

Abstract
Genome-wide association studies have identified a link between genetic variation at the human chromosomal locus 1p13.3 and coronary artery disease. The gene encoding sortilin (SORT1) has been implicated as the causative gene within the locus, as sortilin regulates hepatic lipoprotein metabolism. Here we demonstrated that sortilin also directly affects atherogenesis, independent of its regulatory role in lipoprotein metabolism. In a mouse model of atherosclerosis, deletion of Sort1 did not alter plasma cholesterol levels, but reduced the development of both early and late atherosclerotic lesions. We determined that sortilin is a high-affinity receptor for the proinflammatory cytokines IL-6 and IFN-γ. Moreover, macrophages and Th1 cells (both of which mediate atherosclerotic plaque formation) lacking sortilin had reduced secretion of IL-6 and IFN-γ, but not of other measured cytokines. Transfer of sortilin-deficient BM into irradiated atherosclerotic mice reduced atherosclerosis and systemic markers of inflammation. Together, these data demonstrate that sortilin influences cytokine secretion and that targeting sortilin in immune cells attenuates inflammation and reduces atherosclerosis.
Introduction
SNPs at the 1p13.3 locus are associated with coronary artery disease and myocardial infarction in humans (1, 2) as well as with LDL cholesterol levels (3). Recent studies have implicated SORT1 as the causative gene (4) and revealed several mechanisms whereby its product, sortilin, may affect atherosclerosis through regulation of hepatic lipoprotein secretion and clearance (4–7).
Sortilin is a multiligand receptor enriched in the Golgi compartment. It is a member of the Vps10p domain receptor family, characterized by a 10-bladed β-propeller that forms a cavity for binding of soluble ligands and a C-terminal cytoplasmic tail that contains sorting motifs responsible for subcellular distribution of the receptors (8–10). Sortilin exerts diverse cellular functions including intracellular sorting of proteins, such as acid sphingomyelinase, apolipoprotein B100 (apoB100), and PCSK9, as well as engaging in signaling as a coreceptor in cell surface receptor complexes (5–7, 11–13). Given the broad functional repertoire of sortilin, we hypothesized that it may affect atherogenesis beyond regulating plasma cholesterol levels.
Results and Discussion
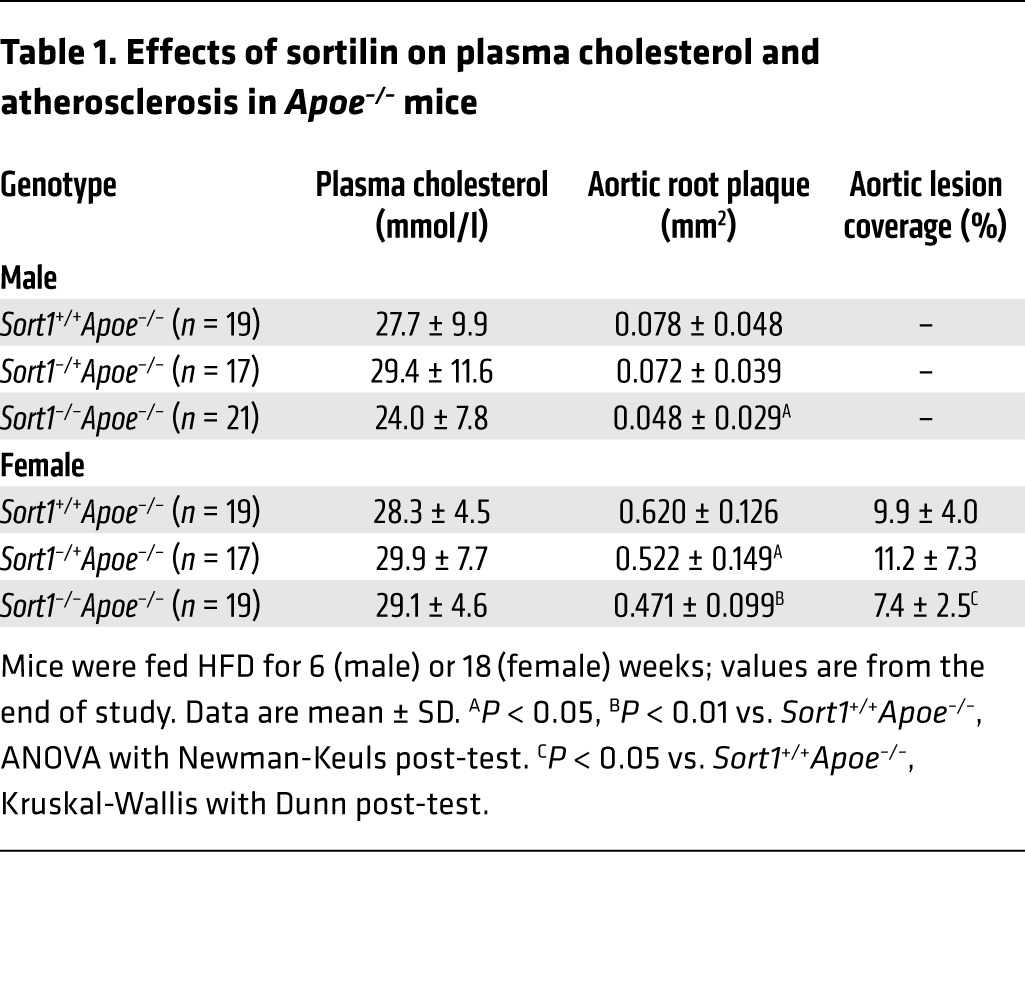
To investigate the involvement of sortilin in atherogenesis, we generated apolipoprotein E–deficient (Apoe–/–) mice that were either homozygous, heterozygous, or WT for a targeted Sort1 deletion (14). Males were fed high-fat diet (HFD) for 6 weeks, and females for 18 weeks. Analyzing the genders at separate time points allowed us to screen for effects in early and late atherosclerosis with sufficient statistical power, albeit at the expense of assessing potential gender-mediated differences. Deletion of sortilin did not significantly alter plasma cholesterol or the distribution of cholesterol across size-fractionated lipoprotein classes, but did reduce atherosclerosis at both time points (Table 1 and Supplemental Figures 1 and 2; supplemental material available online with this article; doi:10.1172/JCI76002DS1). Furthermore, sortilin was expressed in several murine cell types that participate in atherosclerosis (macrophages, Th1 cells, and smooth muscle cells; Supplemental Figure 3), which suggests that the site of action could be within the atherosclerotic lesion. Because the effect of sortilin was evident already at the early time point, at which lesions consist primarily of macrophages and Th1 cells, we further speculated that sortilin may regulate some function of these cell types.
Table 1.
Effects of sortilin on plasma cholesterol and atherosclerosis in Apoe–/– mice
To test the involvement of sortilin in macrophage recruitment, we induced sterile peritonitis with thioglycolate, but found no significant differences between Sort1–/– and Sort1+/+ mice in the number of recruited macrophages after 4 days (13.7 ± 1.1 vs. 12.8 ± 0.8 × 106 cells, mean ± SEM, n = 8 per group; P = NS). Furthermore, we analyzed macrophage foam cell formation by several mechanisms, including scavenger receptor–mediated endocytosis of oxidized LDL, phagocytosis of aggregated LDL, and direct internalization of native LDL at high concentrations, but observed no differences between Sort1–/– and Sort1+/+ BM macrophages (BMMs) (Figure 1, A and B).
Figure 1. Macrophage and Th1 cell function.

(A) Fluorescent LDL-loaded BMMs. Scale bars: 10 μm. (B) BMMs were cultured from Sort1+/+ (n = 10) and Sort1–/– (n = 9) mice and incubated with Atto-633–labeled oxidized (oxLDL), aggregated (agLDL), or native LDL for 20 hours. Foam cell formation was quantified by flow cytometry as mean fluorescence intensity among 10,000 macrophages from each mouse. (C) Activation of Sort1+/+ and Sort1–/– BMMs, induced by LPS for the indicated times and assessed by phosphorylation of MAPKs, revealed no differences. (D–G) After 6 hours of LPS stimulation, the amount of IL-6 was significantly reduced in media from Sort1–/– versus Sort1+/+ BMMs (D), whereas TNF-α secretion (E) and iNOS (F) and Il6 (G) mRNA expression were unaffected. (H and I) Culture medium (CM) from activated Sort1–/– Th1 cells contained less IFN-γ than did Sort1+/+ Th1 cells (H) and was less efficient in priming BMMs to secrete TNF-α after LPS activation (I). Representative results from 3 independent experiments (mean ± SEM; n = 6–9 per group). **P < 0.01, ***P < 0.0001, Student’s t test (D and I) or Mann-Whitney test (I).
Proinflammatory cytokines secreted from activated macrophages and Th1 cells play crucial roles in plaque progression (15). To assess the potential role of sortilin in macrophage function, we first stimulated BMMs toward an M1 phenotype with LPS and studied phosphorylation of MAPKs as markers of activation, but found no differences between Sort1–/– and Sort+/+ macrophages (Figure 1C). Second, we measured a broad range of cytokines secreted from M1 macrophages after 24 hours of LPS stimulation. Interestingly, the level of IL-6 was consistently lower in culture media from BMMs cultured from Sort1–/– (n = 8) versus Sort1+/+ (n = 9) mice, whereas levels of other cytokines were similar between groups (e.g., TNF-α, IL-12, and monocyte chemoattractant protein–1; Supplemental Figure 4, A–G). In follow-up experiments, we found that the secreted level of IL-6 was already reduced after 6 hours of LPS stimulation, whereas the secreted TNF-α level and the cellular expression of Il6 and iNOS mRNA were unaltered (Figure 1, D–G), indicative of similar activation levels. Furthermore, we found a 50% reduction in IFN-γ secretion from Sort1–/– versus Sort1+/+ activated Th1 cells, whereas TNF-α secretion was unaffected, indicative of normal activation (Figure 1H and Supplemental Figure 4H). Consistently, conditioned medium from Sort1–/– Th1 cells was less efficient in priming BMMs to secrete TNF-α upon LPS activation (Figure 1I).
We speculated that sortilin, being an intracellular sorting receptor, might influence IL-6 and IFN-γ secretion by physically interacting in the secretory pathway; therefore, we next assessed binding by surface plasmon resonance analysis. Both IL-6 and IFN-γ, but not TNF-α, rapidly associated with the immobilized extracellular domain of sortilin in a dose-dependent manner with high affinity (Figure 2, A and B). Binding was abolished by sortilin propeptide (Figure 2C and data not shown), which indicated that both cytokines bind to the tunnel of sortilin (10).
Figure 2. Sortilin binds IL-6 and IFN-γ with high affinity.

(A) Surface plasmon resonance curves depict binding of IL-6 to immobilized soluble sortilin receptor (0–600 seconds) at IL-6 concentrations ranging 100–1,000 nM. After 600 seconds, buffer was added and IL-6 dissociated from the immobilized sortilin receptor. IL-6 bound with high affinity to sortilin. KD, 7 nM. (B) Similar experiment showing binding of IFN-γ to the immobilized soluble sortilin receptor (0–600 seconds) at IFN-γ concentrations ranging 250–1,500 nM. KD, 1 nM. (C) GST-fused sortilin propeptide (GST-Pro) was added (200–650 seconds); at 650–1,200 seconds, IL-6, GST-fused sortilin propeptide, or both were added. At 1,200 seconds, buffer was added and ligands dissociated. Binding between IL-6 and sortilin was fully inhibited by the sortilin propeptide. (D) Strong colocalization of IL-6 and sortilin at the plasma membrane after addition of IL-6 to sortilin-transfected HEK293 cells, but not control HEK293 cells. Propeptide abolished this colocalization. Nonpermeabilized cells at 4°C. Scale bars: 10 μm. (E) Different IL-6 secretion between Sort1–/– and Sort1+/+ macrophages was maintained after blocking surface sortilin with propeptide. Representative data from 2 independent experiments (mean ± SEM; n = 7). *P < 0.05, Student’s t test. (F) Coimmunoprecipitation in HEK293 cells transfected with sortilin and/or IL-6. Top: Sortilin coprecipitated with IL-6 when IL-6 was pulled down. Bottom: Cell lysates.
As part of the sortilin pool resides in the plasma membrane, we also assessed binding by incubating HEK293 cells with soluble IL-6. Strong colocalization of sortilin and IL-6 at the plasma membrane was seen in sortilin-transfected, but not control, HEK293 cells, and blocking surface sortilin with propeptide eliminated this colocalization (Figure 2D). Notably, blocking surface sortilin with propeptide in activated Sort1+/+ and Sort1–/– macrophages did not abolish the difference in IL-6 secretion (Figure 2E), which indicated that it involved intracellular binding. Interaction of sortilin and IL-6 was further confirmed by coimmunoprecipitation in HEK293 cells transfected with sortilin and IL-6 (Figure 2F).
The IL-6 and IFN-γ secretion defect observed in activated macrophages and Th1 cells suggested that sortilin deficiency in these immune cells — and potentially others — may be the cause of the reduced atherosclerosis in Sort1–/–Apoe–/– mice. To test this, we subjected Sort1+/+Apoe–/– mice to γ-irradiation and reconstituted them with BM cells from Sort1+/+Apoe–/– or Sort1–/–Apoe–/– mice. Sort1–/–Apoe–/– mice reconstituted with Sort1–/–Apoe–/– BM cells served as global deletion control (Supplemental Figure 5). After 4 weeks, Sort1–/–Apoe–/– chimerism among circulating cells in Sort1+/+Apoe–/– recipients of Sort1–/–Apoe–/– BM was >93%, increasing to >97% at euthanization (Supplemental Figure 6). Absolute numbers of circulating immune cells was similar among groups, and all mice thrived well (Supplemental Figures 7 and 8).
In HFD-fed BM chimeric mice, plasma cholesterol levels and distribution of cholesterol across size-fractionated lipoproteins were similar among groups, except for total cholesterol in female mice, in which the global Sort1 deletion group had significantly lower levels than the other 2 groups (Figure 3, A and B).
Figure 3. Sortilin deficiency in immune cells reduces atherosclerosis.

(A and B) Plasma cholesterol and distribution of cholesterol across size-fractionated lipoprotein classes in male and female BM-transplanted mice. (C and D) Lesions in the aortic arch (C) and in cross-sections of the aortic root (D) were reduced in male Sort1+/+Apoe–/– recipients of Sort1–/–Apoe–/– BM after 9 weeks of HFD. Scale bars: 1 mm (C); 250 μm (D). (E) Sortilin deficiency in immune cells (i.e., Sort1+/+Apoe–/– recipients of Sort1–/–Apoe–/– BM) also reduced aortic lesion coverage in female mice after 19 weeks of HFD. An additional reduction was seen in control Sort1–/–Apoe–/– recipients of Sort1–/–Apoe–/– BM (global sortilin deletion). (F and G) Plasma IL-6 and TNF-α levels were reduced in mice with sortilin-deficient immune cells. Data are mean ± SEM of n = 14–20 per group. *P < 0.05, **P < 0.01, ***P < 0.001, ANOVA with Newman-Keuls post-test.
After 9 weeks of HFD feeding, male Sort1+/+Apoe–/– recipients of Sort1–/–Apoe–/– BM showed a 45% reduction in aortic lesion coverage and a 37% decrease in aortic root lesion area compared with those receiving Sort1+/+Apoe–/– BM (Figure 3, C and D). Interestingly, the deletion of sortilin in immune cells fully accounted for the difference in atherosclerosis between male Sort1–/–Apoe–/– and Sort1+/+Apoe–/– mice. Similar results were obtained in females at the more advanced stage of atherosclerosis, after 19 weeks of HFD: deletion of sortilin in circulating immune cells was followed by a 34% decrease in aortic lesion coverage (Figure 3E). In this experiment, a further reduction was seen in mice that also lacked sortilin in recipient tissues. This could potentially be explained by the difference in plasma cholesterol levels, although no significant correlations between plasma cholesterol and atherosclerosis were detected within any of the BM-transplanted groups. Notably, levels of not only IL-6, but also TNF-α, were substantially reduced in plasma of mice transplanted with Sort1–/– cells (Figure 3, F and G), indicative of general attenuation of inflammation.
Our combined data uncovered a novel site of action for sortilin in atherosclerosis. Previous studies demonstrated that sortilin binds to apoB100, but not apoB48, and described how sortilin regulates secretion and clearance of apoB100-containing lipoproteins in various mouse models (4–7). By working in the Apoe–/– model of atherosclerosis, in which the vast majority of lipoproteins are of the apoB48-containing remnant type (16), we were able to reduce the effect of sortilin on plasma cholesterol and unmask a direct effect of sortilin in inflammation and atherosclerosis.
Recently, Herda et al. demonstrated that sortilin binds and controls exocytotic trafficking of IFN-γ from T cells (17). Here, we confirmed these findings and extended the functional repertoire of sortilin to include macrophage secretion of the important proinflammatory cytokine IL-6. Since IL-6 binding was blocked by propeptide, which is not cleaved from sortilin until the late trans-Golgi network (18), the sorting event must occur between late trans-Golgi and the cell surface.
The amelioration of IFN-γ and IL-6 may exert a synergistic dampening effect on inflammation. IFN-γ is an important stimulator of macrophage activation, consistent with the observed reduced priming efficiency of conditioned medium from Sort1–/– Th1 cells (Figure 1I); conversely, IL-6 facilitates T cell immunity and IFN-γ secretion (19). This reinforcing cross-talk between IL-6– and IFN-γ–mediated effects may explain why the in vivo reduction of IL-6 and TNF-α in mice transplanted with Sort1–/– cells superseded the effect size in any of the isolated cell culture assays.
IL-6 and IFN-γ exert complex effects in atherosclerosis, and although the majority of studies have found an overall facilitating role, experiments in mice with either complete or BM-specific deletion of IL-6, IFN-γ, or IFN-γ receptor genes have not been consistent (20, 21). Perhaps most relevant to the present study, recent studies with inhibition rather than complete abolition of IFN-γ or IL-6 signaling have been shown to inhibit atherosclerosis in mice (22, 23). Furthermore, a functional gene variant that inhibits classical IL-6 signaling is associated with reduced risk of atherosclerotic heart disease in humans (24). Although we find it plausible that the effect in the BM-transplanted mice was mediated by the cytokine secretion defects, our data do not prove this; other (or additional) sortilin-controlled mechanisms may be involved.
In conclusion, our present findings revealed novel regulatory roles for sortilin in cytokine secretion and showed that targeting sortilin in immune cells attenuated the inflammatory response and reduced atherosclerosis. These findings indicate that sortilin may influence atherosclerosis in ways other than regulating LDL turnover. Furthermore, given the widespread effects of IL-6 and IFN-γ in inflammation, they may be of relevance to other inflammatory diseases.
Methods
Further information can be found in Supplemental Methods.
Mice.
Sort1–/– mice, created as previously described (14), were backcrossed onto C57BL/6 mice for >10 generations. Apoe–/– mice were obtained from Taconic. Mice were fed HFD (D12079B, Research Diets) to accelerate atherosclerosis.
BM transplant.
Sort1+/+Apoe–/– and Sort1–/–Apoe–/– mice (aged 8 weeks) were lethally irradiated and rescued with 107 BM cells from age- and sex-matched Sort1+/+Apoe–/– or Sort1–/–Apoe–/– donor mice. Hematopoietic chimerism was measured by quantitative PCR for targeted and WT Sort1 alleles in blood DNA.
Quantification of atherosclerosis.
The proximal aorta was sectioned to quantify aortic root lesions. Aortic lesion coverage was measured en face after staining of lesions with Oil Red O.
Cell culture assays.
BMMs were generated as described previously (25). Foam cell formation was assessed by incubating BMMs with Atto-633–labeled native (1 mg/ml), aggregated (1 mg/ml), or oxidized (20 μg/ml) human LDL for 20 hours. Activation of BMMs was assessed by adding LPS (100 ng/ml) for 0, 15, 30, 60, 120, or 240 minutes and analyzing MAPK phosphorylation by immunoblotting. Cytokine secretion from LPS-activated (100 ng/ml) BMMs or anti-CD3/anti-CD28–activated Th1 cells into cell culture medium was analyzed using 20-plex mouse cytokine Luminex-kits (Invitrogen) or IL-6, TNF-α, and IFN-γ ELISAs (eBioscience). Quantitative PCR was performed with primers in Supplemental Table 1. To assess the ability of Th1-conditioned cell medium to prime macrophages for cytokine secretion, BMMs were incubated with 10% conditioned medium from activated Th1 cells for 12 hours, followed by activation with LPS (1 ng/ml).
Protein binding assays.
Binding of human IL-6, TNF-α, and IFN-γ to the soluble extracellular domain of human sortilin was assessed by surface plasmon resonance (Biacore). Binding of human IL-6 was also analyzed in HEK293 cells transfected with human full-length sortilin by immunofluorescence and coimmunoprecipitation.
Statistics.
P values were calculated using 2-tailed unpaired Student’s t test, 1-way ANOVA with Newman-Keuls post-test, or Kruskal-Wallis nonparametric test with Dunn’s post-test, as indicated. A P value less than 0.05 was considered statistically significant.
Study approval.
All procedures followed the Danish legislation for the protection of animals and were approved by the Danish Animal Experiments Inspectorate.
Supplementary Material
Acknowledgments
We thank Dorte Wilhardt Jørgensen, Lisa Maria Røge, Zahra Partovi Nasr, Anne-Kerstine Thomassen, Anne Marie Bundsgaard, and Benedicte Vestergaard for technical assistance. This work was supported by the Independent Research Council | Health Sciences, the Lundbeck Foundation, Aase og Ejnar Danielsens Fond, Snedkermester Sophus Jacobsen og Hustru Astrid Jacobsens Fond, Grosserer L.F. Foghts Fond, and Fonden for Lægevidenskabens Fremme.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2014;124(12):5317–5322. doi:10.1172/JCI76002.
References
- 1.Samani NJ, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357(5):443–453. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schunkert H, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43(4):333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kathiresan S, et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet. 2008;40(2):189–197. doi: 10.1038/ng.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Musunuru K, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466(7307):714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kjolby M, et al. Sort1, encoded by the cardiovascular risk locus 1p13.3, is a regulator of hepatic lipoprotein export. Cell Metab. 2010;12(3):213–223. doi: 10.1016/j.cmet.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 6.Strong A, et al. Hepatic sortilin regulates both apolipoprotein B secretion and LDL catabolism. J Clin Invest. 2012;122(8):2807–2816. doi: 10.1172/JCI63563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gustafsen C, et al. The hypercholesterolemia-risk gene SORT1 facilitates PCSK9 secretion. Cell Metab. 2014;19(2):310–318. doi: 10.1016/j.cmet.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 8.Petersen CM, et al. Molecular identification of a novel candidate sorting receptor purified from human brain by receptor-associated protein affinity chromatography. J Biol Chem. 1997;272(6):3599–3605. doi: 10.1074/jbc.272.6.3599. [DOI] [PubMed] [Google Scholar]
- 9.Nielsen MS, et al. The sortilin cytoplasmic tail conveys Golgi-endosome transport and binds the VHS domain of the GGA2 sorting protein. EMBO J. 2001;20(9):2180–2190. doi: 10.1093/emboj/20.9.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quistgaard EM, Madsen P, Grøftehauge MK, Nissen P, Petersen CM, Thirup SS. Ligands bind to Sortilin in the tunnel of a ten-bladed beta-propeller domain. Nat Struct Mol Biol. 2009;16(1):96–98. doi: 10.1038/nsmb.1543. [DOI] [PubMed] [Google Scholar]
- 11.Nykjaer A, Willnow TE. Sortilin: a receptor to regulate neuronal viability and function. Trends Neurosci. 2012;35(4):261–270. doi: 10.1016/j.tins.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 12.Wahe A, et al. Golgi-to-phagosome transport of acid sphingomyelinase and prosaposin is mediated by sortilin. J Cell Sci. 2010;123(14):2502–2511. doi: 10.1242/jcs.067686. [DOI] [PubMed] [Google Scholar]
- 13.Larsen JV, et al. Sortilin facilitates signaling of ciliary neurotrophic factor and related helical type 1 cytokines targeting the gp130/leukemia inhibitory factor receptor heterodimer. Mol Cell Biol. 2010;30(17):4175–4187. doi: 10.1128/MCB.00274-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jansen P, et al. Roles for the pro-neurotrophin receptor sortilin in neuronal development, aging and brain injury. Nat Neurosci. 2007;10(11):1449–1457. doi: 10.1038/nn2000. [DOI] [PubMed] [Google Scholar]
- 15.Libby P, Lichtman AH, Hansson GK. Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity. 2013;38(6):1092–1104. doi: 10.1016/j.immuni.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishibashi S, Herz J, Maeda N, Goldstein JL, Brown MS. The two-receptor model of lipoprotein clearance: tests of the hypothesis in “knockout” mice lacking the low density lipoprotein receptor, apolipoprotein E, or both proteins. Proc Natl Acad Sci U S A. 1994;91(10):4431–4435. doi: 10.1073/pnas.91.10.4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herda S, et al. The sorting receptor Sortilin exhibits a dual function in exocytic trafficking of interferon-γ and granzyme A in T cells. Immunity. 2012;37(5):854–866. doi: 10.1016/j.immuni.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 18.Munck Petersen C, et al. Propeptide cleavage conditions sortilin/neurotensin receptor-3 for ligand binding. EMBO J. 1999;18(3):595–604. doi: 10.1093/emboj/18.3.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nurieva R, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448(7152):480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 20.Mallat Z, Taleb S, Ait-Oufella H, Tedgui A. The role of adaptive T cell immunity in atherosclerosis. J Lipid Res. 2009;50(suppl):S364–S369. doi: 10.1194/jlr.R800092-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schuett H, Luchtefeld M, Grothusen C, Grote K, Schieffer B. How much is too much? Interleukin-6 and its signalling in atherosclerosis. Thromb Haemost. 2009;102(2):215–222. doi: 10.1160/TH09-05-0297. [DOI] [PubMed] [Google Scholar]
- 22.Koga M, et al. Inhibition of progression and stabilization of plaques by postnatal interferon-γ function blocking in ApoE-knockout mice. Circ Res. 2007;101(4):348–356. doi: 10.1161/CIRCRESAHA.106.147256. [DOI] [PubMed] [Google Scholar]
- 23.Schuett H, et al. Transsignaling of interleukin-6 crucially contributes to atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2012;32(2):281–290. doi: 10.1161/ATVBAHA.111.229435. [DOI] [PubMed] [Google Scholar]
- 24.IL6R Genetics Consortium Emerging Risk Factors Collaboration et al. Interleukin-6 receptor pathways in coronary heart disease: a collaborative meta-analysis of 82 studies. Lancet. 2012;379(9822):1205–1213. doi: 10.1016/S0140-6736(11)61931-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weischenfeldt J, Porse B.Bone Marrow-Derived Macrophages (BMM): isolation and applications. Cold Spring Harbor Protocols. 20082008:pdb.prot5080. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.