

Abstract
Sevoflurane preconditioning has shown to exert delayed caridioprotection against subsequent ischemia and reperfusion injury, but the mechanisms underlying is unclear. Inhibition of autophagy by 3-methyladenine (3-MA) or knockdown of Beclin 1 leads to enhanced cardiac myocyte survival. Our study aimed to test whether sevoflurane preconditioning provides a second window of anesthetic preconditioning (SWOP) via inhibit Beclin 1-mediated autophagic cell death. H9c2 rat cardiomyocytes were randomly divided into five groups: Control (CON) group; hypoxia/reoxygenation (H/R) group, rat cardiomyocytes was exposed in the airtight container for 2 h followed by 1 h of reoxgenation; SWOP group, rat cardiomyocytes was exposed to 1 h of 2.5% sevoflurane 24 h before H/R; Autophagic inhibitors, 3-methyladenine (3-MA, 10 mM) was added to culture medium 15 min before sevoflurane exposure (3-MA+SWOP group) or cells were treated by 3-MA alone (3-MA group). The cell proliferation was significantly increased in SWOP group (79.49 ± 1.37%, P < 0.05) when compared to H/R group (62.2 ± 6.49%, P < 0.05). 3-MA administered before SWOP significantly attenuated the H/R induced autophagy and cell death. H/R injury up-regulated the expression of LC3-II and Beclin 1 proteins (342 ± 66% and 163 ± 18%, respectively, P < 0.05) compared to the CON group (100%), which were increased in SWOP group (202 ± 77% and 128 ± 8%, respectively, P < 0.05). The expression of LC3-II and Beclin 1 proteins was decreased in 3-MA group (110 ± 28% and 97 ± 6%, respectively) and 3-MA+SWOP group (93 ± 7% and 98 ± 6%, respectively) compared with H/R group, but Bcl-2 was upregulated in 3-MA group (158 ± 4%) and 3-MA+SWOP group (156 ± 5%) compared to H/R group (103 ± 7%). In conclusion, sevoflurane preconditioning confers delayed cardioprotection via inhibition Beclin 1-mediated autophagic cell death in cardiac myocytes 24 h before exposed to H/R injury.
Keywords: Autophagy, Beclin 1, hypoxia/reoxgenation, sevoflurane
Introduction
Anesthetic preconditioning (APC) is a cellular protective mechanism whereby exposure to a volatile anesthetic renders a tissue more resistant to a subsequent ischemic insult. This benefit is well established in models of myocardial protection [1]. The process can elicits a bi-phasic pattern of cardioprotection. An acute window manifests almost immediately following the APC stimulus and lasts for up to 2 h, (termed early or classical phase). A delayed window appears 12-24 h later and lasts for up to 72 h (termed the second window of protection or delayed phase) [2]. It was demonstrated that sevoflurane is capable of producing a delayed window of preconditioning [3]. However, the underlying mechanisms by which sevoflurane confers delayed cardioprotection are needed to elucidate.
Increased autophagy has long been known to occur in hearts after ischemia and reperfusion. The autophagy protein Beclin 1 was initially identified in a yeast two-hybrid system as a Bcl-2 interacting protein [4], and it was later reported that Bcl-2 binding to Beclin 1 disrupted autophagy. In addition, a mutant of Beclin 1 lacking the Bcl-2 binding domain was found to induce excessive autophagy and cell death when overexpressed in cells. The same group found that transgenic mice overexpressing Bcl-2 in the heart had reduced levels of autophagy in the heart compared to wild type in response to starvation, suggesting that Bcl-2 functions as a negative regulator of autophagy by inhibiting Beclin 1 [5]. We have previously demonstrated that sevoflurane preconditioning significantly up-regulated the ischaemia/reperfusion induced changes in Bcl-2 protein expression [6].
To investigate the cardioprotective role of SWOP in cardiomyocytes, a H/R model was employed. The aim of this study was to determine if SWOP mediates delayed cardioprotection via inhibition Beclin 1-mediated autophagic cell death in cardiac myocytes exposed to H/R injury.
Materials and methods
Cell culture
H9c2 cells, a clonal line derived from rats heart were obtained from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences. Rats myocardium-derived H9c2 Cardiac myoblast cell line was maintained in DMEM (Gibco, Grand Island, NY, USA) with 1 g/L glucose supplemented with 10% (v/v) FBS (Gibco, Grand Island, NY, USA) and 1% penicillin/streptomycin (v/v). Cells were routinely grown to subconfluency (> 80% by visual estimate) in 75 cm2 flasks at 37°C in a humidified atmosphere of 5% CO2 for examination by transmission electron microscopy and protein analysis. Treatment with 3-methyladenine (Sigma, St. Louis, MO, USA) was accomplished by adding 15 min before sevoflurane preconditioning.
Cell culture induced by H/R
For induction of H/R injury, plates containing rat cardiomyocytes was placed into a humidified airtight container, continued access to 95% N2/5% CO2 to achieve a oxygen-deficient environment. The sealed chamber was placed into a 37°C incubator for 2 h in low serum media (0.5% FBS). After hypoxia incubation, the cells were provided with fresh medium and then restored to 95% air/5% CO2 for 1 h reoxygenation. The control plates were kept in normoxic conditions for corresponding times.
Experimental design
H9c2 rat cardiomyocytes were randomly divided into five groups (Figure 1): Control (CON) group; H/R group, rat cardiomyocytes was exposed in the airtight container for 2 h followed by 1 h of reoxgenation; SWOP group, rat cardiomyocytes was exposed to 1 h of 2.5% sevoflurane 24 h before H/R; Autophagic inhibitors, 3-methyladenine (3-MA, 10 mM) was added to culture medium 15 min before sevoflurane exposure (3-MA+SWOP group) or cells were treated by 3-MA alone (3-MA group).
Figure 1.
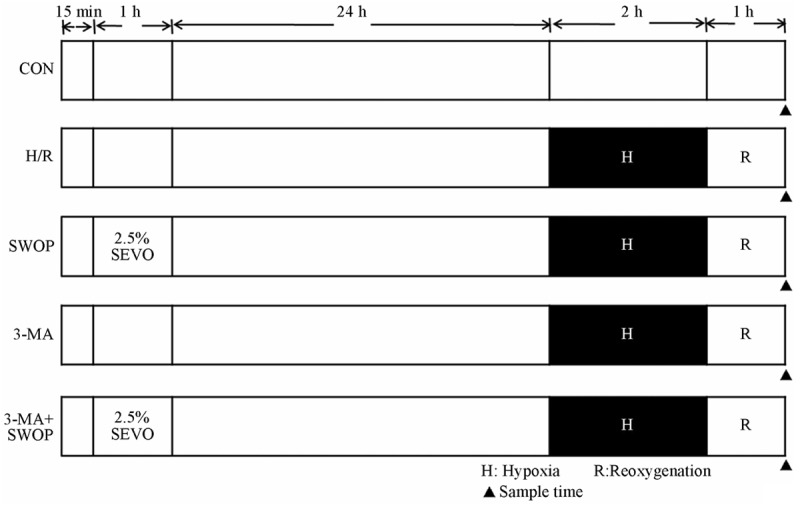
Schematic illustration of experimental protocols. H9c2 rat cardiomyocytes were randomly divided into five groups: Control (CON) group; H/R group, rat cardiomyocytes was exposed in the airtight container for 2 h followed by 1 h of reoxgenation; SWOP group, rat cardiomyocytes was exposed to 1 h of 2.5% sevoflurane 24 h before H/R; Autophagic inhibitors, 3-methyladenine (3-MA, 10 mM) was added to culture medium 15 min before sevoflurane exposure (3-MA+SWOP group) or cells were treated by 3-MA alone (3-MA group).
MTT assay
The H9c2 rat cardiomyocytes were cultured in 96-well plates at a density of 5 × 103 cells per well for 24 h. The samples were maintained under sevoflurane precondition or treated with 3-MA or subjected to H/R as described above. 10 μl of 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT, 5 mg/ml) (Sigma, St. Louis, MO, USA) was added to each well and incubated for an additional 4 h after exposure to H/R. The supernatant was removed. Dimethyl sulfoxide (DMSO; 100 μl/well) was added to dissolve the blue formazan crystals converted from MTT by live cells. Cell viability was assessed by absorbance at 570 nm measured on a μQuant Universal Microplate spectrophotometer. The absorbance (A) values of formazan were calculated as a percentage of the control untreated wells, and transferred to a dose-response curve. The survival rate of cardiomyocytes to each drug with different concentrations was calculated as follows: survival rate = 100% × (A drug treated-A blank)/(A control-A blank). Each of groups was measured in triplicate wells on the same plate in three independent experiments.
Flow cytometry assay
The percentage of cells that underwent apoptosis or necrosis was determined using Annexin V-FITC and propidium iodide (PI) kit according to the manufacturer’s instructions. The cells were harvested, washed twice with cold 1 × phosphate-buffered saline (PBS), and resuspended in 500 μL of cold 1 × binding buffer. The 100 μL of cell suspension was stained with 10 μL of Annexin V-FITC for 15 min in the dark and on ice and then added 380 μL of binding buffer and 10 μL of PI. Flow cytometry analysis was performed. Ten thousand events were analyzed using the Winmdi software.
Transmission electron microscopy (TEM)
Cell were fixed for TEM by removing culture medium, washing one time in flesh medium, and adding 2.5% PBS-buffered glutaraldehyde at 4°C for 1 h, dehydration in a graded ethanol series, and flat embedding in Araldite. Ultrathin sections (40-60 nm) were placed on grids (200 mesh), and double-stained with uranyl acetate and lead citrate. The grids containing the sections were observed on a Philips CM-120 electron microscope.
Immunofluorescence
Cultures were washed in PBS for 3 × 5 min, then for 30 min in PBS containing 4% paraformaldehyde (pH 7.4), washed in PBS for 3 × 5 min, blocked in PBS containing 1% normal bovine serum albumin and 0.1% Triton-X-100 for 1 h at room temperature. To assess the alteration and location of target protein, H9c2 Cardiac myoblast cell were incubated with a mixture of goat polyclonal anti-MAP1-LC3 antibody in the above blocking solution at 4°C overnight. Cultures were subsequently washed and incubated with a mixture of ant-rabbit IgG antibody conjugated to FITC for 1 h at 37°C, then rinsed several times and incubated with 10 mg/ml 4’-6-diamidino-2-phenylin-dole (DAPI) for 5 min at room temperature. After 10 min incubation and several rinses, cultures were mounted on glass slides, cover-slipped with Vectashield mounting medium for fluorescence, and analyzed with a fluorescence microscope.
Western blot
The MAP1-LC3, Beclin 1, Bcl-2, and β-actin proteins were detected with the antibodies rabbit polyclonal anti-MAP-LC3, rabbit polyclonal anti-bcl-2 (Abcam, Cambridgeshire, UK), goat polyclonal anti-beclin 1 (Santa-Cruz, CA, USA), and mouse monoclonal anti-β-actin (Lianke Biological Technology Co., Ltd, Changzhou, China). Protein concentration was determined using a BCA kit (Pierce, Rockford, IL, USA). Fourty micrograms of protein from the homogenized sample was mixed 1:4 with loading buffer, separated by 10% SDS-PAGE and transferred to a nitrocellulose membrane. After blocking the non-specific binding sites with 5% milk powder, the membrane was treated with the first antibodies at 4°C overnight, then washed and incubated for 2 h with horseradish peroxidase-conjugated goat anti-rabbit antibody, donkey anti-goat antibody, and goat anti-mouse antibody, in TBST containing 5% nonfat dry milk. Immunoreatctivety was detected with enhanced chemiluminesence autoradiography. Specific antigen-antibody complex was detected by a standard enhanced chemiluminescence detection system. The amount of detected protein was quantified by Scion Image 4.03 software (Scion Corporation, Frederick, USA) and was expressed as the ratio to β-actin protein.
Statistical analysis
Data are presented as means ± SD for at least three sets of independence experiments. Differences between groups were assessed using One-way analysis of variance (ANOVA) were used for statistical analysis (Prism v5.0; GraphPad Software, San Diego, CA, USA) of data obtained within the same group of rats cardiac myoblast Differences were considered to be statistically significant when P < 0.05.
Results
Cell survival rate
As shown in Figure 2, the cell viability was reduced significantly by H/R injury. The H/R group (62.2 ± 6.49%) and SWOP group (79.49 ± 1.37%) had a significant decrease in cell survival rate versus control (P < 0.05 vs. CON). However, the cells with sevoflurane preconditioning increase cell survival rate compared with H/R alone (79.49 ± 1.37% vs. 62.2 ± 6.49%, P < 0.05).
Figure 2.
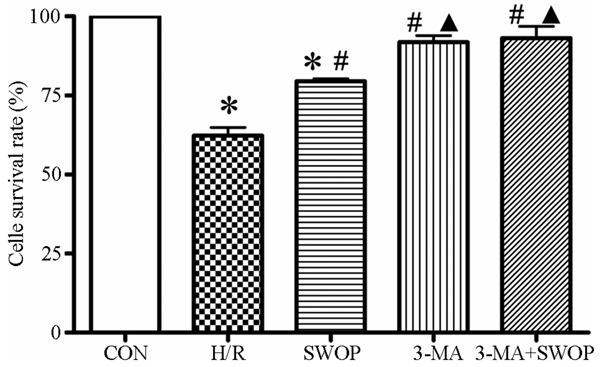
The cell survival rate was meansured after different treatments in the control, H/R injury, sevoflurane preconditioning (SWOP), 3-MA and 3-MA+SWOP groups. Means ± SD for each group. *P < 0.05, H/R and SWOP vs control. #P < 0.05, SWOP, 3-MA and 3-MA+SWOP vs. H/R. ▲P < 0.05, 3-MA and 3-MA+SWOP vs. SWOP.
Apoptotic cells percentage
The calculated apoptosis cell percentage in the control group was as low as (0.99 ± 0.70%). The H/R group (9.68 ± 1.10%), SWOP group (7.30 ± 0.55%), 3-MA group (5.85 ± 1.45%) and 3-MA+SWOP group (4.60 ± 1.99%) significantly increased the apoptotic cells percentage compared with the control group (*P < 0.05, vs. control). However, the sevoflurane preconditioning and 3-MA reduced significantly the apoptotic cells percentage after H/R injury (#P < 0.05, vs. H/R) (Figure 3).
Figure 3.
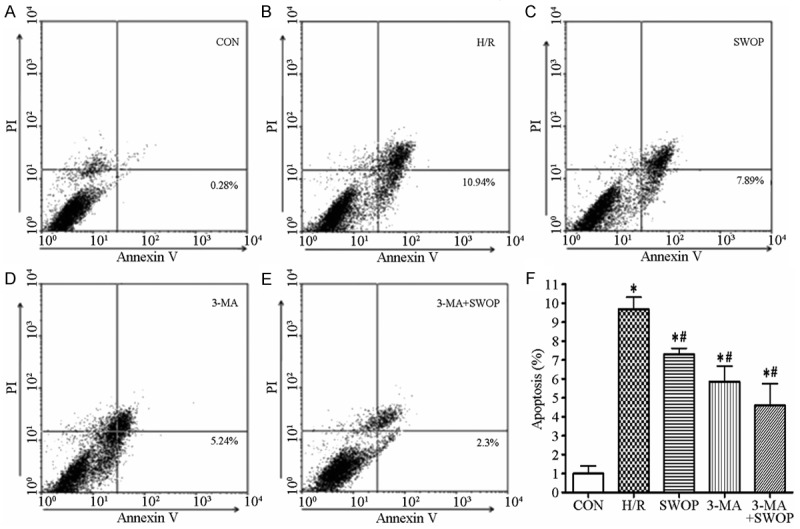
The annexin V-FITC apoptosis detection kit estimated apoptotic cells percentage. A. The apoptotic cell percentage was measured by flow Cytometer after different treatments in the control, H/R injury, sevoflurane preconditioning (SWOP), 3-MA and 3-MA+SWOP groups. B. The flow cytometric histograms of apoptotic rate was showed for each group. *P < 0.05, control vs. H/R, SWOP, 3-MA and 3-MA+SWOP. #P < 0.05, SWOP, 3-MA and 3-MA+SWOP vs. H/R.
Ultrastructural changes in cells
As shown in Figure 4, ultrastructural changes were examined with transmission electron microscope (TEM) in control group, H/R group and SWOP group H9c2 rat cardiomyocytes. TEM images showed normal cytoplasm, mitochondria, nucleus, and chromatin in control H9c2 cardiomyocytes, while few or no autophagosomes and lysosomes were obsereved. In contrast, the TEM images from H/R group cells displayed many autophagosomes at various developmental stages. There was observed a double membrane autophagosomes (Figure 4, as indicated by red arrow) and many lysosome (Figure 4, as indicated by green arrow) in the cytoplasm.
Figure 4.
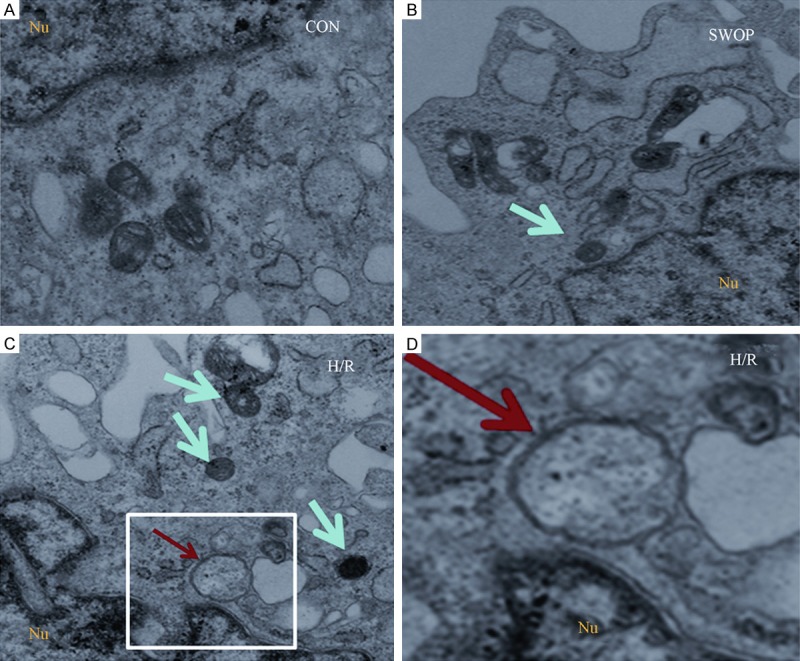
The ultrastructure of the control group, H/R group and SWOP group H9c2 rat cardiomyocytes in the TEM. There was few or no autophagosomes in the control group. At the H/R group, typical autophagosomes with the characteristic double membrane are note (red arrow), the number of lysosomes increased (green arrow). The SWOP group were few of lysosomes apparent in the cytoplasm (green arrow).
Changes in autophagic activity
As shown in Figure 5, we used H9c2 rat cardiomyocytes to explore the role of autophagy during H/R injury. To determine whether autophagic activity is modulated in response to H/R, we first characterized changes in cellular autophagosomal content using fluorescence microscope. During the initiation of autophagy, cytosolic LC3 (LC3-I) is cleaved and lipidated to form LC3-II [7,8]. LC3-II is then recruited to the autophagosomal membrane [9]. Thus, punctuate green fluorescent protein LC3-labeled (GFP-LC3) structures represent autophagosomes, also referred to as autophagic vacuoles. Importantly, overexpression of GFP-LC3 does not affect autophagic activity, and transgenic mice expressing GFP-LC3 display no detectable abnormalities [10,11]. We transfected H9c2 rat cardiomyocytes with GFP-LC3 and compared the abundance of autophagic vacuoles in cells subjected to H/R to sevoflurane preconditioning cells. The results demonstrate that cellular autophagic vacuoles content was increased after H/R injury. Sevoflurane preconditioning and 3-MA treatmented decreased FITC-labeled MAP1-LC3 fluorescence intensity compared with control group.
Figure 5.
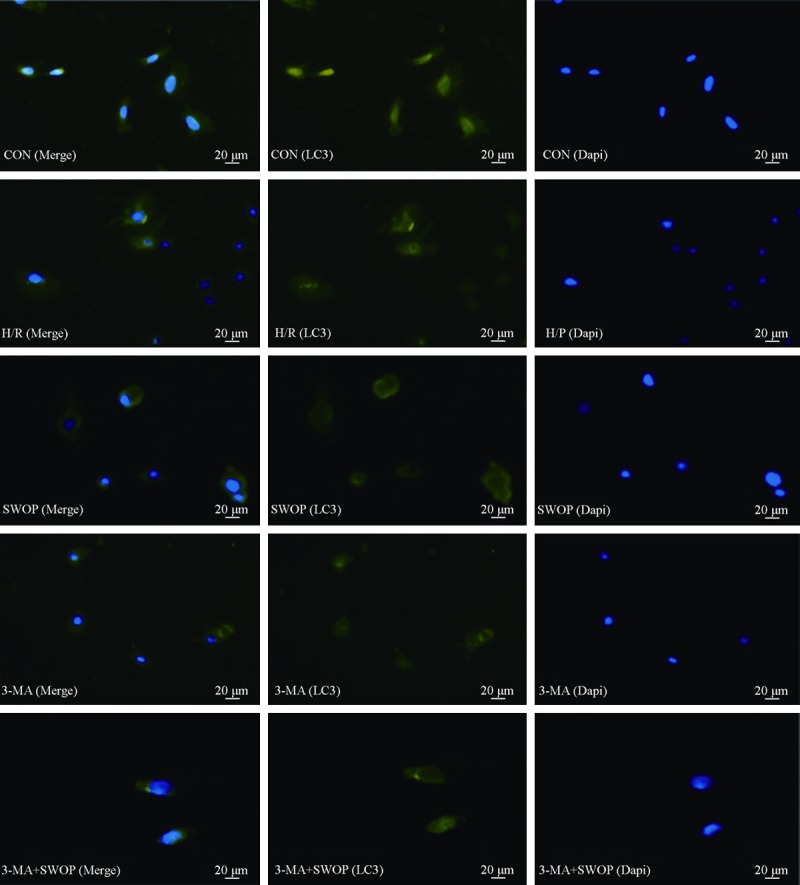
Photomicrographs of H9c2 rat cardiomyocytes stained with MAP1-LC3 and DAPI in control (the first panels), after H/R injury (the second panels), and treatment with sevoflurane preconditioning (the third panels), treatment with 3-MA (the fourth panels), or 3-MA+SWOP (the fifth panels). Scale bar = 20 μm.
LC3, Beclin 1 and Bcl-2 protein expression
H/R injury up-regulated the expression of LC3-II and Beclin 1 proteins (342 ± 66% and 163 ± 18%, respectively, P < 0.05) compared with the CON group (100%), which were increased in SWOP group (202 ± 77% and 128 ± 8%, respectively, P < 0.05). The expression of LC3-II and Beclin 1 proteins was decreased in 3-MA group (110 ± 28% and 97 ± 6%, respectively) and 3-MA+SWOP group (93 ± 7% and 98 ± 6%, respectively) compared with H/R group, but Bcl-2 was upregulated in 3-MA group (158 ± 4%) and 3-MA+SWOP group (156 ± 5%) compared to H/R group (103 ± 7%). N = 5 for each group (Figure 6).
Figure 6.
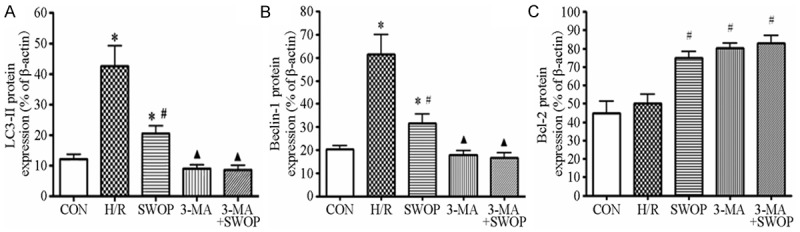
The protein expression of LC3-II (A), Beclin 1 (B) and Bcl-2 (C). H/R injury up-regulated the expression of LC3-II and Beclin 1 proteins when compared to control group, which were lower in SWOP group, 3-MA group and 3-MA+SWOP group than in H/R group, but Bcl-2 expression was upregulated. N = 5 for each group. Data are presented as mean ± SD. *P < 0.05 vs. CON; #P < 0.05 vs. H/R; ▲P < 0.05 vs. SWOP.
Discussion
The results of our study indicated that 2.5% sevoflurane preconditioning 24 h before H/R effectively reduces the survival rate and apoptosis cell percentage of H9c2 rat cardiomyocytes. Our data support the hypothesis that sevoflurane can produce a delayed window of preconditioning. Animal studies indicate that volatile anesthetics offer protection from ischemia-reperfusion injury in heart [3] and brain [12] tissue lasting for up to 48 h. Tonkovic-Capin et al. [13] first reported successful late preconditioning of the heart in a rabbit model using isoflurane at 1% end-tidal concentration. Subsequent studies confirmed these findings in rats [3,14,15] and mice [16]. In accordance with these animal studies, the present study now provides evidence of a late protective phenotype after sevoflurane inhalation in H9c2 rat cardiomyocytes.
It has already been shown that the volatile anesthetic sevoflurane protects the heart against ischemia-induced adenosine triphosphate (ATP) depletion, Ca2+ overload and oxidative stress through activation of protein kinase C (PKC), opening of mitochondrial K+ATP channels (mito K+ ATP) and the production of reactive oxygen species (ROS) [17,18].
However, it is unclear whether autophagy involved in volatile anesthetic-induced late preconditioning. In the present study, we have shown that autophagy accompanies single cycle H/R in H9c2 rat cardiomyocytes. Reduction, by the 3-MA treament, in expression of Beclin 1, a protein known to be important for autophagy, results in a corresponding reduction in the autophagy induced by H/R. SWOP which we have shown previously to protect cardiomyocytes from cell death induced by H/R, also reduces autophagy, at least partly by reducing Beclin 1 expression. These beneficial effects were according with the selective inhibition of autophagy 3-MA, indicating that autophagy is involved in sevoflurane preconditioning induced delayed cardioprotection.
Recently, studies have demonstrated that autophagy is a vital process in the heart, presumably participating in the removal of dysfunctional cytosolic components and serving as a catabolic energy source during times of starvation [19]. Activation of lysosomal pathways was reported more than 30 years ago in neonatal hearts exposed in vitro to hypoxia and glucose deprivation followed by reoxygenation with glucose restoration [20]. Since then, many studies have reported increased autophagic activity in the settings of ischemia and ischemia/reperfusion [21,22]. The inhibition of ischemia/reperfusion-induced autophagy by either 3-methyladenine (3-MA) or by genetic knockdown of Beclin 1 leads to enhanced cardiac myocyte survival in vitro [5]. In according with this study, treated by 3-MA increased cell survival rate in H9c2 rat cardiomyocytes.
Autophagy contributes to the mechanism of autophagic cell death. Furthermore, it has been clarified that the the existence of a biochemical mechanism of crosstalk between apoptosis and autophagic cell death. Recent studies have suggested proteins of interest were the autophagy protein beclin-1 (Atg6) and its interaction with the anti-apoptotic protein bcl-2. Beclin-1 is a bcl-2 interacting protein that has been documented to be an important player in the induction of autophagy. This interaction between these proteins is considered significant as it governs the switch between the induction of autophagy and apoptosis [5,23].
Our observation that sevoflurane preconditioning downregulated the expressions of Beclin 1 and upregulated Bcl-2 while increased in cell survival and decreased the apoptosis cell percentage after H/R injury. The inhibition of autophagy by Bcl-2 has been demonstrated in yeast, in an ex vivo model, and in vivo in mice [24]. This inhibition is dependent on the Beclin 1/Bcl-2 interaction. Interestingly, this interaction is modulated by conditions known to regulate autophagy. Under nutrient-rich conditions, when autophagy is inhibited, Beclin 1 and Bcl-2 interact strongly. In contrast, in starvation, when autophagic rates are high, the interaction between Beclin 1 and Bcl-2 is weak. This suggests that Bcl-2 acts as a rheostat that turns autophagy on or off when required. Under these conditions, it may be possible to regulate Bcl-2 autophagic activity quickly. In our experiments, reduction of Beclin 1 expression by SWOP significantly reduced the number of autophagosomes and lysosome after H/R, confirming a central role for Beclin 1 in autophagocytosis in the heart. Here we demonstrate that SWOP reduces H/R-induced autophagy by reducing expression of Beclin 1.
Here, we used hypoxia-reoxygenation induced autophagy in H9c2 cells, an established in vitro model for mimicking ischemia/reperfusion of cardiomyocytes [25], to examine the cellular mechanisms responsible for the anti-autophagy effects of SWOP. The H9c2 rat cardiomyocytes is an excellent model for studying many aspects of cardiac cell physiology [26]. In our hands H9c2 cells reproducibly underwent PCD in response to simulated H/R via pathways resembling in vivo cardiac H/R injury. In such conditions, nutrient-starved cells massively accumulate autophagic vacuoles in the cytoplasm, as determined by electron microscopy or by following the redistribution of the autophagic vacuoles marker LC3 fused with LC3-GFP [27].
In the study, there was few or no autophagosomes in the control group. At the H/R group, typical autophagosomes with the characteristic double membrane are note, the number of lysosomes increased. The SWOP group was few of lysosomes apparent in the cytoplasm. As mentioned above, massive autophagic vacuolization is observed in some instances of cell death, which has been named “autophagic cell death” (type 2 cell deaths). The mere presence of autophagic vacuoles in the cytoplasm does not necessarily indicate an increased level of autophagy, since a reduction of the fusion between autophagosomes and lysosomes suffices to promote autophagic vacuoles accumulation. Thus, quantitative methods for the detection of cytoplasmic protein turnover should be employed in addition to autophagic vacuoles monitoring, in order to verify augmented levels of autophagy. In order to elucidate the mechanisms of apoptosis, we detected the expression of autophagy-related proteins by Western blot. We found preconditioning with sevoflurane increased the expression level of autophagy-related protein LC3-II and Beclin-1. Similarly, Perucho et al. found isoflurane precondition increased LC3-II expression to increase the Wild-type mice [28].
Several limitations of this study should be noted. Firstly, we did not identify the area of necrosis or apoptosis. Secondly, the sevoflurane concentration used in this study was 2.5%, other concentrations may have greater or lesser effects. Thirdly, further investigation is needed to identify essential components in these complex signal transduction cascades that mediate SWOP in our study.
In summary, our findings suggest that preconditioning with 2.5% sevoflurane preconditioning (SWOP) 24 h before H/R produced delayed myocardial protection on H9c2 rat cardiomyocytes. On the other hand, SWOP reduces the survival rate and apoptosis cell percentage of cells via inhibition Beclin 1-mediated autophagic cell death in cardiac myocytes.
Acknowledgements
This work was supported by grants No. BK20141187 (to Dr. Wang) from the Natural Science Foundation of Jiangsu Province, and by Grants No SYS201473 (to Dr. Qiao) from the Technology Bureau of Suzhou, China. Dr. Wang also received support from the Project of Gusu Health Key Talent. The Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Disclosure of conflict of interest
None.
References
- 1.Zvara DA, Bryant AJ, Deal DD. Anesthetic preconditioning with sevoflurane does not protect the spinal cord after an ischemic-reperfusion injury in the rat. Anesth Analg. 2006;102:1341–1347. doi: 10.1213/01.ane.0000204357.06219.8c. [DOI] [PubMed] [Google Scholar]
- 2.Kuzuya T, Hoshida S, Yamashita N, Fuji H, Oe H, Hori M, Kamada T, Tada M. Delayed effects of sublethal ischemia on the acquisition of tolerance to ischemia. Circ Res. 1993;72:1293–1299. doi: 10.1161/01.res.72.6.1293. [DOI] [PubMed] [Google Scholar]
- 3.Lutz M, Liu H. Inhaled Sevoflurane produces better delayed myocardial protection at 48 versus 24 hours after exposure. Anesth Analg. 2006;102:984–990. doi: 10.1213/01.ane.0000198568.79079.4c. [DOI] [PubMed] [Google Scholar]
- 4.Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2 interacting protein. J Virol. 1998;72:8586–8596. doi: 10.1128/jvi.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 6.Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117:2805–2812. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- 7.Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- 8.Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–668. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirisako T, Baba M, Ishihara N, Miyazawa K, Ohsumi M, Yoshimori T, Noda T, Ohsumi Y. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J Cell Biol. 1999;147:435–446. doi: 10.1083/jcb.147.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Payne RS, Akca O, Roewer N, Schurr A, Kehl F. Sevoflurane induced preconditioning protects against cerebral ischemic neuronal damage in rats. Brain Res. 2005;1034:147–152. doi: 10.1016/j.brainres.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 12.Tonkovic-Capin M, Gross GJ, Bosnjak ZJ, Bosnjak ZJ, Tweddell JS, Fitzpatrick CM, Baker JE. Delayed cardioprotection by isoflurane: role of K(ATP) channels. Am J Physiol Heart Circ Physiol. 2002;283:H61–68. doi: 10.1152/ajpheart.01040.2001. [DOI] [PubMed] [Google Scholar]
- 13.Shi Y, Hutchins WC, Su J, Siker D, Hogg N, Pritchard KA Jr, Keszler A, Tweddell JS, Baker JE. Delayed cardioprotection with isoflurane: role of reactive oxygen and nitrogen. Am J Physiol Heart Circ Physiol. 2005;288:H175–184. doi: 10.1152/ajpheart.00494.2004. [DOI] [PubMed] [Google Scholar]
- 14.Wakeno-Takahashi M, Otani H, Nakao S, Imamura H, Shingu K. Isoflurane induces second window of preconditioning through upregulation of inducible nitric oxide synthase in rat heart. Am J Physiol Heart Circ Physiol. 2005;289:H2585–2591. doi: 10.1152/ajpheart.00400.2005. [DOI] [PubMed] [Google Scholar]
- 15.Tsutsumi YM, Patel HH, Huang D, Roth DM. Role of 12-lipoxygenase in volatile anesthetic-induced delayed preconditioningin mice. Am J Physiol Heart Circ Physiol. 2006;291:H979–983. doi: 10.1152/ajpheart.00266.2006. [DOI] [PubMed] [Google Scholar]
- 16.Kevin LG, Novalija E, Riess ML, Camara AK, Rhodes SS, Stowe DF. Sevoflurane exposure generates superoxide but leads to decreased superoxide during ischemia and reperfusion in isolated hearts. Anesth Analg. 2003;96:949–955. doi: 10.1213/01.ANE.0000052515.25465.35. [DOI] [PubMed] [Google Scholar]
- 17.Novalija E, Kevin LG, Eells JT, Henry MM, Stowe DF. Anesthetic preconditioning improves adenosine triphosphate synthesis and reduces reactive oxygen species formation in mitochondria after ischemia by a redox dependent mechanism. Anesthesiology. 2003;98:1155–1163. doi: 10.1097/00000542-200305000-00018. [DOI] [PubMed] [Google Scholar]
- 18.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 19.Sybers HD, Ingwall J, DeLuca M. Autophagy in cardiac myocytes. Recent Adv Stud Cardiac Struct Metab. 1976;12:453–463. [PubMed] [Google Scholar]
- 20.Yan L, Vatner DE, Kim SJ, Ge H, Masurekar M, Massover WH, Yang G, Matsui Y, Sadoshima J, Vatner SF. Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci U S A. 2005;102:13807–13812. doi: 10.1073/pnas.0506843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281:29776–29787. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- 22.Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, Gottlieb RA, Gustafsson AB. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007;14:146–157. doi: 10.1038/sj.cdd.4401936. [DOI] [PubMed] [Google Scholar]
- 23.Pattingre S, Levine B. Bcl-2 inhibition of autophagy: a new route to cancer? Cancer Res. 2006;66:2885–2888. doi: 10.1158/0008-5472.CAN-05-4412. [DOI] [PubMed] [Google Scholar]
- 24.Cuervo AM. Autophagy: many paths to the same end. Mol Cell Biochem. 2004;263:55–72. doi: 10.1023/B:MCBI.0000041848.57020.57. [DOI] [PubMed] [Google Scholar]
- 25.Gurusamy N, Lekli I, Mukherjee S, Ray D, Ahsan MK, Gherghiceanu M, Popescu LM, Das DK. Cardioprotection by resveratrol: a novel mechanism via mechanism via autophagy involving the mTORC2 pathway. Cardiovasc Res. 2010;86:103–112. doi: 10.1093/cvr/cvp384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park M, Youn B, Zheng XL, Wu D, Xu A, Sweeney G. Globular Adiponectin, Acting via AdipoR1/APPL1, Protects H9c2 Cells from Hypoxia/Reoxygenation-Induced Apoptosis. PLoS One. 2011;6:e19143. doi: 10.1371/journal.pone.0019143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.González-Polo RA, Boya P, Pauleau AL, Pauleau AL, Jalil A, Larochette N, Souquère S, Eskelinen EL, Pierron G, Saftig P, Kroemer G. The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci. 2005;118:3091–3102. doi: 10.1242/jcs.02447. [DOI] [PubMed] [Google Scholar]
- 28.Perucho J, Rubio I, Casarejos MJ, Gomez A, Rodriguez-Navarro JA, Solano RM, De Yébenes JG, Mena MA. Anesthesia with isoflurane increases amyloid pathology in mice models of Alzheimer’s disease. J Alzheimers Dis. 2010;19:1245–1257. doi: 10.3233/JAD-2010-1318. [DOI] [PubMed] [Google Scholar]