

Abstract
Estrogen receptor–positive (ER+) breast cancers adapt to hormone deprivation and become resistant to antiestrogen therapy. Here, we performed deep sequencing on ER+ tumors that remained highly proliferative after treatment with the aromatase inhibitor letrozole and identified a D189Y mutation in the inhibitory SH2 domain of the SRC family kinase (SFK) LYN. Evaluation of 463 breast tumors in The Cancer Genome Atlas revealed four LYN mutations, two of which affected the SH2 domain. In addition, LYN was upregulated in multiple ER+ breast cancer lines resistant to long-term estrogen deprivation (LTED). An RNAi-based kinome screen revealed that LYN is required for growth of ER+ LTED breast cancer cells. Kinase assays and immunoblot analyses of SRC substrates in transfected cells indicated that LYND189Y has higher catalytic activity than WT protein. Further, LYND189Y exhibited reduced phosphorylation at the inhibitory Y507 site compared with LYNWT. Other SH2 domain LYN mutants, E159K and K209N, also exhibited higher catalytic activity and reduced inhibitory site phosphorylation. LYND189Y overexpression abrogated growth inhibition by fulvestrant and/or the PI3K inhibitor BKM120 in 3 ER+ breast cancer cell lines. The SFK inhibitor dasatinib enhanced the antitumor effect of BKM120 and fulvestrant against estrogen-deprived ER+ xenografts but not LYND189Y-expressing xenografts. These results suggest that LYN mutations mediate escape from antiestrogens in a subset of ER+ breast cancers.
Introduction
LYN is a member of the SRC family of protein tyrosine kinases (SFKs), key regulators of several cellular processes, including cancer cell growth, migration, invasion, and survival (1, 2). Overexpression of LYN, as measured by immunohistochemistry (IHC), is associated with an epithelial-to-mesenchymal transition and correlates with a shorter overall survival in breast cancer (3). SRC engages in bidirectional crosstalk with the estrogen receptor α (ERα) (4), in which its kinase phosphorylates ERα at Y537 (5), resulting in an enhancement of ER transcriptional activity (6).
Two-thirds of breast cancers express ER and/or progesterone receptor (PR), biomarkers indicative of hormone dependence (7). Therapies against ER+ breast cancers inhibit ER function by antagonizing ligand binding to ER (tamoxifen), downregulating ER (fulvestrant), or blocking estrogen biosynthesis and reducing circulating estrogen levels (8) (aromatase inhibitors [AIs]). Although AIs produce an objective tumor response rate of 30% to 40% in patients with metastatic ER+ breast cancer, a significant fraction of patients do not respond or progress quickly on this therapy (9). Thus, elucidating the molecular mechanisms underlying this resistance is critical for improving patient outcome. In addition, discovery of biomarkers predictive of clinical benefit from antiestrogens and prospective identification of patients who are resistant to these therapies are needed.
ER blockade with antiestrogens inhibits tumor cell proliferation in hormone-dependent ER+ breast cancers. This can be measured by IHC for the nuclear antigen Ki67, which identifies cells in the G1/S and M phases of the cell cycle (10). The Immediate Preoperative Anastrozole, Tamoxifen, or Combined with Tamoxifen (IMPACT) study showed that the high Ki67 score in tumors after 2 or 12 weeks of antiestrogen therapy predicts a shorter recurrence-free survival (11, 12). These data suggest that a high tumor cell proliferation (i.e., high Ki67) following treatment with an AI can be used to identify ER+ tumors that are resistant to endocrine therapy and as an unbiased approach to discover molecular effectors of such resistance.
The objective of this study is to identify kinase mutations associated with resistance to estrogen deprivation. We performed deep kinome sequencing on 4 ER+/HER2– breast cancers that retained high Ki67 scores (14.8%–24.5%) following 2 weeks of treatment with the AI letrozole. We identified a novel D189Y somatic mutation in LYN in an endocrine-resistant tumor, as defined by the Ki67 score after treatment. Although stable overexpression of WT LYN (referred to herein as LYNWT) or the D189Y mutation in LYN (referred to herein as LYND189Y) accelerated MCF-7 cell growth in estrogen-depleted media, the mutant was more potent than LYNWT at inducing this effect. LYND189Y but not LYNWT exhibited reduced phosphorylation of the inhibitory Y507 residue, suggesting that this substitution limited the ability of LYN to achieve an inactive conformation. Similar results were seen with two other reported SRC homology 2 (SH2) domain mutants of LYN, E159K and K209N. Ectopic expression of LYND189Y also limited the antitumor effect of the ER downregulator fulvestrant and the pan-phosphoinositide 3- kinase (PI3K) inhibitor BKM120 in 3 ER+ breast cancer cell lines. Further, inhibition of SFKs with the small molecule dasatinib enhanced the antitumor effect of BKM120 and fulvestrant against estrogen-deprived parental MCF-7 and MCF-7/LYNWT xenografts in ovariectomized mice but not MCF-7/LYND189Y xenografts. These data suggest the need to develop potent SFK inhibitors, which, in combination with PI3K and ER inhibitors, may be an effective treatment for endocrine-resistant breast cancer.
Results
Deep kinome sequencing identifies a novel D189Y mutation in LYN.
NCT00651976 is an IRB-approved clinical trial at Vanderbilt University, in which postmenopausal women with newly diagnosed ER+/HER2– operable breast cancer consented to treatment with letrozole (2.5 mg/d) for 10 to 21 days prior to surgery (Supplemental Figure 1A; supplemental material available online with this article; doi:10.1172/JCI72573DS1). Tumor cell proliferation was assessed by Ki67 IHC in a core biopsy before treatment and in the surgical specimen (after treatment). From among the first 23 patients enrolled in this study, we selected 4 tumors that retained the highest Ki67 level in the biopsy after treatment (14.8%–24.5%; Supplemental Table 1). Consistent with a previous report (12), we considered an after treatment Ki67 score of >10% as a surrogate of endocrine resistance.
Genomic DNA from 4 formalin-fixed, paraffin-embedded (FFPE) surgically excised tumors and matched normal adjacent breast tissue or histologically confirmed negative lymph nodes was deep sequenced using a capture approach, including 517 kinases and 46 cancer-related genes from the Agilent SureSelect Kinome Kit (Supplemental Table 2). Tumor cellularity ranged from 20% to 50%. Annotated variant call files were provided by Expression Analysis for the sequenced tumors. To prioritize variants for molecular analysis, we filtered all variants against the reference hg19 sequence that (a) were not in the capture-targeted region, (b) were known SNPs (based on the dbSNP database), or (c) were not exonic or were synonymous in their protein translation. These criteria resulted in 671 candidate variations among the 4 tumor/normal pairs. Next, somatic status was determined by comparing each tumor to its matched normal specimen. For these 4 tumors, 28, 15, 14, and 19 somatic mutations were identified, respectively. Resulting hits were manually screened for likely artifacts in the Integrative Genome Viewer for the presence of strand bias or variants lying only at read ends. After further manual filtering for likely artifacts, we identified a series of candidate somatic mutations in kinases (EPHA7, LYN, DYRK1A, DYRK2, MYLK3, PTK2, TSSK4, CSNK1G3, WNK2, and TEC; Table 1). All 4 tumors contained a mutation in PIK3CA, the gene encoding the p110α catalytic subunit of PI3K; 3 were hot-spot activating mutations (Q546E, H1047R); and 1 was T1025A. PIK3CA hot-spot mutations are associated with increased PI3K catalytic activity (13), cell transformation in vitro and in transgenic mouse models, and resistance to anoikis (14, 15). Related to the T1025A variant, PIK3CA with a substitution in the same base, T1025S, has been shown to transform chicken embryo fibroblasts (16).
Table 1.
Deep kinome sequencing of DNA from surgical specimens after treatment
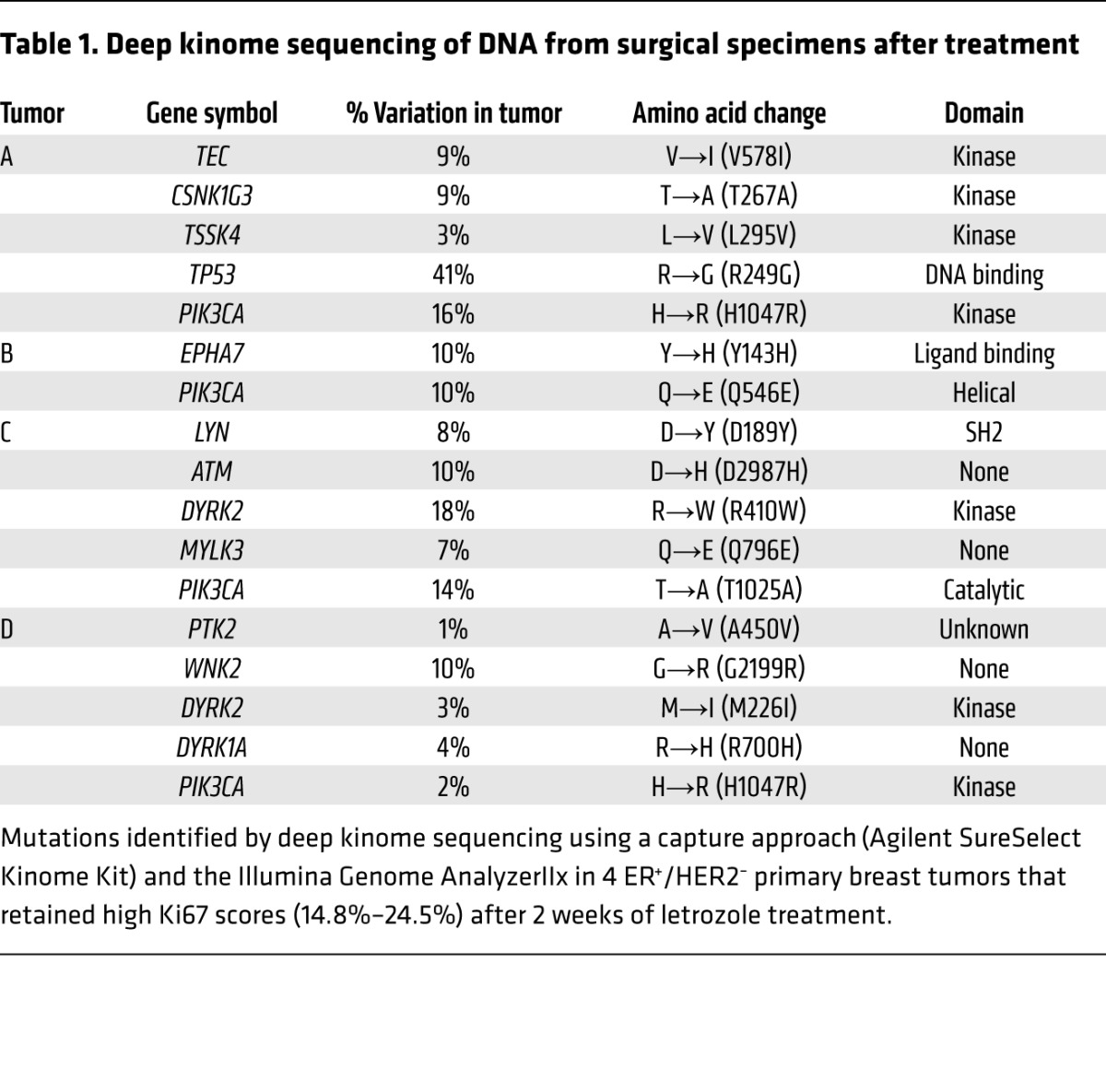
One tumor, referred to as tumor C, showed a previously unreported D189Y somatic mutation in LYN (variant frequency of 8%; Table 1), which was not detected in adjacent normal breast tissue from the same patient (Supplemental Figure 1B). We next confirmed mRNA expression of D189Y LYN by cDNA sequencing in both directions (Supplemental Figure 1C). The allele frequency of D189Y, which is comparable to that of mutant PIK3CA (14%) in the same tumor, probably reflects the clonal nature of this alteration as well as dilution with WT reads in DNA from adjacent normal tissue that could not be macrodissected out. D189Y (565G>T) is located in the LYN SH2 domain, which binds phosphorylated tyrosines (2, 17). The D189Y substitution was predicted to have a high functional affect on LYN, as assessed by mutationassessor.org (http://mutationassessor.org), which estimates the potential consequences of amino acid substitutions in protein function (18). Sanger sequencing confirmed the presence of D189Y LYN in DNA isolated from the tumor before treatment (Figure 1A).
Figure 1. Deep kinome sequencing identifies a D189Y mutation in LYN.

(A) Sanger sequencing of tumor C verified the presence of the D189Y mutation in LYN; the point mutation (G to T) is shown by the arrow. (B) Lysates from 10 ER+/HER2– tumors from patients treated 10–21 days with letrozole prior to surgery were analyzed by RPPA. Antibody signal intensities were log2 normalized and correlated to Ki67 score after letrozole treatment (POST).
We next used the cBio Cancer Genomics Portal (19) to explore LYN mutations in 463 invasive breast carcinomas included in The Cancer Genome Atlas (TCGA; accessed September 9, 2014). There were 4 LYN mutations, E159K, K209N, G418R, and A503D, with the first 2 located in the SH2 domain and the last 2 located in the kinase domain. Despite its low frequency, LYN is the most commonly mutated SFK family member in invasive breast cancers in TCGA (4, 2, 1, and 1 case for LYN, YES1, HCK, and BLK, respectively). LYN mutations have been also reported in low frequency in melanoma and endometrial, lung, and colorectal carcinoma (Supplemental Table 3). Reverse-phase protein array (RPPA) analysis in lysates from 10 tumors of patients enrolled in the NCT00651976 trial (Supplemental Table 4) revealed a significant correlation (P = 0.006) between Y416 phosphorylated SRC (P-SRC) (which detects all activated SFKs) and a high Ki67 score after letrozole (Figure 1B).
RNAi screening identifies LYN as required for hormone-independent ER+ breast cancer growth.
To complement this study, we screened a siRNA library targeting 779 kinases in MCF-7 breast cancer cells resistant to long-term estrogen deprivation (LTED) (20, 21). This screen identified LYN as one of the top hits whose knockdown markedly reduced growth of MCF-7/LTED cells (Figure 2A). Knockdown of LYN with an alternative siRNA (not in the screening library) reduced LYN but not SRC protein levels, as measured by immunoblot analysis (Figure 2B), and significantly inhibited growth of MCF-7/LTED, MDA-361/LTED, HCC-1428/LTED, and ZR75-1/LTED cells (Figure 2C), suggesting that LYN plays a role in hormone-independent ER+ breast cancer growth.
Figure 2. RNAi screen identifies that LYN is required for hormone-independent ER+ breast cancer cell growth.

(A) MCF-7/LTED cells were screened with a siRNA library (Dharmacon) targeting 779 kinases. Cell viability was measured after 4 days, as described in the Methods. Cell growth for each kinase siRNA relative to nonsilencing controls was transformed to a Z-score, and the median Z-score across 4 independent experiments was calculated. (B and C) Cells were transfected with an alternative siRNA targeting LYN (siLYN) (Qiagen; not present in the screening library) or a nonsilencing control (siCon) and reseeded the next day for (B) immunoblot analysis or (C) growth in monolayer. LE, longer exposure. (B) Cells were grown in 10% DCC-FBS for 4 days. Protein lysates were analyzed by immunoblot using LYN, SRC, and actin (control) antibodies. (C) Cells were treated with 10% DCC-FBS for 5 to 10 days and then counted. Data are presented as percentage of control (n = 3, *P < 0.01 vs. siCon).
LYN expression was upregulated in 3 of 4 LTED cell lines compared with their parental counterparts (Figure 3A). These changes were paralleled by LYN mRNA expression in MCF-7/LTED and MDA-361/LTED cells (3.6-fold and 1.5-fold, respectively) compared with their respective parental cells, whereas the other SFKs showed variable changes (Figure 3B). YES1 mRNA was upregulated 1.4-fold in MCF-7/LTED cells, and the other SFKs were downregulated or unchanged. Only LYN mRNA expression was significantly upregulated in MDA-361/LTED cells (Figure 3B).
Figure 3. LYN expression is upregulated in ER+ breast cancer cell lines with acquired resistance to estrogen deprivation.

(A) Cells were treated with 10% DCC-FBS for 24 hours, and lysates were analyzed by immunoblot. (B) Parental and LTED cells were incubated in 10% DCC-FBS for 24 hours. RNA was extracted, reverse transcribed to cDNA, and analyzed by real-time PCR. Threshold cycle values were normalized to the housekeeping gene 36B4. Data are presented as fold vs. parental cells (n = 3; *P < 0.05).
LYND189Y accelerates hormone-independent growth and blocks the antitumor effect of ER and PI3K antagonists.
We next investigated the role of LYND189Y in estrogen-independent growth using ER+/PIK3CA mutant MCF-7 cells stably transduced with GFP (control), WT LYN, or D189Y LYN V5-tagged vectors. The D189Y mutation was verified by Sanger sequencing (Supplemental Figure 2). Expression of LYNWT and LYND189Y in MCF-7 cells was equivalent, as assessed by immunoblot with a V5 antibody (Figure 4A). Treatment with fulvestrant downregulated levels of ER and its target gene product IGF-I receptor (IGF-IR), but this effect was partially abrogated in MCF-7/LYNWT and MCF-7/LYND189Y cells, particularly in cells expressing the mutant. Overexpression of LYND189Y also prevented the complete inhibition of S473 phosphorylated AKT (P-AKT) and phosphorylated PRAS40 (P-PRAS40) levels mediated by BKM120 (Figure 4A), suggesting that mutant LYN contributes to PI3K activation.
Figure 4. LYND189Y accelerates hormone-independent growth and counteracts the effect of PI3K and ER inhibitors.

(A) Cells were treated with 10% DCC-FBS with or without 1 μM BKM120 or 1 μM fulvestrant for 24 hours. Protein lysates were prepared and analyzed by immunoblot with the indicated antibodies. (B and C) MCF-7 cells stably transduced with GFP, LYNWT, or LYND189Y were treated with 10% DCC-FBS with or without 1 μM BKM120 or 1 μM fulvestrant for 12 days. Media and drugs were replenished every 2 to 3 days. (B) Cells were fixed and stained with crystal violet. Representative images and quantitation of integrated intensity (percentage of control) are shown (n = 2; *P < 0.05 vs. respective GFP, #P < 0.01 vs. respective LYNWT). (C) Cells were cultured in Matrigel with or without 1 μM BKM120 or 1 μM fulvestrant and photographed on day 15. The average size of each cellular structure was quantitated using ImageJ. Data are presented as percentage of control (n = 2; *P < 0.05 vs. respective GFP, #P < 0.05 vs. respective LYNWT). Scale bars: 500 μm. (D) Cells were treated overnight in serum-free media with or without 1 μM BKM120 or 1 μM fulvestrant and then analyzed using the Caspase-Glo 3/7 assay. Data are presented as percentage of control (n = 2; *P < 0.05 vs. Con, #P < 0.05 vs. GFP BKM120).
Although stable transduction of either LYNWT or LYND189Y accelerated MCF-7 cell growth in estrogen-depleted media, the mutant was significantly more potent than LYNWT at inducing this effect (P < 0.001). Further, LYND189Y overexpression dampened the growth inhibitory effect of the ER downregulator fulvestrant or the pan-PI3K inhibitor BKM120 (ref. 22 and Figure 4B). More pronounced effects were seen in 3D Matrigel in estrogen-free media: MCF-7/LYND189Y cells formed larger colonies than MCF-7/LYNWT cells (P < 0.0001) and were less sensitive to fulvestrant and BKM120 (P < 0.05; Figure 4C). To extend these results to other cell lines, we stably transduced HCC1428 and ZR75-1 cells with GFP, LYNWT, and LYND189Y vectors (Supplemental Figure 3). Stably transduced HCC1428 and ZR75-1 cells showed similar results in 3D Matrigel in estrogen-free media upon treatment with fulvestrant and BMK120 (Figure 4C). In Supplemental Figure 4, we show these 3D growth data normalized to untreated controls.
We finally examined the effects of LYNWT or LYND189Y on caspase-dependent apoptosis in MCF-7 cells. Treatment with BKM120 significantly increased caspase-7 activity in MCF-7 cells, whereas treatment with fulvestrant did not (Figure 4D). Overexpression of LYND189Y but not LYNWT completely suppressed BKM120-induced caspase-7 activity (Figure 4D), suggesting that LYND189Y can rescue cells from the apoptosis induced by inhibition of PI3K.
SRC inhibitors block growth of MCF-7 cells expressing LYNWT or LYND189Y.
We next investigated whether the gain of function induced by overexpression of WT or mutant LYN is reversed by the small molecule SFK inhibitors dasatinib (LYN IC50, 8.5 nM) (23, 24) and AZD0530 (LYN IC50, 5 nM) (25). Dasatinib but not AZD0530 markedly suppressed Y416 P-SRC in both LYN-transduced cells. AZD0530 was less effective at suppressing phosphorylation of AKT, PRAS40, and S6 in MCF-7/LYND189Y cells (Figure 5A). Inhibition of SFK activity with dasatinib or AZD0530 suppressed the increased phosphorylation of FAK, IGF-IR/InsR, STAT3, EGFR, and MAPK observed upon overexpression of LYNWT or LYND189Y (Figure 5B). Further, addition of dasatinib restored the ability of fulvestrant to reduce ER and phosphorylated IGF-IR/InsR levels in MCF-7/LYND189Y cells (Supplemental Figure 5), suggesting that the mutant stabilizes ER and/or regulates ER transcription. Consistent with previous results, dasatinib and AZD0530 inhibited MCF-7GFP (control) cell growth by 1.8-fold and 2.2-fold, respectively (Figure 5C). MCF-7/LYNWT and MCF-7/LYND189Y cells were still sensitive to both SFK inhibitors, albeit less so to AZD0530 (Figure 5C). In Supplemental Figure 6, we show the data shown in Figure 5C normalized to untreated controls.
Figure 5. Growth of MCF-7 cells overexpressing LYNWT or LYND189Y is reduced by SRC inhibitors.

(A and B) MCF-7 cells stably transduced with GFP, LYNWT, or LYND189Y were treated with 10% FBS with or without 1 μM dasatinib or 1 μM AZD0530 for 24 hours. Protein lysates were prepared and analyzed by immunoblot with the indicated antibodies. (C) MCF-7 cells stably transduced with GFP, LYNWT, or LYND189Y were incubated in 10% DCC-FBS with or without 1 μM dasatinib or 1 μM AZD0530 and counted after 5 days (n = 3; *P < 0.001 vs. Con, #P < 0.05 vs. respective GFP).
LYND189Y exhibits increased catalytic activity and signaling output compared with that of LYNWT.
The differences in the cellular effects of LYNWT and LYND189Y (Figure 4) suggested the mutation conferred LYN a gain of function. Consistent with this notion, overexpression of LYNWT or LYND189Y resulted in increased phosphorylation of FAK, IGF-IR/InsR, EGFR, HER3, STAT3, AKT, and MAPK in MCF-7 cells maintained in FBS-containing media and following serum starvation (Figure 6, A and B). Activated SFK, as measured by Y416 P-SRC levels, was similar in LYNWT and LYND189Y cells. However, overexpression of LYND189Y increased phosphorylation of FAK (a downstream target of SFKs) at Y576/577 and Y925, EGFR at the SRC site Y845, HER3, and IRS-1 above levels induced by ectopic expression of LYNWT (Figure 6, A and B), suggesting that LYND189Y increases the catalytic activity of LYN. On the other hand, HCC1428 and ZR75-1 cells stably transduced with LYND189Y but not LYNWT contained markedly higher levels of Y416 P-SRC compared with their respective parental cells (Figure 6C).
Figure 6. LYND189Y exhibits a gain of function compared with LYNWT.

(A) MCF-7 cells stably transduced with GFP, LYNWT, or LYND189Y were plated in 10% FBS. Cell lysates were harvested and analyzed by immunoblot using the indicated antibodies against phosphorylated SRC substrates. (B) MCF-7 or (C) HCC-1428 and ZR 75-1 cells stably transduced with GFP, LYNWT, or LYND189Y were incubated in serum-free media for 24 or 48 hours, and then protein lysates were prepared and analyzed by immunoblot using the indicated antibodies. (D) For in vitro kinase assays, lysates from cells in A were precipitated with a V5 antibody, as described in the Methods. Kinase reactions were done in the presence or absence of 0.2 mM ATP and then analyzed by immunoblot with pTyr and V5 antibodies. (E) Cells from A were incubated in serum-free media (SFM) or 10% FBS for 48 hours. Lysates were precipitated with a V5 antibody, and pulldowns were analyzed by immunoblot with Y507 P-LYN and V5 antibodies. Densitometry analysis was performed using ImageJ; the ratios of pTyr/V5 bands in D and P-LYNY507/V5 bands in E are shown below the blots.
To interrogate a biochemical difference between WT and mutant LYN, we performed in vitro kinase assays. V5-tagged LYNWT and LYND189Y were precipitated from stably transduced MCF-7 cells. LYND189Y showed a 2.2-fold increase in kinase activity compared with LYNWT in the absence of added ATP (Figure 6D). However, based on the location of the mutation in the SH2 domain of LYN, we speculated that D189Y would mainly prevent an inactive conformation of the kinase. SFKs are phosphorylated by C-terminal SRC kinase (CSK) in a C-terminal regulatory tyrosine, which keeps the SFKs in an inactive conformation involving an intramolecular interaction between this residue and the SH2 domain (17). Upon dephosphorylation of the C-terminal tyrosine (Y507 in LYN), SFKs adopt an open conformation in which Y416 (Y396 in LYN) in the activation loop is phosphorylated and the kinase is fully activated. We speculated that D189Y would reduce the interaction between the regulatory tyrosine and the SH2 domain, thus preventing the inactive conformation of LYN. Indeed, LYND189Y overexpression markedly decreased phosphorylation of LYN at the Y507 C-terminal regulatory site compared with LYNWT in MCF-7, HCC-1428, and ZR75-1 cells (Figure 6, B and C). To corroborate these data, we precipitated V5-tagged LYNWT and LYND189Y from MCF-7 cell lysates and performed immunoblot analysis with an antibody that recognizes LYN phosphorylated in Y507 (P-LYN). Y507 P-LYN content in LYNWT and LYND189 was corrected for V5 expression in the V5 pulldowns, as measured by densitometry. Compared with LYNWT, LYND189Y contained 71% lower levels of Y507 P-LYN in 10% FBS and 14% lower levels of Y507 P-LYN in serum-free media (Figure 6E).
We next extended these studies to other LYN mutants identified in TCGA. V5-tagged lentiviral vectors encoding LYNE159K, LYNK209N, LYNA370T, LYNG418R, and LYNA503D were prepared and stably transduced into MCF-7 cells (Figure 7A). We were unable to express the A503D variant. Similar to LYND189Y, the 2 other SH2 domain mutants (LYNE159K and LYNK209N) expressed lower levels of Y507 P-LYN (Figure 7, A and B). Compared with MCF-7/LYNWT cells, LYNE159K and LYNK209N cells exhibited enhanced growth in estrogen-free media and were less sensitive to the inhibitory effect of fulvestrant and BKM120 (Figure 7C). In Supplemental Figure 7, we show these growth data normalized to untreated controls.
Figure 7. TCGA LYN mutations.

(A) MCF-7 cells stably transduced with GFP, LYNWT, LYND189Y or other LYN mutants described in TCGA (E159K, K209N, A370T, and G418R) were plated in 10% FBS. Cell lysates were harvested and analyzed by immunoblot using the indicated antibodies. Densitometry analysis was performed using ImageJ; the ratio of P-LYNY507/V5 bands is shown below each cell line column. PAR, parental. (B) Cells described in A were incubated in serum-free media for 24 or 48 hours. Protein lysates were then prepared and analyzed by immunoblot using the indicated antibodies. Calnex, calnexin (an antibody used in the immunoblot analysis). (C) Cells described in A were cultured in Matrigel with or without 1 μM BKM120 or 1 μM fulvestrant. On day 15, colonies were stained with MTT and photographed with a Gelcount reader. The average size of each cellular structure was quantitated and expressed relative to untreated GFP controls (n = 3; *P < 0.05 vs. respective treatments of GFP controls, #P < 0.001 vs. respective treatment of LYNWT cells). Scale bars: 500 μm.
Pharmacological inhibition of the SFKs enhances the antitumor effect of BKM120 and fulvestrant in vivo.
The results of the RNAi screen in LTED ER+ breast cancer cells (Figure 2), the higher levels of LYN in 3 of 4 LTED cell lines (Figure 3), and the growth advantage conferred by LYNWT (Figure 4) all suggested that overexpression of SFKs, such as LYN, can mediate escape from endocrine therapy. If so, addition of a SFK inhibitor should improve the effect of this therapy and/or delay acquired drug resistance. We examined this possibility in ER+/PIK3CA mutant MCF-7 xenografts. We recently reported that growth of these xenografts established in ovariectomized mice and in the absence of added estrogens is synergistically inhibited by a combination of fulvestrant and BKM120 (21). Inhibition of the SFKs with dasatinib modestly increased the growth inhibitory effect of BKM120 in MCF-7/LYND189Y cells, while it significantly increased the effect of fulvestrant (Figure 8A). In Supplemental Figure 8, we show these growth data normalized to untreated controls.
Figure 8. Pharmacological inhibition of the SFKs potentiates the antitumor effect of BKM120 and fulvestrant in vivo.

(A) MCF-7 cells stably transduced with GFP, LYNWT, or LYND189Y were treated with 10% DCC-FBS with or without 1 μM dasatinib, 1 μM BKM120, or 1 μM fulvestrant. Media and drugs were replenished every 3 days. Cells were counted after 5 days. Data are presented as percentage of control (n = 3; *P < 0.0001 vs. respective Con, #P < 0.01 vs. Fulv). (B) MCF-7 cells were injected s.c. into athymic ovariectomized mice supplemented with short-term 14-day release 17β-estradiol pellets. Mice bearing tumors ≥150 mm3 were randomized to vehicle, dasatinib (15 mg/kg/d, p.o.), BKM120 (30 mg/kg/d, p.o.) and fulvestrant (5 mg/wk, s.c.), or BKM120, fulvestrant, and dasatinib for 7 weeks. Data are presented as log2 of mean tumor volume (*P < 0.0001 vs. vehicle, #P < 0.01 vs. BKM and Fulv or dasatinib). (C) Xenografts from B were homogenized 4 hours after the last dose of BKM120 or dasatinib and 24 hours after the last dose of fulvestrant. Tumor lysates were analyzed by immunoblot using the indicated antibodies. (D) IHC for Y416 P-SRC. Representative images of tumors from A and quantitative comparison of membrane histoscores, as described in the Methods (H-score; *P < 0.0001). Scale bars: 200 μm.
We next assessed the effects of combined inhibition of ER, PI3K, and SFKs on tumor growth in vivo. MCF-7 xenografts were established in ovariectomized athymic mice. Treatment with BKM120 and fulvestrant or BKM120, fulvestrant, and dasatinib, but not dasatinib alone, inhibited growth of established tumors compared with vehicle (P < 0.0001; Figure 8B). Treatment with triple combination induced near-complete tumor regression and was significantly more effective than BKM120 and fulvestrant or dasatinib alone (P < 0.01; Figure 8B). Treatment with BKM120 and fulvestrant as well as BKM120, fulvestrant, and dasatinib decreased ER, PR, IGF-IR, P-AKT, P-PRAS40, phosphorylated S6 (P-S6), and phosphorylated MAPK (P-MAPK) tumor levels. Treatment with dasatinib alone reduced P-AKT, P-S6, and P-MAPK levels as well as those of ER, PR, and IGF-IR (Figure 8C), suggesting that, in untreated tumors, activated SFKs stabilize ER and its transcriptional output in vivo. Consistent with drug-mediated target inhibition, dasatinib-treated tumors exhibited lower levels of Y416 P-SRC, as measured by IHC (Figure 8D). Overall, the drug combinations were well-tolerated, with <10% weight loss (Supplemental Figure 9). These results suggest that pharmacological inhibition of SFKs enhances the antitumor effect of BKM120 and fulvestrant against estrogen-deprived MCF-7 xenografts in vivo.
Finally, we established MCF-7/LYNWT and MCF-7/LYND189Y xenografts and treated them with fulvestrant and BKM120 with or without dasatinib. Similar to parental MCF7 tumors, these xenografts were sensitive to fulvestrant and BKM120 but did not show complete responses. Treatment with dasatinib added statistically to the effect of fulvestrant and BKM120 against MCF-7/LYNWT but not MCF-7/LYND189Y xenografts (Figure 9, A and B). Of note, dasatinib partially inhibited Y416 P-SRC in MCF-7/LYNWT xenografts but not in MCF-7/LYND189Y xenografts, as measured by IHC (Figure 9C), also consistent with increased catalytic activity of the mutant over the WT enzyme.
Figure 9. Pharmacological inhibition of SFKs enhances the effect of BKM120 and fulvestrant against MCF-7/LYNWT xenografts but not MCF-7/LYND189Y xenografts.

(A) MCF-7 cells stably transduced with LYNWT or LYND189Y were injected s.c. into athymic ovariectomized mice, as described in the Methods. Mice bearing tumors ≥150 mm3 were randomized and treated as in Figure 8B. Data are presented as log2 of mean tumor volume (*P < 0.0001 vs. vehicle, #P < 0.05 vs. BKM plus Fulv). (B) Xenografts from A were harvested 4 hours after the last dose of BKM120 or dasatinib and 24 hours after the last dose of fulvestrant. Tumor lysates were analyzed by immunoblot using the indicated antibodies. (C) IHC for Y416 P-SRC. Representative images of tumors from A and quantitative comparison of membrane histoscores, as described in the Methods (H-score; *P < 0.05). Scale bars: 200 μm.
Discussion
Although endocrine therapies are effective against ER+ breast cancers, de novo and acquired drug resistance remain a significant clinical problem. The factors mediating resistance to antiestrogens have been the subject of considerable investigation and include alterations in pathways such as E2F/cyclin D/CDKs (21), PI3K/mTOR (20), and FGFR (26). To further explore genetic alterations that may lead to intrinsic endocrine resistance, we performed deep kinome sequencing on 4 ER+/HER2– human breast cancers that retained high Ki67 levels (a cellular marker of endocrine resistance) following short-term treatment with the AI letrozole. In one of these tumors, we identified a novel D189Y mutation in the SH2 domain of LYN. Analysis of TCGA breast cancer data revealed 4 LYN mutations, 2 in the SH2 domain of LYN. Further, RNAi screening identified LYN as being required for hormone-independent ER+ breast cancer growth, and knockdown of LYN inhibited growth of 4 of 4 LTED cell lines.
We next studied the functional role of LYN mutations in several ER+ human breast cancer cell lines. Overexpression of LYNWT or LYND189Y in MCF-7 cells increased phosphorylation of SFKs (at Y416) and known targets of SFKs, including FAK, IGF-IR/InsR, EGFR, HER3, STAT3, AKT, and MAPK. However, LYND189Y expression resulted in higher phosphorylation of FAK, EGFR, HER3, and IRS-1 compared with LYNWT. Although stable transduction of either LYNWT or LYND189Y accelerated MCF-7, HCC1428, and ZR75-1 cell growth in estrogen-depleted media, the mutant was substantially more potent than LYNWT at inducing this effect.
We also showed a concurrent decrease in phosphorylation of LYN at the C-terminal regulatory site Y507 upon expression of the LYND189Y mutant in MCF-7, HCC1428, and ZR75-1 cells (Figure 6, B and C). Phosphorylation of Y507 by c-SRC tyrosine kinase (CSK) maintains LYN in an inactive conformation, which requires an intramolecular interaction between Y507 and the SH2 domain. Upon dephosphorylation, LYN adopts an open conformation in which the activation loop Y396 is phosphorylated and the kinase is activated (17). Thus, we speculated that the LYND189Y mutation may prevent/hinder the interaction between Y507 and the SH2 domain, thus abrogating LYN inactivation and potentially increasing signaling duration and/or amplitude. However, molecular modeling of autoinhibited SRC did not indicate how the mutation might alter the Y507-mediated conformation of the protein due to its distal location. Additionally, modeling of an activating peptide substrate did not suggest a direct role for D189 in the interaction between the SH2 domain and the activating peptide. Since no active SFK structures that include the SH2 domain are available, determining whether a tyrosine substitution at D189 might alter intermolecular or intramolecular interactions in the active state of the protein will require additional structural experiments beyond the scope of this study. Moreover, the potential exists that unidentified proteins may interact at the solvent-accessible D189 interface of the LYN SH2 domain.
All 4 sequenced tumors harbored an activating mutation in PIK3CA (H1047R, T1025A, or Q546E; Table 1). The PI3K pathway has been causally associated with resistance to endocrine therapy (20, 27–31). A tumor protein signature of PI3K activation predicts poor outcome following adjuvant endocrine therapy (20). Upon acquisition of hormone independence, ER+ breast cancer cells increase their dependence on PI3K/AKT signaling (20). However, the clinical significance of activating PIK3CA mutations, which occur in approximately 40% of breast cancers, remains to be fully established (27). Parallel sequencing on tumor DNA accrued from patients treated with AIs was recently reported (32). In this study, Ellis et al. found that PIK3CA was mutated at a frequency of 41.3% but was not associated with clinical or Ki67 response (32). These investigators also identified a LYNA370T mutation in an AI-resistant tumor and suggested the SFK FYN as a hub of connectivity highly associated with Ki67 status in breast tumors treated with AIs (32). The LYND189Y mutation identified in this study was present in a tumor also harboring an activating PIK3CAT1025A mutation, suggesting a potential link between aberrant LYN and PI3K signaling in endocrine resistance. Consistent with the notion of LYN limiting the full effect of ER and PI3K inhibitors, pharmacological inhibition of the SFKs with dasatinib enhanced the antitumor effects of BKM120 plus fulvestrant against estrogen-deprived parental MCF-7 xenografts and MCF-7 xenografts overexpressing LYNWT.
The prototype SFK SRC has previously been shown to play a role in endocrine resistance. Elevated SRC kinase activity promotes invasion and motility in tamoxifen-resistant MCF-7 cells (33), and combined inhibition of SRC and ER impairs breast cancer growth in vitro and in vivo (33–37). Although LYN has been shown to be important in triple-negative breast cancer (3), to our knowledge, we are the first to identify a possible role for LYN in ER+ breast cancers resistant to estrogen deprivation.
Knockdown of CSK, a negative regulator of SRC, has been shown to suppress fulvestrant-induced proteasomal degradation of ER (38). Since phosphorylation by CSK inhibits SFK activity (17), these data suggest the converse, that is, that derepressed SFKs, as a result of loss of CSK or activating mutations in LYN, block fulvestrant-induced downregulation of ER and growth inhibition (Figure 4). Consistent with this hypothesis, treatment with dasatinib restored the effect of fulvestrant on ER levels (Supplemental Figure 5) and increased the growth inhibitory effects of fulvestrant (Figure 8A). We have shown previously that treatment with BKM120 and fulvestrant induces regression of estrogen-deprived MCF-7 xenografts (21). We expanded upon this by showing that treatment with the combination of dasatinib, BKM120, and fulvestrant was more effective at suppressing ER+/PIK3CA mutant MCF-7 xenograft growth compared with BKM120 and fulvestrant (Figure 8B). LYND189Y appears to hyperactivate LYN (Figure 6D), diminishing the effectiveness of dasatinib in vivo (Figure 9A). We postulate that resistance to dasatinib in LYN mutant xenografts is due to an inability of LYND189Y to achieve an inactive conformation (Figure 6, B and E, and Figure 7A), without changing the conformation and/or affinity of the ATP-binding site. Thus, higher doses may restore the “competitiveness” of dasatinib for the ATP-binding site. Given the lack of detectable inhibition of Y416 P-SRC by dasatinib in MCF-7/LYND189Y tumors, we speculate that higher doses would be needed to achieve adequate target inhibition LYN in vivo. These data also suggest that SFKs compensate for the combined effect of PI3K and ER inhibitors against ER+ breast cancers. Phase I and II clinical studies are currently investigating dasatinib in combination with letrozole or fulvestrant (plus an IGF-IR antibody) in patients with ER+ breast cancer (39).
In summary, ER+ breast cancers harbor multiple molecular alterations capable of mediating hormone-independent growth. We have identified LYN mutations as a potential mechanism of escape from estrogen deprivation in ER+ breast cancers. These results support (a) further sequencing studies of LYN in ER+ tumors refractory to endocrine therapy and (b) the need to develop potent SFK inhibitors, which, in combination with PI3K and ER inhibitors, may be an effective treatment for endocrine-resistant breast cancer.
Methods
Deep kinome sequencing.
Sequencing was performed on DNA extracted from 4 FFPE surgically removed breast tumors that retained a high Ki67 score after 2 weeks of therapy with letrozole (clinical trial NCT00651976). Patient-matched normal adjacent FFPE tissue was also sequenced. Genomic DNA isolated from these tumors and from adjacent normal breast tissue was fragmented into 200-bp sections using a Covaris E210, followed by purification using AMPure XP beads (Agencourt). Library preparation and capture was performed via specifications set by the Agilent SureSelect Human Kinome Kit (Agilent); libraries were normalized to 147 ng/μl prior to capture. Enrichment of kinase regions was performed by adding 500 ng of prepared library along with hybridization buffer, blocking mix, and capture library provided with the kit. Post-capture samples were normalized to 2 nM and then pooled in a 6-plex; clustering was performed on a cBot (Illumina) using the TruSeq PE Cluster Kit v2 (Illumina) and samples were loaded onto an Illumina GAIIx for sequencing, using the TruSeq SBS Kit v4 GA (Illumina) for 72-bp PE sequencing.
RPPAs.
Lysates were prepared from frozen core biopsies of the surgical specimens and analyzed by RPPA as described previously (20, 40, 41). FFPE tumor sections before and after treatment were used for Ki67 IHC (DAKO no. M7240) and scored as described previously (12).
siRNA screen.
MCF-7/LTED cells were transfected with the Dharmacon RTF Protein Kinase siRNA library as described previously (21, 42).
Cell growth assays.
Cells with or without dasatinib (LC Laboratories), AZD0530 (Selleck Chemicals), BKM120 (Selleck Chemicals), or fulvestrant (ICI 182780, Tocris Biosciences) were seeded in 10% dextran-coated charcoal–treated FBS (DCC-FBS) for proliferation in 2D growth assay and counted or fixed/stained with crystal violet as described previously (20). For siRNA experiments, cells in 100-mm dishes were transfected with a siRNA targeting LYN (Hs LYN 13, Qiagen) or a nonsilencing control using HiPerfect Transfection Reagent according to the manufacturer’s protocol (Qiagen). The next day, cells were reseeded in 10% DCC-FBS for proliferation assays and immunoblot analyses (20). 3D growth assays were conducted in growth factor–reduced Matrigel (BD Biosciences) as described previously (43) with 10% DCC-FBS–containing media. Phase-contrast pictures were taken using an Olympus CK40 microscope and Gelcount scanning.
Immunoblot analysis and immunoprecipitation.
Cell lysates were subjected to immunoblot analysis as described previously (20). Immunoprecipitation was performed using Dynal protein G beads (Invitrogen) and V5 (Invitrogen) or 4G10 pTyr (Millipore) antibodies (44). In vitro kinase assays were performed in immune complexes precipitated with a V5 antibody as described previously (45).
Apoptosis assay.
Cells (2 × 103 cells per well) were plated in white 96-well plates, treated overnight in serum-free media with or without BKM120 or fulvestrant, and then subjected to the Caspase-Glo 3/7 Assay according to the manufacturer’s protocol (Promega).
Xenograft studies.
Female ovariectomized athymic mice were implanted s.c. with a 14-day-release 17β-estradiol pellet (0.17 mg) and 107 MCF-7 parental cells or stably transfected with LYNWT or LYNE159K. After 4 weeks, mice bearing tumors ≥150 mm3 were randomly assigned to treatment with vehicle (80 mM sodium citrate buffer, pH 3), dasatinib (15 mg/kg/d, per os [p.o.]), BKM120 (30 mg/kg/d, p.o.) and fulvestrant (5 mg/wk, s.c.), or BKM120, fulvestrant, and dasatinib. Tumor diameters were measured using calipers twice per week, and volume in mm3 was calculated with the formula: volume = width2 × length/2. Tumors were harvested and snap frozen in liquid N2 or fixed in 10% formalin prior to paraffin embedding for IHC.
Statistics.
In cell proliferation assays, significant differences were determined by repeated-measures ANOVA with Bonferroni post-hoc test, except as noted below. Paired 2-tailed t tests were used to determine significant differences in siRNA proliferation assays, Caspase-Glo 3/7 assay, real-time qPCR assays, and IHC histoscores. A P value of less than 0.05 was considered statistically significant. Bar graphs show mean ± SD, unless otherwise stated in the figure legends.
Study approval.
Breast tumor blocks were from a cohort of patients who had consented to the use of any deidentified tissues for research purposes under the auspices of an IRB-approved protocol (Vanderbilt IRB no. NCT00651976). Inclusion criteria for this study were postmenopausal women with newly diagnosed ER+/HER2– operable breast cancer treated with letrozole (2.5 mg/d) for 10 to 21 days prior to surgery. Tumor cell proliferation was assessed by Ki67 IHC in a core biopsy before treatment and in the surgical specimen (after treatment). Experiments with mice were approved by the Vanderbilt Institutional Animal Care and Use Committee, protocol no. M/10/111.
Additional details are provided in the Supplemental Methods.
Supplementary Material
Acknowledgments
We thank William C. Hahn and David E. Root for the pDONR223-LYN (Addgene plasmid 23905), pENTRY-GFP (Addgene plasmid 15301), and pLX302 (Addgene plasmid 25896) vectors. We acknowledge that library and deep kinome sequencing services were provided through a grant from Expression Analysis and Illumina (Research Acceleration Grant [In Kind] from the American Cancer Society). This work was supported by postdoctoral fellowship grant no. PF-10-184-01-TBE from the American Cancer Society (to E.M. Fox), Research Acceleration Grant (In Kind) from the American Cancer Society (to E.M. Fox), Susan G. Komen for the Cure postdoctoral fellowship PDF12229712 (to J.M. Balko), Vanderbilt Breast SPORE grant P50 CA098131, Vanderbilt-Ingram Cancer Center support grant P30 CA68485, a Breast Cancer Research Foundation grant (to C.L. Arteaga), MD Anderson Cancer Center support grant P30 CA016672, Susan G. Komen for the Cure Foundation grant SAC100013 (to C.L. Arteaga), and Instituto Nacional de Enfermedades Neoplásicas, Lima, Perú (to L.J. Schwarz).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2014;124(12):5490–5502. doi:10.1172/JCI72573.
References
- 1.Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004;23(48):7906–7909. doi: 10.1038/sj.onc.1208160. [DOI] [PubMed] [Google Scholar]
- 2.Kim LC, Song L, Haura EB. Src kinases as therapeutic targets for cancer. Nat Rev Clin Oncol. 2009;6(10):587–595. doi: 10.1038/nrclinonc.2009.129. [DOI] [PubMed] [Google Scholar]
- 3.Choi YL, et al. LYN is a mediator of epithelial-mesenchymal transition and a target of dasatinib in breast cancer. Cancer Res. 2010;70(6):2296–2306. doi: 10.1158/0008-5472.CAN-09-3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fox EM, Andrade J, Shupnik MA. Novel actions of estrogen to promote proliferation: integration of cytoplasmic and nuclear pathways. Steroids. 2009;74(7):622–627. doi: 10.1016/j.steroids.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arnold SF, Vorojeikina DP, Notides AC. Phosphorylation of tyrosine 537 on the human estrogen receptor is required for binding to an estrogen response element. J Biol Chem. 1995;270(50):30205–30212. doi: 10.1074/jbc.270.50.30205. [DOI] [PubMed] [Google Scholar]
- 6.Sun J, Zhou W, Kaliappan K, Nawaz Z, Slingerland JM. ERalpha phosphorylation at Y537 by Src triggers E6-AP-ERalpha binding, ERalpha ubiquitylation, promoter occupancy, and target gene expression. Mol Endocrinol. 2012;26(9):1567–1577. doi: 10.1210/me.2012-1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Forbes JF, Cuzick J, Buzdar A, Howell A, Tobias JS, Baum M. Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 100-month analysis of the ATAC trial. Lancet Oncol. 2008;9(1):45–53. doi: 10.1016/S1470-2045(07)70385-6. [DOI] [PubMed] [Google Scholar]
- 8.Geisler J, Haynes B, Anker G, Dowsett M, Lonning PE. Influence of letrozole and anastrozole on total body aromatization and plasma estrogen levels in postmenopausal breast cancer patients evaluated in a randomized, cross-over study. J Clin Oncol. 2002;20(3):751–757. doi: 10.1200/JCO.20.3.751. [DOI] [PubMed] [Google Scholar]
- 9.Smith IE, et al. Neoadjuvant treatment of postmenopausal breast cancer with anastrozole, tamoxifen, or both in combination: the Immediate Preoperative Anastrozole, Tamoxifen, or Combined with Tamoxifen (IMPACT) multicenter double-blind randomized trial. J Clin Oncol. 2005;23(22):5108–5116. doi: 10.1200/JCO.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 10.Urruticoechea A, Smith IE, Dowsett M. Proliferation marker Ki-67 in early breast cancer. J Clin Oncol. 2005;23(28):7212–7220. doi: 10.1200/JCO.2005.07.501. [DOI] [PubMed] [Google Scholar]
- 11.Dowsett M, et al. Short-term changes in Ki-67 during neoadjuvant treatment of primary breast cancer with anastrozole or tamoxifen alone or combined correlate with recurrence-free survival. Clin Cancer Res. 2005;11(2):951s–958s. [PubMed] [Google Scholar]
- 12.Dowsett M, et al. Prognostic value of Ki67 expression after short-term presurgical endocrine therapy for primary breast cancer. J Natl Cancer Inst. 2007;99(2):167–170. doi: 10.1093/jnci/djk020. [DOI] [PubMed] [Google Scholar]
- 13.Carson JD, et al. Effects of oncogenic p110alpha subunit mutations on the lipid kinase activity of phosphoinositide 3-kinase. Biochem J. 2008;409(2):519–524. doi: 10.1042/BJ20070681. [DOI] [PubMed] [Google Scholar]
- 14.Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A. 2005;102(3):802–807. doi: 10.1073/pnas.0408864102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Samuels Y, et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell. 2005;7(6):561–573. doi: 10.1016/j.ccr.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 16.Gymnopoulos M, Elsliger MA, Vogt PK. Rare cancer-specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci U S A. 2007;104(13):5569–5574. doi: 10.1073/pnas.0701005104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ingley E. Src family kinases: regulation of their activities, levels and identification of new pathways. Biochim Biophys Acta. 2008;1784(1):56–65. doi: 10.1016/j.bbapap.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 18.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39(17):e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cerami E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller TW, et al. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest. 2010;120(7):2406–2413. doi: 10.1172/JCI41680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller TW, et al. ERα-dependent E2F transcription can mediate resistance to estrogen deprivation in human breast cancer. Cancer Discov. 2011;1(4):338–351. doi: 10.1158/2159-8290.CD-11-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maira SM, et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther. 2012;11(2):317–328. doi: 10.1158/1535-7163.MCT-11-0474. [DOI] [PubMed] [Google Scholar]
- 23.Lombardo LJ, et al. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47(27):6658–6661. doi: 10.1021/jm049486a. [DOI] [PubMed] [Google Scholar]
- 24.Nam S, et al. Action of the Src family kinase inhibitor, dasatinib (BMS-354825), on human prostate cancer cells. Cancer Res. 2005;65(20):9185–9189. doi: 10.1158/0008-5472.CAN-05-1731. [DOI] [PubMed] [Google Scholar]
- 25.Green TP, et al. Preclinical anticancer activity of the potent, oral Src inhibitor AZD0530. Mol Oncol. 2009;3(3):248–261. doi: 10.1016/j.molonc.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turner N, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010;70(5):2085–2094. doi: 10.1158/0008-5472.CAN-09-3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller TW, Balko JM, Arteaga CL. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. J Clin Oncol. 2011;29(33):4452–4461. doi: 10.1200/JCO.2010.34.4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller TW, et al. Loss of Phosphatase and Tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer. Cancer Res. 2009;69(10):4192–4201. doi: 10.1158/0008-5472.CAN-09-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor α: a new model for anti-estrogen resistance. J Biol Chem. 2001;276(13):9817–9824. doi: 10.1074/jbc.M010840200. [DOI] [PubMed] [Google Scholar]
- 30.Crowder RJ, et al. PIK3CA and PIK3CB inhibition produce synthetic lethality when combined with estrogen deprivation in estrogen receptor-positive breast cancer. Cancer Res. 2009;69(9):3955–3962. doi: 10.1158/0008-5472.CAN-08-4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Block M, et al. Inhibition of the AKT/mTOR and erbB pathways by gefitinib, perifosine and analogs of gonadotropin-releasing hormone I and II to overcome tamoxifen resistance in breast cancer cells. Int J Oncol. 2012;41(5):1845–1854. doi: 10.3892/ijo.2012.1591. [DOI] [PubMed] [Google Scholar]
- 32.Ellis MJ, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486(7403):353–360. doi: 10.1038/nature11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hiscox S, Morgan L, Green TP, Barrow D, Gee J, Nicholson RI. Elevated Src activity promotes cellular invasion and motility in tamoxifen resistant breast cancer cells. Breast Cancer Res Treat. 2006;97(3):263–274. doi: 10.1007/s10549-005-9120-9. [DOI] [PubMed] [Google Scholar]
- 34.Chen Y, et al. Combined Src and aromatase inhibition impairs human breast cancer growth in vivo and bypass pathways are activated in AZD0530-resistant tumors. Clin Cancer Res. 2009;15(10):3396–3405. doi: 10.1158/1078-0432.CCR-08-3127. [DOI] [PubMed] [Google Scholar]
- 35.Chen Y, et al. Combined Src and ER blockade impairs human breast cancer proliferation in vitro and in vivo. Breast Cancer Res Treat. 2011;128(1):69–78. doi: 10.1007/s10549-010-1024-7. [DOI] [PubMed] [Google Scholar]
- 36.Hiscox S, Morgan L, Green T, Nicholson RI. Src as a therapeutic target in anti-hormone/anti-growth factor-resistant breast cancer. Endocr Relat Cancer. 2006;13(suppl 1):S53–S59. doi: 10.1677/erc.1.01297. [DOI] [PubMed] [Google Scholar]
- 37.Hiscox S, et al. Dual targeting of Src and ER prevents acquired antihormone resistance in breast cancer cells. Breast Cancer Res Treat. 2009;115(1):57–67. doi: 10.1007/s10549-008-0058-6. [DOI] [PubMed] [Google Scholar]
- 38.Yeh WL, Shioda K, Coser KR, Rivizzigno D, McSweeney KR, Shioda T. Fulvestrant-induced cell death and proteasomal degradation of estrogen receptor α protein in MCF-7 cells require the CSK c-Src tyrosine kinase. PLoS One. 2013;8(4):e60889. doi: 10.1371/journal.pone.0060889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Montero JC, Seoane S, Ocana A, Pandiella A. Inhibition of SRC family kinases and receptor tyrosine kinases by dasatinib: possible combinations in solid tumors. Clin Cancer Res. 2011;17(17):5546–5552. doi: 10.1158/1078-0432.CCR-10-2616. [DOI] [PubMed] [Google Scholar]
- 40.Tibes R, et al. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther. 2006;5(10):2512–2521. doi: 10.1158/1535-7163.MCT-06-0334. [DOI] [PubMed] [Google Scholar]
- 41.Hennessy B, et al. A technical assessment of the utility of reverse phase protein arrays for the study of the functional proteome in non-microdissected human breast cancers. Clin Proteomics. 2010;6(4):129–151. doi: 10.1007/s12014-010-9055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fox EM, et al. A kinome-wide screen identifies the insulin/IGF-I receptor pathway as a mechanism of escape from hormone dependence in breast cancer. Cancer Res. 2011;71(21):6773–6784. doi: 10.1158/0008-5472.CAN-11-1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiang B, Muthuswamy SK. Using three-dimensional acinar structures for molecular and cell biological assays. Methods Enzymol. 2006;406:692–701. doi: 10.1016/S0076-6879(06)06054-X. [DOI] [PubMed] [Google Scholar]
- 44.Engelman JA, et al. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc Natl Acad Sci U S A. 2005;102(10):3788–3793. doi: 10.1073/pnas.0409773102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang SE, et al. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell. 2006;10(1):25–38. doi: 10.1016/j.ccr.2006.05.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.