

Abstract
Innate immunity to viral infection involves induction of the type I IFN response; however, dysfunctional regulation of this pathway leads to inappropriate inflammation. Here, we evaluated a nonconsanguineous family of mixed European descent, with 4 members affected by systemic inflammatory and autoimmune conditions, including lupus, with variable clinical expression. We identified a germline dominant gain-of-function mutation in TMEM173, which encodes stimulator of type I IFN gene (STING), in the affected individuals. STING is a key signaling molecule in cytosolic DNA-sensing pathways, and STING activation normally requires dimerization, which is induced by 2′3′ cyclic GMP-AMP (cGAMP) produced by the cGAMP synthase in response to cytosolic DNA. Structural modeling supported constitutive activation of the mutant STING protein based on stabilized dimerization. In agreement with the model predictions, we found that the STING mutant spontaneously localizes in the Golgi of patient fibroblasts and is constitutively active in the absence of exogenous 2′3′-cGAMP in vitro. Accordingly, we observed elevated serum IFN activity and a type I IFN signature in peripheral blood from affected family members. These findings highlight the key role of STING in activating both the innate and adaptive immune responses and implicate aberrant STING activation in features of human lupus.
Introduction
The type I IFN response is a key component of innate immunity to viral infection. A tight control of this pathway is required to avoid disease-causing inflammation. This is well illustrated by the elucidation of the genetic basis of Aicardi-Goutières syndrome (AGS), an inflammatory phenotype sometimes mimicking pathogenic viral infection, which is related to enhanced type I IFN signaling as a consequence of mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR (1–4). All of these proteins normally participate in regulating nucleic acid metabolism, and their dysfunction leads to an accumulation in the cytosol of nucleic acid, which is then detected to instigate a prototypic antiviral response. Recently, gain-of-function mutations in IFIH1 (also known as MDA-5), encoding a cytosolic viral RNA receptor, were reported in patients demonstrating a spectrum of inflammatory phenotypes, including classical AGS (5). Similarly, cytosolic DNA sensors have been described as leading to type I IFN and inflammatory cytokine induction (e.g., IL-1 and IL-18) (6, 7). In particular, stimulator of type I IFN gene (STING) has been identified as a central mediator of responses to cytosolic DNA upon induced dimerization (8–11) and as the receptor of 2′3′ cyclic GMP-AMP (cGAMP) produced by the recently described DNA sensor cGAMP synthase (12, 13). In this report, we describe an inherited dominant TMEM173 mutation leading to constitutive activation of STING and thereby to an inflammatory and a lupus-like disease.
Results and Discussion
Clinical presentation and familial history.
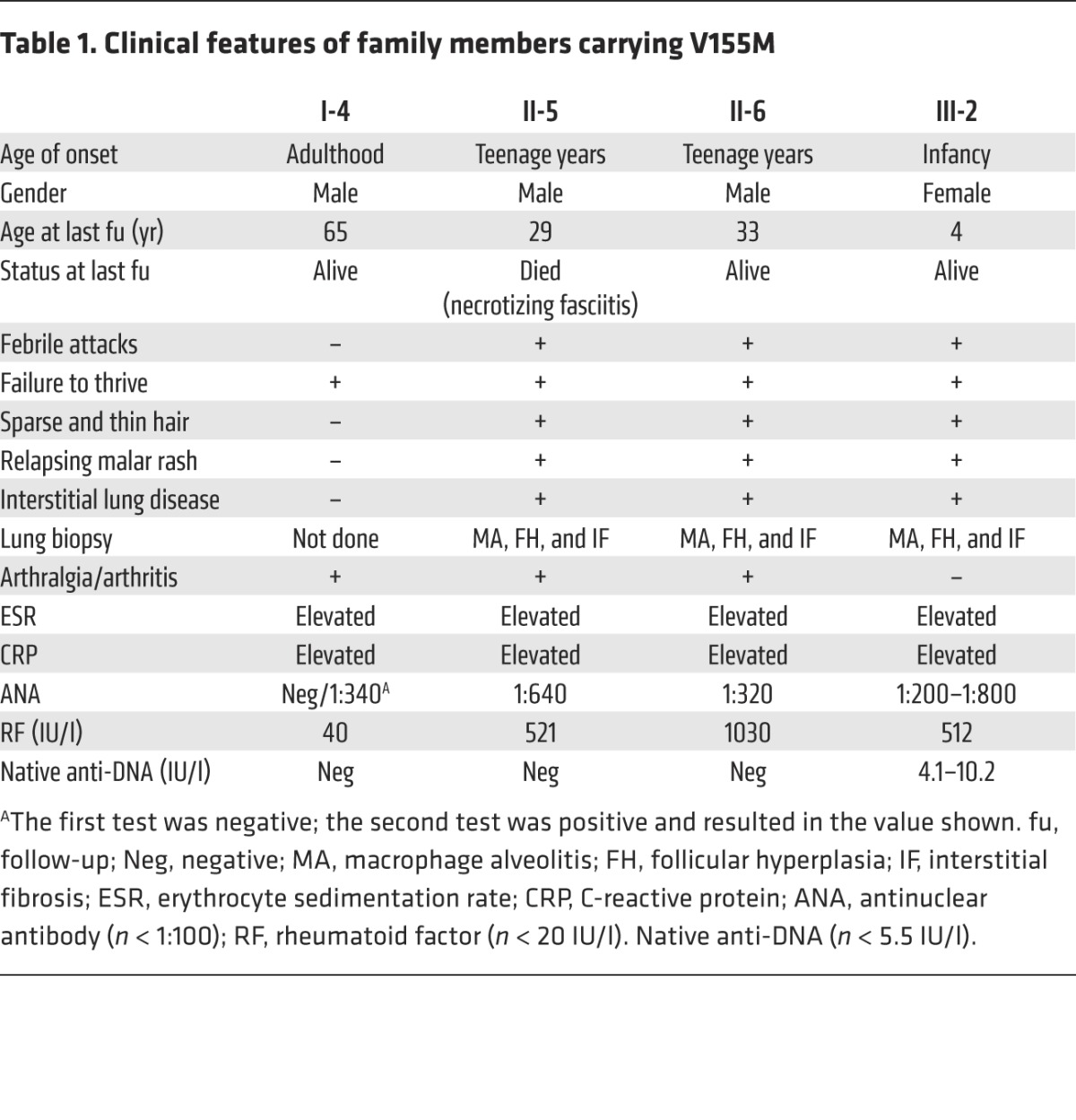
We report 4 members of a single kindred (Figure 1, A and B) exhibiting a complex systemic inflammatory syndrome associated with pulmonary fibrosis (Figure 1, B and C) and autoimmunity (Table 1, Supplemental Methods, and Supplemental Table 1; supplemental material available online with this article; doi:10.1172/JCI79100DS1). The index case had early-onset febrile attacks, malar rash, lung disease, and failure to thrive. The clinical history suggested a familial syndrome, with variable clinical expression. The proband’s father (II-5) and paternal uncle (II-6) are monozygotic twins and presented with a similar inflammatory syndrome, with lung disease and arthritis (see Supplemental Methods for details). The paternal grandfather of the proband (I-4) is currently 65 years old, and no clinically relevant lung disease has been observed (Table 1).
Figure 1. Familial STING mutation associates with inflammatory and autoimmune condition.

(A) Pedigree of the family carrying the STING V155M heterozygous mutation. Affected individuals are indicated by black boxes and circles; deceased individuals are indicated by diagonal bars; patient III-2 is indicated by an arrow. (B) Familial segregation of the V155M mutation. (C) Lung CT scan from patient III-2, showing interstitial lung disease in a parenchymal view (left) and fibrosis in a mediastinal view (right). (D) Lung biopsy from patient III-2 (Masson’s trichrome; original magnification, ×10) and immunohistochemistry (hematoxylin; original magnification, ×10; left), with anti-CD20 (middle) and anti CD68 (right) antibodies, showing macrophage alveolar infiltration, follicular hyperplasia, and mild interstitial fibrosis.
Table 1.
Clinical features of family members carrying V155M
Genetic analysis reveals a TMEM173/STING mutation.
We undertook whole-exome sequencing on DNA from the 2 available patients (proband [III-2] and II-6) and the healthy mother of the proband (II-4) and looked for variants present in both patients and absent in the mother. After exclusion of common polymorphisms described in publically available libraries (dbSNP, 1000 genomes project, Exome Variant Server) and an in-house database of 4,047 exomes, we identified a rare variant c.463G>A (resulting in a p.V155M substitution) in TMEM173, encoding STING (Figure 1B). This variant was confirmed by Sanger sequencing (Supplemental Table 2). The 3 affected surviving members tested (III-2, II-6, I-4) all carried the same p.V155M variant (Figure 1B), which was considered as likely pathogenic on the basis of species conservation, in silico (sorting intolerant from tolerant and Polyphen2) predictions, and the previous observation that mutagenesis at this position (V155R) profoundly affects STING function by impairing its dimerization (14).
3D modeling of the V155M mutant.
In order to gain further insight into the effect of the p.V155M substitution on protein activity, we examined the experimental 3D structure of the C-terminal cytosolic domain (CTD) of STING (14–16). The V155 residue is located in the first highly hydrophobic helix (α5) of the CTD of STING that forms intermolecular hydrophobic interactions also involving helix α7 (Supplemental Figure 1). Within the dimer, p.V155 is at the center of a hydrophobic network, in the vicinity of the closest contact between the 2 subunits (p.G158) (Supplemental Figure 1B). This network is similar whatever the conformational state of the protein (bound or not to cyclic dinucleotides). There is only one major possibility for accommodating the methionine side chain of the p.V155M substitution (Supplemental Figure 1C), which is predicted to lead to the establishment of a strikingly tighter network of interactions. The p.V155M substitution is likely to stabilize the position of p.M271 from the same subunit, which forms a strong sulfur-aromatic interaction with p.W161 from the other subunit. This should reinforce the stability of the dimer. It is thus possible that the p.V155M mutant mimics the effect of ligand binding.
Constitutive activation of the V155M mutant in vitro.
To examine the activity of the p.V155M mutated STING protein, we used a luciferase reporter assay that includes the IFN-β gene promoter. In this assay, overexpression of wild-type STING protein activated the IFNB promoter 2-fold over control in the absence of ligand (Figure 2A). Stimulation with synthetic 2′3′-cGAMP enhanced promoter activation in a dose-dependent manner, and this induction was not observed in the absence of STING (Figure 2A). In contrast, the p.V155M mutant induced reporter activity in the absence of synthetic ligand stimulation, and stimulation with synthetic 2′3′-cGAMP did not further activate reporter activity to a significant level. These data further suggest that the p.V155M mutation mutation provides constitutive activation independent of ligand binding. Interestingly, mutant STING expression was decreased in comparison with wild-type STING expression (Figure 2B). Thus, the observed enhancement of activity was not due to overexpression of the protein and might suggest that the mutant is less stable than the wild-type protein or that the mutant protein is more likely to be degraded.
Figure 2. Constitutive activation of the mutant STING in vitro.

(A) Luciferase induction in HEK293FT cells cotransfected with empty vector (EV), wild-type STING, V155M STING, and a luciferase plasmid under the control of the IFNB promoter and stimulated with increasing amounts of synthetic 2′3′-cGAMP complexed to lipofectamine (maximal dose, 4 μg/ml; dose-response dilution factor, 3) (n = 3, mean and SEM; paired t test, *P < 0.05). (B) Immunoblot of STING expression in HEK293FT cells transfected with wild-type and V155M STING (representative of 3 independent experiments).
Abnormal STING intracellular localization in cells from patient III-2.
We next analyzed STING intracellular localization in fibroblasts from the proband and a control by confocal microscopy (Figure 3). At steady state, STING localized mainly in the Golgi and in perinuclear punctiform vesicles of patient fibroblasts (Figure 3C and Supplemental Figure 5). Such localization has been reported to correspond with STING activation (9). In contrast, STING was uniformly expressed in the cytoplasm of control fibroblasts (Figure 3A). Upon 2′3′-cGAMP activation, mutant STING remained localized to the Golgi and the perinuclear vesicles (Figure 3D), whereas wild-type STING was mostly observed in the latter structures (Figure 3B). These findings indicate that mutant STING is likely activated in vivo in patient cells independently of ligand addition and that there is only limited further activation following 2′3′-cGAMP stimulation.
Figure 3. Intracellular localization of p.V155M STING.

STING localization in primary fibroblasts assessed by confocal microscopy. (A) Healthy control and (C) patient fibroblasts (III-2) at steady state. (B) Healthy control and (D) patient fibroblasts (III-2) after 8-hour stimulation with 4 μg/ml 2′3′-cGAMP. Images are representative of 3 independent experiments. Scale bar: 15 μm.
Functional consequences of the V155M STING mutant.
Activation of STING is known to induce type I IFN production through TBK-1 phosphorylation and IRF3 phosphorylation (9). In order to study the functional consequences of the V155M STING variant, we assessed the expression of 6 gene transcripts known to be overexpressed in PBMCs of patients with AGS (IFI44L, SIGLEC1, RSAD2, IFI27, IFIT1, and ISG15) (17). Positive controls were generated by treatment of healthy control PBMCs with IFN-α (1,000 U/ml) for 6 hours (18). PBMCs from the patient (III-2) and her grandfather (I-4) exhibited an overexpression of all 6 tested genes as compared with control PBMCs and PBMCs from the healthy mother (II-4) (Supplemental Figure 2). Similar overexpression was also seen in in vitro–activated T cells (Supplemental Figure 3). In contrast, a set of non–type I IFN–regulated genes remained normally expressed in peripheral blood compared with control samples (Supplemental Figure 4). These results suggest that cells with the V155M variant spontaneously express type I IFN. Accordingly, serum-type I IFN activity was found to be elevated in patient blood (III-2).
Collectively, the results reported here suggest a gain-of-function consequence of the V155M variant. The constitutively active mutant thus results in upregulated type I IFN production, as measured by serum IFN activity, and a type I IFN signature in whole blood. The very same mutation and 2 other mutations at neighboring residues have been recently reported to be associated with inflammation and severe vasculopathy in humans (19). The present work thus confirms the gain-of-function nature of the V155M mutation, provides a structural model accounting for constitutive mutant STING activity, and extends the spectrum of clinical phenotypic expression.
As predicted by the structural analysis, substitution by a methionine residue at position 155 strengthens the hydrophobic network established by the α5 helix at the dimer interface and consequently may enhance the stability of the STING dimer and/or mimic the effect of ligand binding. The ligand-independent STING activity, as observed in functional assays, indicates a constitutive in vitro activity and suggests spontaneous dimerization. This was further confirmed by the intracellular localization of the mutant STING in the Golgi and in perinuclear vesicles in fibroblasts at steady state. Furthermore, activation by 2′3′-cGAMP did not significantly modify the intracellular localization or IFN-β promoter activity (20).
Whether the V155M TMEM173 is constitutively active in vivo in all cell types is not known. It remains possible that mutant activity varies from one cell type to another as a consequence of variable expression of inhibitors or differential regulation of STING expression. An additional level of complexity might also lie downstream of the type I IFN responses. Interestingly, the V155M TMEM173 mutation is associated with chronic inflammation of highly variable expression. At one end of the spectrum, it has been found to be associated with an early-onset severe cutaneous vasculopathy accompanied with extensive tissue loss (19) and, on the other, it has been found to be associated with mild inflammation, without important clinical consequences (I-4). In addition, the V155M STING can even result in a severe chronic inflammation characterized by pulmonary inflammation and fibrosis, associated with a malar rash and autoantibody production reminiscent of systemic lupus erythematosus (III-4). The most severe clinical presentations appear to be associated with de novo TMEM173 mutations (19), whereas the familial inheritance of the V155M STING is associated with milder clinical phenotypes. The observed phenotypic variability could be driven by a modifier intervening downstream of STING or by viral infections that might contribute to setting a tonic IFN signal (21).
Our findings highlight the central role of STING in type I IFN production and demonstrate its causal role in inflammatory syndromes variably accompanied with small vessel vasculopathy, pulmonary fibrosis, and autoimmunity reminiscent of SLE. Hence, TMEM173 gain-of-function mutations should be screened for as a monogenic cause of this broad spectrum of diseases. STING could represent a new therapeutic target in these disorders as well as other more common inflammatory diseases triggered by cytosolic DNA stimulation of microbial or endogenous origin, resulting in dysregulated type I IFN production.
Methods
Further details are provided in the Supplemental Methods.
Statistics.
Analyses were performed with PRISM software (version 4 for Macintosh, GraphPad Inc.). Statistical hypotheses were tested using 2-tailed t test. A P value of less than 0.05 was considered significant.
Study approval.
Written informed consent (parental consent, in case of minors) was obtained from all participants of the study. The study and protocols conform to the 1975 Declaration of Helsinki and were approved by the comité de protection des personnes Ile de France II and the French advisory committee on data processing in medical research.
Supplementary Material
Acknowledgments
The authors wish to thank the patients and family members for their cooperation in this study. This work was funded by grants from the Institut National de la Santé et de la Recherche Médicale, the European Research Council (ERC PIDIMMUN no. 249816 to A. Fischer; GA 309449 Fellowship to Y.J. Crow), and Agence Nationale de la Recherche (ANR-14-CE14-0026-01 “Lumugene” to F. Rieux-Laucat). This program has received a state subsidy managed by the National Research Agency under the “Investments for the Future” program bearing the reference ANR-10-IAHU-01 and IDEX Sorbonne Paris Cité (SPC/JFG/2013 - 031 to F. Rieux-Laucat). N. Jeremiah is supported by the Fondation Arthritis (France). Exome sequencing was funded by the Fondation Maladie Rares (France). The Leica SP8 was funded by Fondation ARC (France). N. Manel was supported by the ATIP-Avenir program, Agence Nationale de Recherche sur le SIDA (ANRS), Ville de Paris Emergence program, European FP7 Marie Curie Actions, LABEX VRI, LABEX DCBIOL, ACTERIA Foundation, Fondation Schlumberger pour l’éducation et la Recherche, and European Research Council grant 309848 HIVINNATE.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2014;124(12):5516–5520. doi:10.1172/JCI79100.
References
- 1.Crow YJ, et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet. 2006;38(8):917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 2.Crow YJ, et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet. 2006;38(8):910–916. doi: 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- 3.Rice GI, et al. Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet. 2009;41(7):829–832. doi: 10.1038/ng.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rice GI, et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet. 2012;44(11):1243–1248. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rice GI, et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet. 2014;46(5):503–509. doi: 10.1038/ng.2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458(7237):509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hornung V, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458(7237):514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455(7213):674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461(7265):788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun W, et al. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci U S A. 2009;106(21):8653–8658. doi: 10.1073/pnas.0900850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abe T, et al. STING recognition of cytoplasmic DNA instigates cellular defense. Mol Cell. 2013;50(1):5–15. doi: 10.1016/j.molcel.2013.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339(6121):786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu J, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339(6121):826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ouyang S, et al. Structural analysis of the STING adaptor protein reveals a hydrophobic dimer interface and mode of cyclic di-GMP binding. Immunity. 2012;36(6):1073–1086. doi: 10.1016/j.immuni.2012.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shu C, Yi G, Watts T, Kao CC, Li P. Structure of STING bound to cyclic di-GMP reveals the mechanism of cyclic dinucleotide recognition by the immune system. Nat Struct Mol Biol. 2012;19(7):722–724. doi: 10.1038/nsmb.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shang G, et al. Crystal structures of STING protein reveal basis for recognition of cyclic di-GMP. Nat Struct Mol Biol. 2012;19(7):725–727. doi: 10.1038/nsmb.2332. [DOI] [PubMed] [Google Scholar]
- 17.Rice GI, et al. Assessment of interferon-related biomarkers in Aicardi-Goutieres syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol. 2013;12(12):1159–1169. doi: 10.1016/S1474-4422(13)70258-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bennett L, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197(6):711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371(6):507–518. doi: 10.1056/NEJMoa1312625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Konno H, Konno K, Barber GN. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell. 2013;155(3):688–698. doi: 10.1016/j.cell.2013.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gough DJ, Messina NL, Clarke CJ, Johnstone RW, Levy DE. Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity. 2012;36(2):166–174. doi: 10.1016/j.immuni.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.