

Abstract
Pathological cardiac hypertrophy, a common response of the heart to a variety of cardiovascular diseases, is typically associated with myocytes remodeling and fibrotic replacement, cardiac dysfunction. Exercise preconditioning (EP) increases the myocardial mechanical load and enhances tolerance of cardiac ischemia-reperfusion injury (IRI), however, is less reported in pathological cardiac hypertrophy. To determine the effect of EP in pathological cardiac hypertrophy, Male 10-wk-old Sprague-Dawley rats (n=30) were subjected to 4 weeks of EP followed by 4-8 weeks of pressure overload (transverse aortic constriction, TAC) to induce pathological remodeling. TAC in untrained controls (n=30) led to pathological cardiac hypertrophy, depressed systolic function. We observed that left ventricular wall thickness in end diastole, heart size, heart weight-to-body weight ratio, heart weight-to-tibia length ratio, cross-sectional area of cardiomyocytes and the reactivation of fetal genes (atrial natriuretic peptide and brain natriuretic peptide) were markedly increased, meanwhile left ventricular internal dimension at end-diastole, systolic function were significantly decreased by TAC at 4 wks after operation (P < 0.01), all of which were effectively inhibited by EP treatment (P < 0.05), but the differences of these parameters were decreased at 8 wks after operation. Furthermore, EP treatment inhibited degradation of IκBα, and decreased NF-κB p65 subunit levels in the nuclear fraction, and then reduced IL2 levels in the myocardium of rats subject to TAC. EP can effectively attenuate pathological cardiac hypertrophic responses induced by TAC possibly through inhibition of degradation of IκB and blockade of the NF-κB signaling pathway in the early stage of pathological cardiac hypertrophy.
Keywords: Exercise preconditioning, pathological cardiac hypertrophy, pressure-overload, IκB, NF-κB
Introduction
Pathological cardiac hypertrophy, accompanying with hypertension, aortic stenosis and valve defects, is typically associated with myocytes remodeling and fibrotic replacement, cardiac dysfunction, and increases risk of heart failure and sudden death [1-3]. A pathological stimulus causing pressure overload (e.g. aortic stenosis, hypertension) produces an increase in systolic wall stress which results in concentric hypertrophy. If the chronic stimulus is not relieved, the hypertrophied heart will dilate, contractile function will fall and then heart failure will develop. At present there is no good way to cure heart failure, and long term survival following heart failure remains poor, with one third of patients dying within a year of diagnosis [4-7]. Thus, recent studies have focused on how to prevent or reverse cardiac hypertrophy and transition to heart failure, and identifying the molecular mechanisms and finding new therapeutic targets. It is well known that physiological stimuli (e.g. regular aerobic exercise) enhance cardiac function. Exercise preconditioning (EP), a physiologic favorable adaptation in many organs, including the heart, increases the myocardial mechanical load and enhances tolerance of cardiac ischemia-reperfusion injury (IRI), however, is less reported in pathological cardiac hypertrophy, accompanied by a positive alteration in stroke volume and sympathetic nervous system activity, therefore, reduces the incidence of arrhythmias [8,9] and myocardial apoptosis [10], and improves heart function [11].
Nuclear transcription factor(NF)-κB is a ubiquitous transcription factor which regulates relative genes involved cardiac remodeling, antiapoptotic [12] and inflammatory responses [13]. It can play an important role in the pathophysiology of myocardial ischemia/reperfusion injury, atherosclerosis and heart failure [14-19]. The NF-κB family has five subunits, including p65, RelB, c-Rel, p50, and p52, which form homo- or hetero-dimers [12,20]. Under sedentary conditions, inactive NF-κB dimers are bound to inhibitor of κB (IκB) in the cytoplasm, whereas on stimulation, IκB kinase (IκK)-mediated IκB phosphorylation results in IκB ubiquitination and nuclear translocation of NF-κB. The activation of the NF-κB pathway plays a key role in the development of cardiac hypertrophy, and the inhibition of NF-κB pathway could promote the regression of cardiac hypertrophy induced by pressure overload [21,22].
The transverse aortic constriction (TAC) model is performed by banding the aorta between the innominate artery and the left carotid artery and is a convincing method for producing pressure overload-induced pathological hypertrophy and heart failure [23]. Similar to aortic stenosis and hypertension, TAC initially leads to compensated hypertrophy of the heart, which often is associated with a temporary enhancement of cardiac contractility. Over time, however, the response to the chronic hemodynamic overload becomes maladaptive, resulting in cardiac dilatation and heart failure [24]. Hence, in light of this and the fact that NF-κB is a potential critical mediator of cardiac hypertrophy, we tested in the present study the hypothesis that EP could attenuate pathological cardiac hypertrophy induced by pressure overload through the inhibition of the NF-κB pathway. Our data presented below unveils EP can effectively suppress the activation of the NF-κB pathway in myocardium, as well as TAC-induced cardiac hypertrophic responses in rats.
Materials and methods
Experimental animals
The experimental protocols were approved by the Committee on Animal Research at the Second Military Medical University. Male 10-wk-old Sprague-Dawley rats (n=90, weight 120-140 g) were obtained from Shanghai S&P - Shall Kay Laboratory Animal Co., LTD (Shanghai, China), and were cared for according to the Guiding Principles for the Care and Use of Animals based on the Helsinki Declaration in Second Military Medical University laboratory animals center. All rats were maintained on a 12 h:12 h light: dark cycle and received food and water ad libitum.
Experimental protocol
All animals were randomly divided into three groups (30 rats per group): sham group, TAC group and EP-treated TAC group (EP+TAC group). According to the Bedford standard [25], rats of EP group were subjected to moderate-intensity exercise (about 60% of maximal aerobic velocity) [26] on a motor-driven treadmill (Hangzhou, China) for 4 wks. In the first 5 days, these rats were trained for 0% grade, 40 min/day at 15 m/min, and duration and intensity increased daily until the animals were trained for 60 min at 18 m/min, 0% grade. Thereafter, exercise intensity was kept constant at moderate level at each training period (5 days per week). All post training experiments in trained rats were performed 48 h after the last exercise training bout to avoid acute effects of exercise. The other two groups rats remained at sedentary for the training period. After EP, sixty rats of EP+TAC and TAC group were performed by tying a 3-0 silk suture over an 8 gauge needle to produce pressure overload-induced pathological cardiac hypertrophy as previously described [19,27-29]. The sham group procedure was performed identically but without the aortic ligation. No further exercise training was undertaken after TAC. Four wks and 8 wks after surgery, sham, TAC and EP+TAC rats were examined with echocardiography, and were weighed and decapitated under anesthesia, followed by collection of cardiac tissues and tibia length. Heart weight-to-body weight (HW/BW) ratios and the heart weight-to-tibia length ratio (HW/TL) were recorded at the time of tissue collection.
Echocardiography
At 4 wks and 8 wks after operation, Rats (n=10, per group) were anesthetized by isoflurane and transthoracic echocardiographic measurement was carried out by an animal specific instrument (Visual Sonics Vevo770, Visual Sonics Inc, Toronto, Canada) [30,31]. LV wall thicknesses, LV chamber dimensions, LV volumes, and fractional shortening were determined from M-mode tracings. All measurements were performed by two experienced persons and taken in a double-blind manner.
Morphology and histological analyses
Excised hearts (n=5, per group) were weighed, perfused with PBS and fixed with 4% polyformaldehyde for global morphometry and then with 10% formalin for further histological analysis. Paraffin embedded hearts were sectioned at 4-6 μm thickness and stained with hematoxylin and eosin (H-E). Cardiomyocyte morphology and histology of LV free wall was visualized under a high magnification to assess cross-sectional area (CSA) using a video camera (Leica Qwin 3) attached to a micrometer. Images were analyzed using an Image J (NIH, USA). CSA of cardiomyocyte measured from 5 sections for one heart and 5 hearts (4 wks after TAC) examined, and five randomly chosen fields were evaluated from each cross section of the LV free wall.
Quantitative real-time polymerase chain reaction analysis
Total RNA was isolated from the LV free wall of animal models (n=6, per group) using TRIzol reagent (Invitrogen). SYBR Green qRT-PCR (Takara Bio Inc, Otsu, Japan) was carried out with reverse primer and forward primers designed specifically for each of the mRNAs (Table 1), qRT-PCR primers were compounded and purchased from Sangon Biological (Shanghai, China). Gene expression was normalized to that of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) for mRNA. The expression of target genes was quantified by qRT-PCR by using One Step SYBR Prime Script RT-PCR Kit II (Takara) on an ABI StepOne Real-Time PCR System (Life Technologies) with conventional protocols. The 2-ΔΔCt method was used to calculate the expression of target genes relative to the reference gene GAPDH.
Table 1.
Primers for quantitative real-time polymerase chain reaction
| Gene | Forward | Reverse |
|---|---|---|
| GAPDH | 5’- GCCATCACTGCCACTCAGAA-3’ | 5’- GGCATGTCAGATCCACAACG-3’ |
| ANP | 5’- AGAGAGTGAGCCGAGACAGC-3’ | 5’-TGGACACCGCACTGTATACG-3’ |
| BNP | 5’-CCGGATCCAGGAGAGACTTC-3’ | 5’-TCTGCAGCCAGGAGGTCTTC-3’ |
Western blot analysis
Total protein lysates were prepared from left ventricles (n=6, per group) as previously described [32]. Isolation of nuclear protein was performed according to the manufacturer’s instructions using Nuclear Extraction Kit from Merck. Individual protein lysates (10 mg/lane) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (7.5%, 15%, or gradient 4~20%; Bio-Rad and Pierce) and transferred to nitrocellulose membranes (GE Healthcare Amersham Biosciences, Oslo, Norway). After incubation in SuperBlock T20 PBS Blocking Buffer (Thermo Fisher Scientific Inc, Waltham, Mass), the membranes were incubated with individual antibodies according to manufacturer instructions. Antibodies for protein detection were as follows: atrial natriuretic peptide (ANP) (Santa Cruz, CA, USA, dilution 1:200), brain natriuretic peptide (BNP) (Abcam, UK, dilution 1:1000), IкBα (Santa Cruz, CA, USA, dilution 1:200), NF-κB p65 (Santa Cruz, CA, USA, dilution 1:200), limin B2 (Abcam, UK, dilution 1:1000) and interleukin 2 (IL2) (Santa Cruz, CA, USA, dilution 1:200). Blots were incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG (EarthOx LLC, CA, USA; dilution 1:5000) and developed with ECL (GE Healthcare Biosciences). Images were acquired and analyzed using a BioDocIt Imaging System (UVP, USA). Relative amounts of proteins were expressed as the percent increase over sham values.
Statistics
Data were expressed as the means ± standard deviation (means ± SD). All computations were carried out using the SPSS version 18.0 software for Windows (SPSS Inc, IL, USA). The statistical significance of differences among experimental groups was evaluated by two-way analysis of variance (ANOVA), When the ANOVA revealed significant between-group differences, post hoc analysis was performed using Bonferroni’s method of multiple comparisons. Single between-group comparisons were made by Student’s t-tests. A two-tailed P value of 0.05 was considered statistically significant.
Results
Elimination and mortality rate of the experimental animals
At EP stage, 2 rats (6.7%) were eliminated because they could not adapt to the treadmill exercise training. Four weeks after operation, the mortality rate of TAC and EP+TAC group were 13.3% (2/15) and 7.1% (1/14), respectively, and the mortality rate went up separately to 33.3% (5/15) and 21.4% (3/14) at 8 wks after TAC. There was not death in sham group.
EP attenuated pathological cardiac hypertrophy induced by pressure overload
At 4 wks after TAC, there were no significant differences in heart rate and body weight (BW) between TAC and EP+TAC rats (Figure 1A). the parameters for assessing cardiac hypertrophy, including LV wall thickness in end diastole (LVWD), heart rise, HW/BW, HW/TL (Figure 1B) and cross-sectional area of cardiomyocytes (CSA) (Figure 1C) were increased , meanwhile LV internal dimension at end-diastole (LVIDd), systolic function (Figure 1A) were decreased in TAC rats, however, this increases were significantly attenuated by EP treatment. At 8 wks after TAC, compared with sham group, the above parameters for assessing cardiac hypertrophy of two surgical groups were increased (P < 0.05), while, between TAC and EP+TAC group, these parameters revealed no significant differences.
Figure 1.
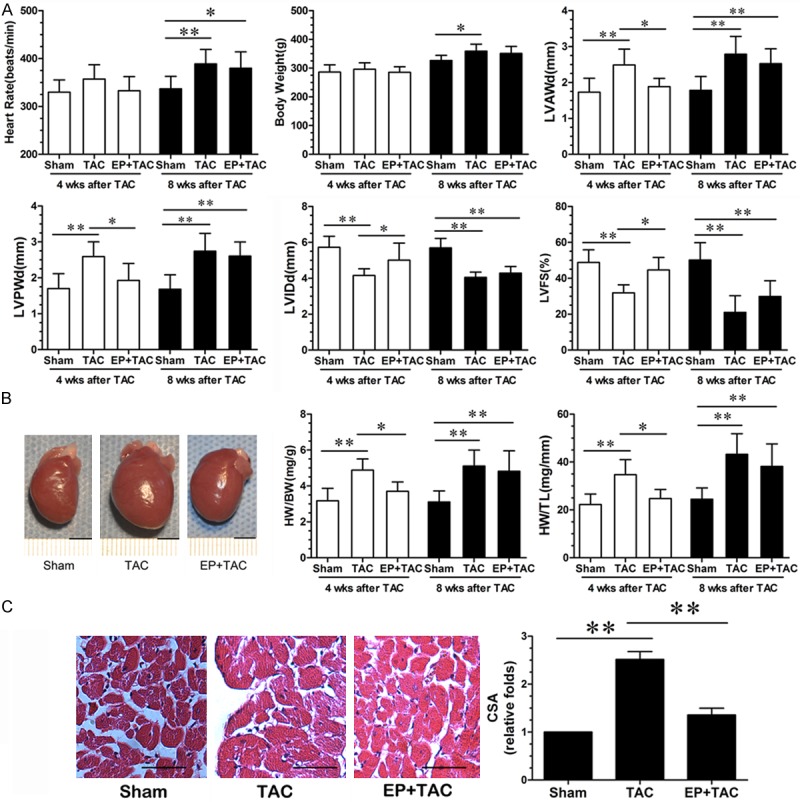
EP attenuated pathological cardiac hypertrophy induced by pressure overload. A: Echocardiographic analysis with representative M-mode tracings from 10 rats. All echocardiography data are shown as mean 6 S.E.M from 10 rats; LVAWd, LV anterior wall thickness at end-diastole; LVPWd, LV posterior wall thickness at end-diastole; LVIDd, LV internal dimension at end-diastole; LVFS, LV fraction shortening; B: Heart morphology and weight; representative global heart photographs of 5 rats (4 wks after TAC) (scale bar: 5 mm); heart weigh to body weight radio (HW/BW) and heart weight-to-tibia length ratio (HW/TL) measured from 5 rats; C: H-E stained LV sections of rats; scale bar: 100 μm; cross sectional area (CSA) of cardiomyocyte measured from 5 sections for one heart and 5 hearts (4 wks after TAC) examined; *P < 0.05, **P < 0.01.
Effects of EP on the ventricular myocardial expression of fetal genes in the hypertrophic hearts
Many genes such as ANP and BNP, which are expressed in fetal heart, are no longer expressed in the adult heart. The expression of these so-called fetal genes is reactivated when the heart undergoes pathological insults such as pathological cardiac hypertrophy. The reactivation of the fetal gene program has been extensively used as an important marker at the gene expression level for cardiac pathological responses. Hence, we evaluated the level of ANP and BNP expression by quantitative RT-PCR and Western blotting. QRT-PCR and western blotting were thus performed to assess the effects of EP on the relative expression levels of ANP and BNP mRNA and protein in the cardiac tissue. Compared with sham group, TAC induced increases in the mRNA and protein levels of both ANP (Figure 2A, 2B) and BNP (Figure 2C, 2D) and the increases were considerably less in EP+TAC at 4 wks after TAC (P < 0.01). At 8 wks after TAC, compared with the sham group, the mRNA and protein levels of both ANP and BNP of two surgical groups were increased (P < 0.01), however, the differences between TAC and EP+TAC group were decreased than those at 4 wks after TAC. These data suggest that EP can effectively suppress the reactivation of the fetal gene program induced by pressure overload, but the protective effect of EP to on pressure overload-induced pathological cardiac hypertrophy is not sustained over time.
Figure 2.

Examination of ANP and BNP expression levels in the LV myocardium. The expressions of ANP and BNP were confirmed by qPCR and Western blot. A: The mRNA expression and of ANP was quantified as the ratio of ANP to GAPDH and expressed as 100% of sham. B: Representative photographs of the protein expression of ANP; β-actin in whole cell lysate used as the loading control. C: The mRNA expression and of BNP was quantified as the ratio of BNP to GAPDH and expressed as 100% of sham. D: Representative photographs of the protein expression of BNP; β-actin in whole cell lysate used as the loading control. Data were shown as mean ± SE from three individual experiments *P < 0.05, **P < 0.01.
Effects of EP on the NF-κB pathway in the heart during pathological cardiac hypertrophy
NF-κB was well known to act, in general, worsening cardiac remodeling or dysfunction by activation of proinflammatory pathway, previous studies showed that the activation of NF-κB is essential for the development of cardiac hypertrophy induced by pressure overload in vivo [33]. Our data show that compared with the sham group, myocardial IкBα protein levels were significantly decreased in the TAC group but were markedly increased in the EP+TAC group, (Figure 3A) at 4 weeks after TAC, but the differences between TAC and EP+TAC group were significantly decreased at 8 wks after TAC than those of at 4 wks after TAC.
Figure 3.

The effect of EP on myocardial NF-κB signaling pathway in rats subject to TAC. A: Detection of cell lysate IκBα expression by Western blot; representative photographs of the protein expression of IκBα; expression of IκBα was quantified as the ratio of IκBα to β-actin and expressed as 100% of sham. B: Detection of cytoplasm NF-κB p65 subunit expression by Western blot; cytoplasm NF-κB p65 subunit levels; expression of NF-κB p65 subunit was quantified as the ratio of NF-κB p65 to β-actin and expressed as 100% of sham. C: Detection of nuclear NF-κB p65 subunit expression by Western blot; nuclear NF-κB p65 subunit levels; expression of NF-κB p65 subunit was quantified as the ratio of NF-κB p65 to lamin B2 and expressed as 100% of sham. D: Detection of cell lysate IL2 expression by Western blot; representative photographs of the protein expression of IL2; expression of IL2 was quantified as the ratio of IL2 to β-actin and expressed as 100% of sham. Data were shown as mean ± SE from six individual experiments *P < 0.05, **P < 0.01.
To further investigate the effects of EP on the nuclear translocation of NF-κB in hypertrophic cardiac tissue, which is an important step in the activation of NF-κB pathway, NF-κB p65 subunit levels in the cytoplasm(Figure 3B) and nucleus(Figure 3C) were explored by western blotting analysis. At 4 wks after TAC, compared with sham rats, nuclear NF-κB p65 subunit levels were increased in the TAC, which were significantly inhibited by EP treatment. IL2 protein levels were significantly lower in the EP+TAC group compared with those in the sham or the TAC groups (Figure 3D).
Discussion
Pathological cardiac hypertrophy is an independent risk factor for cardiovascular morbidity and mortality in humans [34], hence, it is of high clinical significance to attenuate pathological cardiac hypertrophy. In this study, we demonstrated that EP can effectively reduce cardiac hypertrophic responses induced via TAC possibly through inhibition of degradation of IκB and blockade of the NF-κB pathway.
EP, as a physiologic favorable adaptation, increases the myocardial mechanical load, accompanied by a positive alteration in stroke volume and sympathetic nervous system activity. Moreover, exercise modulates important features of maladaptive cardiac remodeling induced by chronic pressure overload, resulting in an improved cardiac phenotype [35-38]. Here we show that LVWD, systolic function, LVIDd, HW/BW and HW/TL were remarkably increased during cardiac hypertrophy induced by TAC-triggered pressure overload at 4 wks after TAC, however, this increases were significantly attenuated by EP treatment. We also found that the differences of these parameters between TAC and EP+TAC group were decreased at 8 wks after operation. These data suggest that the effect of EP on pressure overload-induced pathological cardiac hypertrophy is significant in the early stage of pathological cardiac hypertrophy, but at 8 wks after operation, the protective effect was lost.
In the process of cardiac hypertrophy, several fetal genes, including ANP and BNP, are reactivated. Some evidence suggests that ANP and BNP as cardiac failure marker has been increasingly used in the diagnosis of cardiac failure, differential diagnosis, risk stratification, prognosis and treatment guideline [39-43]. One proposed mechanism for increased BNP gene expression in cardiac hypertrophy in vivo is the activation of the NF-κB pathway [44]. Our data also showed that the relative expression levels of ANP and BNP mRNA and protein in the hypertrophic cardiac tissue were inhibited by EP treatment, especially in the early stages. Furthermore, according to the above research, because of timeliness of EP, we find the effects of EP on the TAC model were not sustained over time. However, confirmation of critical molecular mechanisms relative EP is essential for the development of novel and viable therapeutic intervention strategies for patients with cardiovascular disease who are either unable or unwilling to undertake regular physical activity intervention programs.
Numerous stimuli and signaling pathways can be involved in the process of cardiac hypertrophy, and the NF-κB pathway has been shown to mediate cardiac hypertrophy and maladaptive remodeling [45]. It has been reported that persistent myocyte NF-κB p65 subunit activation in cardiac failure exacerbates cardiac remodelling by imparting pro-inflammatory, pro-fibrotic, and pro-apoptotic effects [21,46]. In addition, NF-κB p65 subunit is a mainly classical signal pathway, and plays an important role in the development of inflammation [14]. To better understand the mechanism of EP on cardiac hypertrophy, we investigated the NF-κB pathway in this research. TAC stimuli, activates IκK, the upstream kinase of IκB. Upon activation by inflammatory factors, IκK phosphorylates IκB, causing rapid IκB polyubiquitination and degradation by proteosomes, however, this abnormality was reversed by exercise training [47]. Degradation of IκB allows the cytoplasmic NF-κB to translocate into the nucleus where NF-κB activates its target genes such as IL2; hence, inhibition of degradation of IκB may block the NF-κB signaling pathway [48,49]. We discovered that myocardial protein levels of IκBα, a pivotal negative regulator of NF-κB activation, were decreased in rats subject to TAC but the decrease was effectively reduced by EP treatment. Since EP suppressed degradation of IκB in the pressure-overload early stage, we then verified changes in NF-κB signaling. An on-off experiment suggested that among the two groups in the TAC cohort, the EP treated groups showed a lower nuclear NF-κB p65 subunit protein level and a lower protein level of myocardial IL2, an inflammatory cytokine that is stimulated by NF-κB activation [50]. Taken together, these findings provide strong evidence that EP treatment inhibits the NF-κB signaling, including the degradation of IκB, NF-κB p65 subunit activation and express of myocardial IL2 in rat hearts undergoing cardiac hypertrophy induced by TAC, which may contribute to the antihypertrophy effects of EP, specifically in early pathological cardiac hypertrophy.
In the present study, we demonstrates that EP can effectively attenuate cardiac hypertrophic responses induced by TAC through echocardiographic, morphological and histological parameters for assessing cardiac hypertrophy and the level of ANP and BNP expression, especially in the early stage of pathological cardiac hypertrophy. Furthermore, we found the possibly mechanism is that EP treatment inhibits the NF-κB signaling, including the degradation of IκB, NF-κB p65 subunit activation and express of myocardial IL2 in rat hearts undergoing cardiac hypertrophy induced by TAC. The beneficial effects of regular physical activity on cardiovascular health are well established. However, the key molecular mechanisms underlying the cardioprotective effects of exercise are not well defined. Therefore, although the effects of EP on NF-κB signaling in the TAC model were not sustained over time, identification of critical pathways that are activated by exercise may lead to novel and viable therapeutic intervention strategies.
This study has some limitations. First, a missing element is preoperative echocardiography on these experimental animals. Second, there is yet no scientific consensus in the critical point of intensity and duration of EP protective effect.
Acknowledgements
This work was supported by Science foundation of Shanghai Bureau of Health (20124361).
Disclosure of conflict of interest
None.
References
- 1.Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322:1561–1566. doi: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- 2.Weber KT, Brilla CG, Janicki JS. Myocardial fibrosis: functional significance and regulatory factors. Cardiovasc Res. 1993;27:341–348. doi: 10.1093/cvr/27.3.341. [DOI] [PubMed] [Google Scholar]
- 3.Cohn JN, Bristow MR, Chien KR, Colucci WS, Frazier OH, Leinwand LA, Lorell BH, Moss AJ, Sonnenblick EH, Walsh RA, Mockrin SC, Reinlib L. Report of the National Heart, Lung, and Blood Institute Special Emphasis Panel on Heart Failure Research. Circulation. 1997;95:766–770. doi: 10.1161/01.cir.95.4.766. [DOI] [PubMed] [Google Scholar]
- 4.Bleumink GS, Knetsch AM, Sturkenboom MC, Straus SM, Hofman A, Deckers JW, Witteman JC, Stricker BH. Quantifying the heart failure epidemic: prevalence, incidence rate, lifetime risk and prognosis of heart failure The Rotterdam Study. Eur Heart J. 2004;25:1614–1619. doi: 10.1016/j.ehj.2004.06.038. [DOI] [PubMed] [Google Scholar]
- 5.Cowie MR, Wood DA, Coats AJ, Thompson SG, Suresh V, Poole-Wilson PA, Sutton GC. Survival of patients with a new diagnosis of heart failure: a population based study. Heart. 2000;83:505–510. doi: 10.1136/heart.83.5.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zannad F, Briancon S, Juilliere Y, Mertes PM, Villemot JP, Alla F, Virion JM. Incidence, clinical and etiologic features, and outcomes of advanced chronic heart failure: the EPICAL Study. Epidemiologie de l’Insuffisance Cardiaque Avancee en Lorraine. J Am Coll Cardiol. 1999;33:734–742. doi: 10.1016/s0735-1097(98)00634-2. [DOI] [PubMed] [Google Scholar]
- 7.McMurray JJ, Pfeffer MA. Heart failure. Lancet. 2005;365:1877–1889. doi: 10.1016/S0140-6736(05)66621-4. [DOI] [PubMed] [Google Scholar]
- 8.Hamilton KL, Quindry JC, French JP, Staib J, Hughes J, Mehta JL, Powers SK. MnSOD antisense treatment and exercise-induced protection against arrhythmias. Free Radic Biol Med. 2004;37:1360–1368. doi: 10.1016/j.freeradbiomed.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 9.Hajnal A, Nagy O, Litvai A, Papp J, Parratt JR, Vegh A. Nitric oxide involvement in the delayed antiarrhythmic effect of treadmill exercise in dogs. Life Sci. 2005;77:1960–1971. doi: 10.1016/j.lfs.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 10.Quindry JC, French J, Hamilton KL, Lee Y, Selsby J, Powers S. Exercise does not increase cyclooxygenase-2 myocardial levels in young or senescent hearts. J Physiol Sci. 2010;60:181–186. doi: 10.1007/s12576-009-0082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hydock DS, Lien CY, Schneider CM, Hayward R. Exercise preconditioning protects against doxorubicin-induced cardiac dysfunction. Med Sci Sports Exerc. 2008;40:808–817. doi: 10.1249/MSS.0b013e318163744a. [DOI] [PubMed] [Google Scholar]
- 12.Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: balancing life and death--a new approach to cancer therapy. J Clin Invest. 2005;115:2625–2632. doi: 10.1172/JCI26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen LF, Greene WC. Shaping the nuclear action of NF-kappaB. Nat Rev Mol Cell Biol. 2004;5:392–401. doi: 10.1038/nrm1368. [DOI] [PubMed] [Google Scholar]
- 14.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 15.Frantz S, Tillmanns J, Kuhlencordt PJ, Schmidt I, Adamek A, Dienesch C, Thum T, Gerondakis S, Ertl G, Bauersachs J. Tissue-specific effects of the nuclear factor kappaB subunit p50 on myocardial ischemia-reperfusion injury. Am J Pathol. 2007;171:507–512. doi: 10.2353/ajpath.2007.061042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frantz S, Hu K, Bayer B, Gerondakis S, Strotmann J, Adamek A, Ertl G, Bauersachs J. Absence of NF-kappaB subunit p50 improves heart failure after myocardial infarction. FASEB J. 2006;20:1918–1920. doi: 10.1096/fj.05-5133fje. [DOI] [PubMed] [Google Scholar]
- 17.Kawano S, Kubota T, Monden Y, Tsutsumi T, Inoue T, Kawamura N, Tsutsui H, Sunagawa K. Blockade of NF-kappaB improves cardiac function and survival after myocardial infarction. Am J Physiol Heart Circ Physiol. 2006;291:H1337–1344. doi: 10.1152/ajpheart.01175.2005. [DOI] [PubMed] [Google Scholar]
- 18.Jia LL, Kang YM, Wang FX, Li HB, Zhang Y, Yu XJ, Qi J, Suo YP, Tian ZJ, Zhu Z, Zhu GQ, Qin DN. Exercise training attenuates hypertension and cardiac hypertrophy by modulating neurotransmitters and cytokines in hypothalamic paraventricular nucleus. PLoS One. 2014;9:e85481. doi: 10.1371/journal.pone.0085481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu S, Zhao C, Yang C, Li X, Huang H, Liu N, Li S, Wang X, Liu J. Gambogic acid suppresses pressure overload cardiac hypertrophy in rats. Am J Cardiovasc Dis. 2013;3:227–238. [PMC free article] [PubMed] [Google Scholar]
- 20.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 21.Zou J, Le K, Xu S, Chen J, Liu Z, Chao X, Geng B, Luo J, Zeng S, Ye J, Liu P. Fenofibrate ameliorates cardiac hypertrophy by activation of peroxisome proliferator-activated receptor-alpha partly via preventing p65-NFkappaB binding to NFATc4. Mol Cell Endocrinol. 2013;370:103–112. doi: 10.1016/j.mce.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 22.Islam KN, Bae JW, Gao E, Koch WJ. Regulation of nuclear factor kappaB (NF-kappaB) in the nucleus of cardiomyocytes by G protein-coupled receptor kinase 5 (GRK5) J Biol Chem. 2013;288:35683–35689. doi: 10.1074/jbc.M113.529347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J, Wu J, Li L, Zou YZ, Zhu DL, Gao PJ. Effect of an acute mechanical stimulus on aortic structure in the transverse aortic constriction mouse model. Clin Exp Pharmacol Physiol. 2011;38:570–576. doi: 10.1111/j.1440-1681.2011.05544.x. [DOI] [PubMed] [Google Scholar]
- 24.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 25.Bedford TG, Tipton CM, Wilson NC, Oppliger RA, Gisolfi CV. Maximum oxygen consumption of rats and its changes with various experimental procedures. J Appl Physiol Respir Environ Exerc Physiol. 1979;47:1278–1283. doi: 10.1152/jappl.1979.47.6.1278. [DOI] [PubMed] [Google Scholar]
- 26.Lawler JM, Powers SK, Hammeren J, Martin AD. Oxygen cost of treadmill running in 24-month-old Fischer-344 rats. Med Sci Sports Exerc. 1993;25:1259–1264. [PubMed] [Google Scholar]
- 27.deAlmeida AC, van Oort RJ, Wehrens XH. Transverse aortic constriction in mice. J Vis Exp. 2010 doi: 10.3791/1729. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu Y, Yin X, Wijaya C, Huang MH, McConnell BK. Acute myocardial infarction in rats. J Vis Exp. 2011 doi: 10.3791/2464. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang S, Weinheimer C, Courtois M, Kovacs A, Zhang CE, Cheng AM, Wang Y, Muslin AJ. The role of the Grb2-p38 MAPK signaling pathway in cardiac hypertrophy and fibrosis. J Clin Invest. 2003;111:833–841. doi: 10.1172/JCI16290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chorianopoulos E, Heger T, Lutz M, Frank D, Bea F, Katus HA, Frey N. FGF-inducible 14-kDa protein (Fn14) is regulated via the RhoA/ROCK kinase pathway in cardiomyocytes and mediates nuclear factor-kappaB activation by TWEAK. Basic Res Cardiol. 2010;105:301–313. doi: 10.1007/s00395-009-0046-y. [DOI] [PubMed] [Google Scholar]
- 31.Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M, Shimizu I, Asahara T, Hamada H, Tomita S, Molkentin JD, Zou Y, Komuro I. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–448. doi: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- 32.Bhuiyan MS, Shioda N, Shibuya M, Iwabuchi Y, Fukunaga K. Activation of endothelial nitric oxide synthase by a vanadium compound ameliorates pressure overload-induced cardiac injury in ovariectomized rats. Hypertension. 2009;53:57–63. doi: 10.1161/HYPERTENSIONAHA.108.118356. [DOI] [PubMed] [Google Scholar]
- 33.Li Y, Ha T, Gao X, Kelley J, Williams DL, Browder IW, Kao RL, Li C. NF-kappaB activation is required for the development of cardiac hypertrophy in vivo. Am J Physiol Heart Circ Physiol. 2004;287:H1712–1720. doi: 10.1152/ajpheart.00124.2004. [DOI] [PubMed] [Google Scholar]
- 34.Yang M, Lim CC, Liao R, Zhang X. A novel microfluidic impedance assay for monitoring endothelin-induced cardiomyocyte hypertrophy. Biosens Bioelectron. 2007;22:1688–1693. doi: 10.1016/j.bios.2006.07.032. [DOI] [PubMed] [Google Scholar]
- 35.Boissiere J, Eder V, Machet MC, Courteix D, Bonnet P. Moderate exercise training does not worsen left ventricle remodeling and function in untreated severe hypertensive rats. J Appl Physiol (1985) 2008;104:321–327. doi: 10.1152/japplphysiol.00442.2007. [DOI] [PubMed] [Google Scholar]
- 36.Emter CA, McCune SA, Sparagna GC, Radin MJ, Moore RL. Low-intensity exercise training delays onset of decompensated heart failure in spontaneously hypertensive heart failure rats. Am J Physiol Heart Circ Physiol. 2005;289:H2030–2038. doi: 10.1152/ajpheart.00526.2005. [DOI] [PubMed] [Google Scholar]
- 37.Garciarena CD, Pinilla OA, Nolly MB, Laguens RP, Escudero EM, Cingolani HE, Ennis IL. Endurance training in the spontaneously hypertensive rat: conversion of pathological into physiological cardiac hypertrophy. Hypertension. 2009;53:708–714. doi: 10.1161/HYPERTENSIONAHA.108.126805. [DOI] [PubMed] [Google Scholar]
- 38.Lachance D, Plante E, Bouchard-Thomassin AA, Champetier S, Roussel E, Drolet MC, Arsenault M, Couet J. Moderate exercise training improves survival and ventricular remodeling in an animal model of left ventricular volume overload. Circ Heart Fail. 2009;2:437–445. doi: 10.1161/CIRCHEARTFAILURE.108.845487. [DOI] [PubMed] [Google Scholar]
- 39.Richards AM. Brain natriuretic peptide-guided management of chronic heart failure: first do no harm. Eur J Heart Fail. 2013;15:832–834. doi: 10.1093/eurjhf/hft109. [DOI] [PubMed] [Google Scholar]
- 40.Eurlings LW, van Pol PE, Kok WE, van Wijk S, Lodewijks-van der Bolt C, Balk AH, Lok DJ, Crijns HJ, van Kraaij DJ, de Jonge N, Meeder JG, Prins M, Pinto YM. Management of chronic heart failure guided by individual N-terminal pro-B-type natriuretic peptide targets: results of the PRIMA (Can PRo-brain-natriuretic peptide guided therapy of chronic heart failure IMprove heart fAilure morbidity and mortality?) study. J Am Coll Cardiol. 2010;56:2090–2100. doi: 10.1016/j.jacc.2010.07.030. [DOI] [PubMed] [Google Scholar]
- 41.Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, Jessup M, Konstam MA, Mancini DM, Michl K, Oates JA, Rahko PS, Silver MA, Stevenson LW, Yancy CW. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation. 2009;119:e391–479. doi: 10.1161/CIRCULATIONAHA.109.192065. [DOI] [PubMed] [Google Scholar]
- 42.Richards AM. The natriuretic peptides in heart failure. Basic Res Cardiol. 2004;99:94–100. doi: 10.1007/s00395-004-0461-z. [DOI] [PubMed] [Google Scholar]
- 43.Iqbal N, Alim KS, Aramin H, Iqbal F, Green E, Higginbotham E, Maisel AS. Novel biomarkers for heart failure. Expert Rev Cardiovasc Ther. 2013;11:1155–1169. doi: 10.1586/14779072.2013.832476. [DOI] [PubMed] [Google Scholar]
- 44.Liang F, Gardner DG. Mechanical strain activates BNP gene transcription through a p38/NF-kappaB-dependent mechanism. J Clin Invest. 1999;104:1603–1612. doi: 10.1172/JCI7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Q, Chen Y, Auger-Messier M, Molkentin JD. Interaction between NFkappaB and NFAT coordinates cardiac hypertrophy and pathological remodeling. Circ Res. 2012;110:1077–1086. doi: 10.1161/CIRCRESAHA.111.260729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hamid T, Guo SZ, Kingery JR, Xiang X, Dawn B, Prabhu SD. Cardiomyocyte NF-kappaB p65 promotes adverse remodelling, apoptosis, and endoplasmic reticulum stress in heart failure. Cardiovasc Res. 2011;89:129–138. doi: 10.1093/cvr/cvq274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sriwijitkamol A, Christ-Roberts C, Berria R, Eagan P, Pratipanawatr T, DeFronzo RA, Mandarino LJ, Musi N. Reduced skeletal muscle inhibitor of kappaB beta content is associated with insulin resistance in subjects with type 2 diabetes: reversal by exercise training. Diabetes. 2006;55:760–767. doi: 10.2337/diabetes.55.03.06.db05-0677. [DOI] [PubMed] [Google Scholar]
- 48.Liu F, Zhang Y, Wu ZQ, Zhao T. Analysis of CCL5 expression in classical Hodgkin’s lymphoma L428 cell line. Mol Med Rep. 2011;4:837–841. doi: 10.3892/mmr.2011.515. [DOI] [PubMed] [Google Scholar]
- 49.Xu G, Li Y, Yoshimoto K, Chen G, Wan C, Iwata T, Mizusawa N, Duan Z, Liu J, Jiang J. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced inflammatory activation is mediated by intracellular free calcium in microglial cells. Toxicology. 2013;308:158–167. doi: 10.1016/j.tox.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 50.Zheng H, Ye L, Fang X, Li B, Wang Y, Xiang X, Kong L, Wang W, Zeng Y, Ye L, Wu Z, She Y, Zhou X. Torque teno virus (SANBAN isolate) ORF2 protein suppresses NF-kappaB pathways via interaction with IkappaB kinases. J Virol. 2007;81:11917–11924. doi: 10.1128/JVI.01101-07. [DOI] [PMC free article] [PubMed] [Google Scholar]