachache and abdominal distension, rapidly became comatose, and died. The aim of this case report is to present our experience of the different clinical signs of SPTCL to expedite its early diagnosis in future. We summarize the main clinical characteristics of SPTCL as a rapidly progressing and increasingly painful wound with necrotic tissue, involving a multisystem disorder, which is easily misdiagnosed, responds poorly to corticosteroid and chemotherapy treatments, and has a high mortality rate. The pathological characteristics are early inflammation, advancing to profuse infiltration of the subcutaneous adipose tissues by CD3+ and/or CD8+ T-cell lymphoma cells. Clinicians must cooperate with pathologists and oncologists to diagnose this disease as soon as possible and to avoid a misdiagnosis. The use of antibiotic and painkillers should minimize the patient’s discomfort and control rapid wound development. Future studies are required to investigate the optimal wound treatment and whether the necrotic tissue should be removed.
Keywords: Wound, subcutaneous panniculitis T-cell lymphoma, pathology, clinical characteristic
Introduction
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is a very rare type of cutaneous lymphoma, which predominantly localizes in the subcutaneous adipose tissue, with no palpable involvement of the lymph nodes. It most frequently presents in the early stage as multiple, painless, subcutaneous nodules on the extremities and trunk. In 2005, the World Health Organization (WHO) defined SPTCL as a primary subcutaneous T-cytotoxic-lymphocyte lymphoma, in which the subcutaneous tissue is infiltrated by large numbers of pleomorphic T cells and macrophages, mainly involving the lower extremities and hemophagocytic syndrome (HPS) [1]. The European-American Lymphoma Classification and WHO classification define SPTCL as a primary cutaneous lymphoma, with internal organ involvement, rapid progression, a poor prognosis, and a high mortality rate, which is easily misdiagnosed because it can present in various forms [2,3].
In February-April 2014, we treated a patient with a refractory, progressive painful wound he had suffered for 7 months. After several histopathological examinations and a multidisciplinary consultation, the patient was finally diagnosed with SPTCL and died 50 days after diagnosis. Here, we analyze the clinical and pathological features of this disease and review the relevant literature to improve its recognition.
Case report
A 56-year-old man presented with refractory, progressive inflamed wound near the left axilla, which had been treated surgically as a subcutaneous nodule 7 months earlier. The wound was covered with necrotic tissue and became increasingly larger after several rounds of debridement by a surgeon. It was very painful and responded poorly to systemic antibiotics. The patient and his family were very anxious and worried. On February 19, 2014, he was transferred to the Wound Care Center of Jinling Hospital for treatment of this chronic and refractory wound. The initial assessment was of a full-thickness wound of 4 cm × 10 cm, wholly covered with necrotic tissue, and involving the fatty layer and part of the muscle layer (Figure 1A). The patient reported wound-related pain (WRP) [4] of 8-9 points on a visual analogue scale (VAS) [5], which affected his sleep and daily living. During his illness, the patient had lost 20 kg in weight. An examination indicated a multisystem disorder and high fever (39°C). The results of chest computed tomography (CT) were: 1. clear flaky shadow in part of the subcutaneous fat layer of the left axilla, and slightly enlarged lymph nodes in the left axilla; 2. interstitial inflammation in the bilateral pulmonary and mediastinal small lymph nodes; and 3. right-side pleural thickening and pericardial effusion. Abdominal CT revealed splenomegaly and spleen calcification. Two biopsies of the wound tissue reported an “inflammatory reaction”, whereas an immunohistochemical examination of the third biopsy, taken after 7 months, found “highly proliferative T lymphocytes with dysplasia and heteromorphous cells.” An immunohistochemical examination of the fourth biopsy, taken after 8 months, found that the tissues were “CD3+, CD8+ positive” (Figure 2). SPTCL was diagnosed based on the histopathological examinations and a multidisciplinary consultation at the Wound Care Center, including pathologists, oncologists, and dermatologists.
Figure 1.
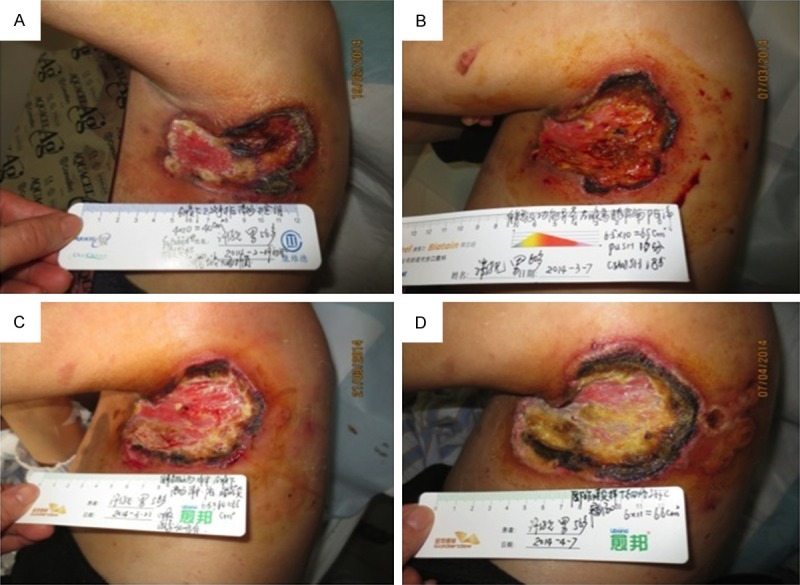
The initial assessment was of a full-thickness wound of 4 cm × 10 cm, wholly covered with necrotic tissue, and involving the fatty layer and part of the muscle layer (A). After treatment for 2 weeks, the wound tissue showed 25% fresh granulation, but still tended to increase in size (B), and the WRP score had decreased to 5-6 points. After 4 weeks of treatment, inflammation was controlled, the pain had eased further, and the area of black necrosis had decreased (C). Fifty days after chemotherapy with corticosteroid therapy and wound care, the wound became seriously painful and increasingly necrotic (D).
Figure 2.
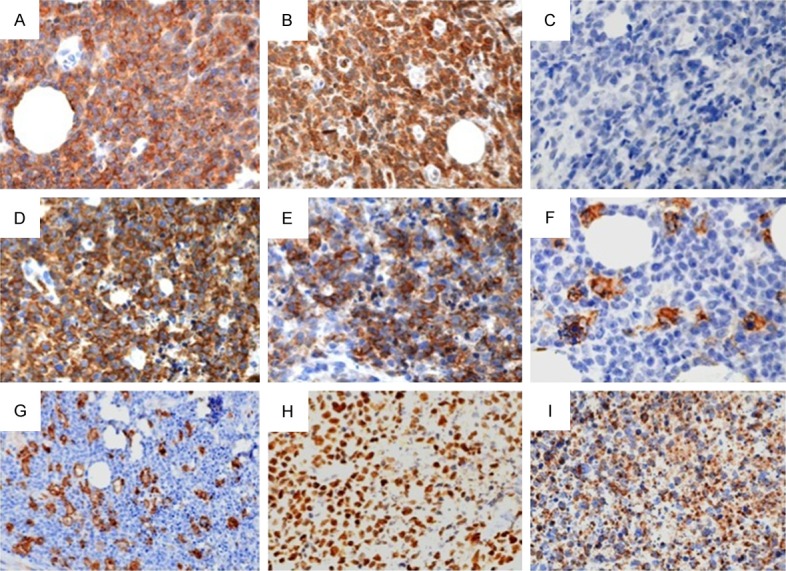
The immunohistochemical staining was used in diagnosis of SPTCL. A: CD2 (3+); B: CD3 (3+); C: CD20 (-); D: CD43 (3+); E: CD56 (3+); F: MPO (-); G: CD163 (+); H: Ki67 (90%+); I: GranB (3+).
Wound care and systemic treatment
After the patient gave his informed consent inform, a certified wound ostomy and continence nurse (CWOCN) at the Wound Care Center took a wound secretion sample to the hospital laboratory for bacterial culture. When the growth of Escherichia coli was reported, the nurse washed the wound bed with saline and covered the wound with topical anti-inflammatory dressings, such as hydrofiber silver dressings (ConvaTec, USA). The dressings were changed and the wound size and pain score were reassessed every 48 h. Photographs of the wound were taken once a week, with the patient’s agreement, to monitor the effects of the wound care, regular procedures, and dressings. To diagnose and treat the illness as soon as possible, we contacted a pathologist, immunologist, respiratory experts, and an oncologist for a multidisciplinary consultation. The patient’s humoral and cellular immune indices and autoantibodies were abnormal (Table 1), so he was treated by a physician with a systemic CHOP chemotherapy regimen (vincristine, epirubicin 80 mg 1 d + 2 mg 1 d + 1.2 g 1 d + cyclophosphamide prednisone 100 mg 1-5 d) and systemic corticosteroid therapy [6]. The wound was treated by the CWOCN with conservative sharp wound debridement and hydrofiber silver dressings for the topical control of inflammation. After treatment for 2 weeks, the wound tissue showed 25% fresh granulation, but still tended to increase in size (Figure 1B), and the WRP score had decreased to 5-6 points. After 4 weeks of treatment, inflammation was controlled, the pain had eased further, and the area of black necrosis had decreased (Figure 1C). After 1 week of systemic chemotherapy with corticosteroid therapy, the wound increased in size again and became more painful, with WRP scores of 10 on VAS. Fifty days after chemotherapy with corticosteroid therapy and wound care, the wound became seriously painful and increasingly necrotic (Figure 1D). The patient developed a serious stomachache and abdominal distension, rapidly became comatose, and died.
Table 1.
Results of laboratory tests (blood)
| Test | Results | Normal range |
|---|---|---|
| C-reactive protein | 18.7 mg/L↑ | < 8 mg/L |
| High-sensitivity C-reactive protein | 16.8 mg/L↑ | < 8 mg/L |
| Hemoglobin | 120 g/L | 120-160 g/L (male) |
| Immunoglobulin E | 129 IU/mL↑ | 0-100 IU/mL |
| Immunoglobulin G | 19.1 g/L ↑ | 7-16 g/L |
| Immunoglobulin kappa light chain | 15.30 g/L↑ | 6.29-13.5 g/L |
| Immunoglobulin lambda light chain | 11.60 g/L↑ | 3.13-7.23 g/L |
| Rheumatoid factor | 24.1 IU/mL↑ | < 20 IU/mL |
| b2-Microglobulin | 4.50 mg/L↑ | 0.7-2.9 mg/L (tumor marker) |
| Ferritin | 858.8 mg/L↑ | 23.9-336.2 mg/L (tumor marker) |
| Anti-nuclear antibody titer | 1:640↑ | |
| Anti-Ro52 antibody | (+) | (–) |
Discussion
Research has shown that the clinical manifestations of SPTCL are complex, with few consensus characteristics [6,7]. In our patient, a poor response to systemic antibiotics and chemotherapy with corticosteroid therapy was one of the clinical features, but this differs from research in Germany and Switzerland, which indicated that systemic corticosteroid is an excellent first-line single-agent therapy for SPTCL [6]. In Japan, a 14-year-old girl suffering from SPTCL with HPS was successfully treated with high-dose chemotherapy and autologous peripheral-blood stem-cell transplantation [7]. A probable explanation of these discrepancies is that our patient had a delayed diagnosis (8 months after his initial presentation) and a multisystemic disorder. This may also explain why the patient responded poorly to wound care, with a progressively larger and more painful wound, and why he rapidly succumbed to coma and died. With an earlier diagnosis, the outcomes could have been much better. Therefore, it is important to establish a multidisciplinary approach with which to study the clinical and histological features of SPTCL to ensure early diagnoses in the future.
In reviewing the recent relevant literature, we found that most studies of SPTCL have been retrospective analyses or case reports. The Asian Medical Center at South Korea Ulsan Medical University retrospectively analyzed eight patients with SPTCL with skin lesions [4]. They were characterized by multiple nodular or diffuse soft-tissue lesions, involving the subcutaneous fat and fascia, with skin damage caused by venous congestion, but to different degrees: 37.5% of patients had whole-layer subcutaneous fat damage, 37.5% had partial damage, 25% had varying levels of damage, and 37.5% had mild fascia damage. One patient suffered moderate damage, one patient severe damage, and the unsteady accounted for 25% of patients. Lesions associated with venous congestion occurred in 87.5% of patients. The Rheu- matology Union Hospital of Beijing Medical University performed a retrospective analysis of eight SPTCL patients with skin lesions. These patients showed characteristic red nodules, nodular panniculitis, and systemic vasculitis. The Tumor Hospital of Beijing Medical University retrospectively analyzed 10 patients with SPTCL and characteristic skin lesions, from 1999 to 2009. They found that 40% of patients had multiple recurrent advanced nodular lesions, 70% had multiple subcutaneous nodules on the limbs and trunk, 30% had nodular ulcers, and 50% had lesions with inconstant pain. Xi’an Tangdu Hospital of the Fourth Military Medical University Department of Hematology in China reported one male patient with SPTCL features, manifesting as skin nodules on the chest [5]. Japan Kochi University reported one male patient with rheumatoid arthritis complicated by features of SPTCL, with multiple axillary and inguinal subcutaneous nodules [6]. Japan Tokyo Women’s Medical University, Department of Dermatology, reported one female patient with SPTCL, with lesions in the lower limbs and the abdominal fat layer [7]. The Children’s Hospital in Detroit, Michigan, USA, reported a 17-year-old female patient with SPTCL, with skin damage lasting more than 3 years and multiple subcutaneous lumps [8]. The Turkey Ankara University Medical Center reported a 20-year-old female SPTCL patient with multiple itching purple nodules on both upper limbs [9]. A 19-month-old boy with SPTCL and characteristic multiple deep skin nodules on his face, legs, upper limbs, and back was reported in Croatia [10]. Two universities in Germany and the Switzerland Hospital Department of Dermatology jointly reported five patients with SPTCL, identified over a period of 13 years, who showed multiple skin nodules in different parts of their bodies in the early stage of the disease, later manifesting as eczema and panniculitis [11]. In summary, the early characteristics of SPTCL patients tend to differ, although multiple nodules on the skin seem to be universal, and the manifestations of skin damage change and differ during the course of the disease. This lack of consistency increases the difficulty of recognizing SPTCL. Therefore, it is very important to explore the individual differences and identify characteristics that can be used for the early diagnosis of SPTCL. Increasingly progressive wound pain was reported for the first time in our patient, but it is unclear whether this is characteristic of SPTCL-related skin damage. This issue is worthy of attention and discussion.
In the past 10 years, the European Organization for Research Mechanism and Treatment of Cancer Cutaneous Lymphoma Group has further classified SPTCL into the α/β SPTCL subtype (SPTCL-AB) and γ/δ SPTCL subtype (SPTCL-GD). The two subtypes differ in their clinical manifestations, pathology, immunohistochemistry, treatment, and prognoses. At the America Mali Lanzhou Baltimore Medical Center, Kao reported one patients with fatal SPTCL who had multisystem involvement, a poor response to treatment, and rapid disease progression, who was attended by a multidisciplinary team of experts, including dermatologists, skin pathologists, hematology experts, oncologists, and expert physicians [12]. Chinese reports found that 87.5% of eight SPTCL patients displayed fever, 50% had swollen lymph nodes, 37.5% had hepatomegaly, 37.5% had splenomegaly, 37.5% had HPS, and 25% had pneumonia and pulmonary infiltration, with rapid disease progression to death. In another report of the clinical manifestations of 10 patients with SPTCL, 40% were diagnosed with lymph-node disease or visceral involvement, 40% had abnormal liver function, 10% had HPS, and 20% had lung infiltration, and their condition progressed rapidly to death. The Beijing Liberation Army General Hospital Department of Hematology of China reported that 10 SPTCL patients mainly manifested with solitary or multiple subcutaneous lesions. Eight patients had high fever, weight loss, liver and bone-marrow involvement, and their disease progressed rapidly, with a mortality rate of 90% and a median survival time of 10 months. At Japan Kochi University, one patient with rheumatic arthritis was diagnosed with SPTCL, manifesting as high fever, back pain, and a poor response to treatment [8]. The clinical manifestations in one patient with SPTCL at Hyderabad India Apollo Hospitals [13] included fever, chills, weight loss, rapidly progressing illness, and death. Eight cases of mortality (20%) in patients with SPTCL were reported by the Asian Medical Center in South Korea Ulsan Medical University [4]. From a comprehensive analysis of the literature, the clinical characteristics of SPTCL include liver, spleen, lung, blood, immune system, and other organ involvement, a poor response to treatment, rapid disease progression, and a high mortality rate (20%-90%).
Analysis of the clinical features of our patient showed that his illness developed rapidly, with a temperature of 39.5°C, and the emergence of multisystem involvement, manifested as left axillary lymphadenopathy, bilateral pulmonary interstitial inflammation, multiple mediastinal lymph nodes, right pleural thickening, pericardial effusion, splenomegaly, and splenic calcification. A patient with multisystem involvement may show a poor response to treatment with hormone and CHOP regimens, and a rapidly developing illness, and this may be the leading cause of death. The period from diagnosis to death was 10 months in our patient, consistent with Wang’s report of a median survival time of 10 months. Therefore, any effective treatment scheme must delay the progression of the disease and reduce the mortality rate, which are key elements of this disease.
The clinical manifestations and pathological features of SPTCL and benign panniculitis are similar, especially in the early stages. Because these symptoms are similar to those of other more common diseases, SPTCL is frequently misdiagnosed as benign panniculitis, eczema, dermatitis, psoriasis, or cellulitis [3,6,14]. Domestic research has reported eight cases of SPTCL diagnosed early because the patients displayed red nodules and nodular panniculitis, and although systemic vasculitis was misdiagnosed as rheumatism, pathological examinations and immunohistochemical confirmation ultimately led to the correct diagnosis. The average period from onset to SPTCL diagnosis is 28.6 months (4-84 months). Our patient presented with early atypical clinical manifestations, and the pathological examination of two subcutaneous nodule tissue biopsies reported a benign lesion, so the disease was misdiagnosed as chronic wound infection and given anti-inflammatory treatment. Because this treatment was ineffective, the lesion increased in size, and the patients displayed an abnormal antibody profile, abnormal cellular and humoral immune indices (Table 1), fever (39.5°C), splenomegaly, and liver function damage. After a multidisciplinary consultation and repeated pathological examinations, the patient was diagnosed with SPTCL. The period from onset to diagnosis was 20 months, approximately 28.6 months shorter than the average period reported in the literature.
Subcutaneous tissue infiltration and/or infiltration by atypical cells, CD3+, and/or CD8+ cells is characteristic of the pathological changes that occur in SPTCL. The detection of lymphocytes with a pathological examination is the main method of diagnosing SPTCL. From our review of the literature, it appears that different countries report different pathological results, increasing the difficulty of identifying the disease. For instance, the Union Hospital of Beijing Medical University Rheumatology Department reported eight patients with SPTCL, each undergoing 2.75 pathological examinations (on average), which confirmed the dense lymphocytic infiltration of the lesion in the subcutaneous fat, with obvious atypical and CD3+ cells. Ten patients with SPTCL at the Beijing Medical University Cancer Hospital included seven with pathological CD56+ T-lymphocyte infiltration, two with CD56– T-lymphocyte infiltration, and one with unknown histopathological results [14]. One reported female patient with SPTCL had multiple foreign nodules. A pathological examination showed that these were surrounded by fat cells and neoplastic lymphocytes. An immunohistochemical evaluation showed that the cells were immunopositive for CD3 and CD5 but immunonegative for CD30 and CD20. This case shows that the pathological features of the lesion/s are important for the diagnosis of SPTCL. The Maryland National Cancer Institute reported 16 patients, aged from 5 months to 21 years, who were diagnosed with SPTCL in 1999-2011, with multiple lesions and pathological results positive for CD3, CD4, CD56, and CD8 and negative for CD5 in 63% of patients [15]. In America, Pincus et al. reported that five patients with lupus erythematosus had the same pathological features as patients with SPTCL, and the main cytotoxic T-cell markers present were CD3 and CD8 [16]. In Italy, Tomasini et al. reported one patient with SPTCL and characteristic CD3+ T cells, who was also CD8 positive and CD4 negative [17].
Los Angeles Santa Monica Medical Center at California University reported one female patient with SPTCL, who displayed enlarged inguinal lymph nodes and was histopathologically positive for CD8 [18]. The skin biopsy report for a 19-month-old male with SPTCL in Croatia showed atypical, small- to medium-sized lymphocytes infiltrating the deep layer of the skin and subcutaneous fat [10]. Immunohistochemical staining showed that the tumor lymphocytes were positive for CD2, CD3, CD5, CD7, and CD8, and negative for CD20, CD34, and CD56. Japan Kochi Medical University reported in a male SPTCL patient with an inguinal nodule, the biopsy of which showed that the subcutaneous fat had excessive CD8+ T-lymphocytic infiltration [6]. The Kerala Medical Center in India reported one female patient with SPTCL, in whom immunohistochemistry detected CD8+ and CD56– lymphocytes [19]. In summary, there is a lack of universal pathological changes characteristic of SPTCL and a dearth of consistent diagnostic criteria for this disease. However, the pathological changes can be summarized as follows: the infiltration of subcutaneous tissue by lymphocytes, atypical lymphocytes, or CD3+ and/or CD8+ cells. During the period from disease onset to diagnosis in our patient, four biopsies of the wound tissue were taken for pathological examination. The first biopsy was 7 months after disease onset, when the pathology report at the local hospital described an inflammatory reaction. After transfer to the Wound Care Center at our hospital at 7 months, a third tissue pathology report cited T-lymphocyte hyperplasia with atypia. The fourth biopsy revealed highly aggressive CD3+ and CD8+ T-cell lymphoma, and SPTCL was diagnosed after hospitalization for 3 weeks. From the discovery of the subcutaneous nodules, which were considered benign, to the detection of the characteristic precancerous and cancerous tissue pathology. Cooperative clinical and laboratory studies will be required to improve the rate of early SPTCL diagnosis.
The clinical manifestations of SPTCL vary and it is easily misdiagnosed. Its diagnosis must be based on a histopathological examination. In future, clinical laboratory data and a multidisciplinary consultation will be required to improve the early diagnosis rate and to avoid misdiagnosis and incorrect early treatment. Delaying the development of the disease is extremely important.
Current reports on the treatment of SPTCL offer only tentative therapies, including the use of hormones, the control of environmental bacteria [20], CHOP chemotherapy [4-6,11], and high-dose chemotherapy with autologous peripheral-blood stem-cell transplantation. Although some of these treatments can delay the progression of the disease or cure it, the evidence of their efficacy is insufficient because most reports have been case studies. A future study must summarize a standard and consistent treatment regimen to improve the response and cure rates and to reduce the mortality rate.
Wound infection, increasing necrosis, and failure to heal are the clinical features of SPTCL. Ineffective debridement of the necrotic tissue causes the wound to expand, and local anti-infectious dressings and systemic antibiotics do not effectively control the further expansion of the wound. We must explore an effective treatment for SPTCL skin damage, especially regarding the evaluation and timing of debridement, and methods to reduce infection, inflammation, and pain.
Acknowledgements
This work was supported in part by the National Natural Science Foundation of China (81371611).
Disclosure of conflict of interest
None.
References
- 1.Willemze R, Jaffe ES, Burg G, Cerroni L, Berti E, Swerdlow SH, Ralfkiaer E, Chimenti S, Diaz-Perez JL, Duncan LM, Grange F, Harris NL, Kempf W, Kerl H, Kurrer M, Knobler R, Pimpinelli N, Sander C, Santucci M, Sterry W, Vermeer MH, Wechsler J, Whittaker S, Meijer CJ. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768–3785. doi: 10.1182/blood-2004-09-3502. [DOI] [PubMed] [Google Scholar]
- 2.Liau JY, Chuang SS, Chu CY, Ku WH, Tsai JH, Shih TF. The presence of clusters of plasmacytoid dendritic cells is a helpful feature for differentiating lupus panniculitis from subcutaneous panniculitis-like T-cell lymphoma. Histopathology. 2013;62:1057–1066. doi: 10.1111/his.12105. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi Y, Takata K, Kato S, Sato Y, Asano N, Ogino T, Hashimoto K, Tashiro Y, Takeuchi S, Masunari T, Hiramatsu Y, Maeda Y, Tanimoto M, Yoshino T. Clinicopathological analysis of 17 primary cutaneous T-cell lymphoma of the gammadelta phenotype from Japan. Cancer Sci. 2014;105:912–923. doi: 10.1111/cas.12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim JW, Chae EJ, Park YS, Lee HJ, Hwang HJ, Lim C, Chung HW. Radiological and clinical features of subcutaneous panniculitis-like T-cell lymphoma. J Comput Assist Tomogr. 2011;35:394–401. doi: 10.1097/RCT.0b013e3182106585. [DOI] [PubMed] [Google Scholar]
- 5.Chen R, Liu L, Liang YM. Treatment relapsed subcutaneous panniculitis-like T-cell lymphoma together HPS by Cyclosporin A. Hematol Rep. 2010;2:e9. doi: 10.4081/hr.2010.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nemoto Y, Taniguchi A, Kamioka M, Nakaoka Y, Hiroi M, Yokoyama A, Enzan H, Daibata M. Epstein-Barr virus-infected subcutaneous panniculitis-like T-cell lymphoma associated with methotrexate treatment. Int J Hematol. 2010;92:364–368. doi: 10.1007/s12185-010-0642-5. [DOI] [PubMed] [Google Scholar]
- 7.Mitsuhashi K, Momose M, Masuda A, Tsunemi Y, Motoji T. Positron emission tomography revealed diffuse involvement of the lower legs and occult extracutaneous lesions in subcutaneous panniculitis-like T-cell lymphoma. Clin Nucl Med. 2013;38:209–211. doi: 10.1097/RLU.0b013e31827087ca. [DOI] [PubMed] [Google Scholar]
- 8.Rodriguez VR, Joshi A, Peng F, Rabah RM, Stockmann PT, Savasan S. Positron emission tomography in subcutaneous panniculitis-like T-cell lymphoma. Pediatr Blood Cancer. 2009;52:406–408. doi: 10.1002/pbc.21805. [DOI] [PubMed] [Google Scholar]
- 9.Soylu S, Gul U, Kilic A, Heper AO, Kuzu I, Minareci BG. A case with an indolent course of subcutaneous panniculitis-like T-cell lymphoma demonstrating Epstein-Barr virus positivity and simulating dermatitis artefacta. Am J Clin Dermatol. 2010;11:147–150. doi: 10.2165/11311060-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 10.Rajic L, Bilic E, Femenic R, Mestrovic D, Ilic I, Lasan-Trcic R, Dubravcic K, Husar K, Kardum-Skelin I, Tesovic G, Culig Z, Konja J. Subcutaneous panniculitis-like T-cell lymphoma in a 19 month-old boy: a case report. Coll Antropol. 2010;34:679–682. [PubMed] [Google Scholar]
- 11.Guenova E, Schanz S, Hoetzenecker W, DeSimone JA, Mehra T, Voykov B, Urosevic-Maiwald M, Berneburg M, Dummer R, French LE, Kerl K, Kamarashev J, Fierlbeck G, Cozzio A. Systemic corticosteroids for subcutaneous panniculitis-like T-cell lymphoma. Br J Dermatol. 2014;171:891–894. doi: 10.1111/bjd.13053. [DOI] [PubMed] [Google Scholar]
- 12.Kao GF, Resh B, McMahon C, Gojo I, Sun CC, Phillips D, Zhao XF. Fatal subcutaneous panniculitis-like T-cell lymphoma gamma/delta subtype (cutaneous gamma/delta T-cell lymphoma): report of a case and review of the literature. Am J Dermatopathol. 2008;30:593–599. doi: 10.1097/DAD.0b013e318182c7bf. [DOI] [PubMed] [Google Scholar]
- 13.Swain M, Swarnalata G, Bhandari T. Subcutaneous panniculitis-like T-cell lymphoma in a case of carcinoma cervix. Indian J Med Paediatr Oncol. 2013;34:104–106. doi: 10.4103/0971-5851.116192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vijaya B, Sunila MD, Manjunath GV. Subcutaneous panniculitic-like T-cell lymphoma: a red alert! The role of a vigilant histopathologist. Indian J Pathol Microbiol. 2011;54:376–378. doi: 10.4103/0377-4929.81647. [DOI] [PubMed] [Google Scholar]
- 15.Huppmann AR, Xi L, Raffeld M, Pittaluga S, Jaffe ES. Subcutaneous panniculitis-like T-cell lymphoma in the pediatric age group: a lymphoma of low malignant potential. Pediatr Blood Cancer. 2013;60:1165–1170. doi: 10.1002/pbc.24462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pincus LB, LeBoit PE, McCalmont TH, Ricci R, Buzio C, Fox LP, Oliver F, Cerroni L. Subcutaneous panniculitis-like T-cell lymphoma with overlapping clinicopathologic features of lupus erythematosus: coexistence of 2 entities? Am J Dermatopathol. 2009;31:520–526. doi: 10.1097/DAD.0b013e3181a84f32. [DOI] [PubMed] [Google Scholar]
- 17.Tomasini D, Berti E. Subcutaneous panniculitis-like T-cell lymphoma. G Ital Dermatol Venereol. 2013;148:395–411. [PubMed] [Google Scholar]
- 18.Levine BD, Seeger LL, James AW, Motamedi K. Subcutaneous panniculitis-like T-cell lymphoma: MRI features and literature review. Skeletal Radiol. 2014;43:1307–1311. doi: 10.1007/s00256-014-1879-5. [DOI] [PubMed] [Google Scholar]
- 19.Francis A, Criton S, Acharya S, Shojan A, Philip RM. Subcutaneous panniculitis-like T-cell lymphoma. Indian J Dermatol. 2010;55:290–292. doi: 10.4103/0019-5154.70707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sakurai E, Satoh T, Akiko YA, Maesawa C, Tsunoda K, Endo M, Akasaka T, Masuda T. Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) with hemophagocytosis (HPS): successful treatment using high-dose chemotherapy (BFM-NHL & ALL-90) and autologous peripheral blood stem cell transplantation. J Clin Exp Hematop. 2013;53:135–140. doi: 10.3960/jslrt.53.135. [DOI] [PubMed] [Google Scholar]