

Abstract
Idiopathic pulmonary fibrosis (IPF) is a fatal fibrotic lung disorder with no effective medical treatments available. The generation of myofibroblasts, which are critical for fibrogenesis, requires both a mechanical signal and activated TGF-β; however, it is not clear how fibroblasts sense and transmit the mechanical signal(s) that promote differentiation into myofibroblasts. As transient receptor potential vanilloid 4 (TRPV4) channels are activated in response to changes in plasma membrane stretch/matrix stiffness, we investigated whether TRPV4 contributes to generation of myofibroblasts and/or experimental lung fibrosis. We determined that TRPV4 activity is upregulated in lung fibroblasts derived from patients with IPF. Moreover, TRPV4-deficient mice were protected from fibrosis. Furthermore, genetic ablation or pharmacological inhibition of TRPV4 function abrogated myofibroblast differentiation, which was restored by TRPV4 reintroduction. TRPV4 channel activity was elevated when cells were plated on matrices of increasing stiffness or on fibrotic lung tissue, and matrix stiffness–dependent myofibroblast differentiation was reduced in response to TRVP4 inhibition. TRPV4 activity modulated TGF-β1–dependent actions in a SMAD-independent manner, enhanced actomyosin remodeling, and increased nuclear translocation of the α-SMA transcription coactivator (MRTF-A). Together, these data indicate that TRPV4 activity mediates pulmonary fibrogenesis and suggest that manipulation of TRPV4 channel activity has potential as a therapeutic approach for fibrotic diseases.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a scarring disorder of the lungs without any effective medical treatment available. As such, the roughly 100,000 patients in the USA with IPF have a median survival of only 2 to 3 years (1). Experience with decades of less than successful therapeutic drug trials has shown that only through a better understanding of the fibrogenic process can we hope to ameliorate this fatal disorder (1–4). Overwhelming evidence demonstrates the significance of myofibroblast differentiation and persistence to normal and pathological wound healing in the lung, heart, skin, skeletal muscle, and liver (5, 6). During the repair process, myofibroblasts localize to fibrotic lesions, synthesize and remodel extracellular matrix proteins in part through their contractile properties, and express cytokines and chemokines that can drive pathologic fibrosis (5–7). Myofibroblasts are characterized by their expression of α-smooth muscle actin (α-SMA) and its incorporation into actin stress fibers that function to stabilize cell shapes and generate intracellular contractile force. Optimal myofibroblast differentiation is regulated by active TGF-β, the presence of the fibronectin ED-A domain, and a mechanical force signal (5–9). Classic studies show that fibroblasts in floating collagen gels, a condition in which they are incapable of generating intracellular tension, will fail to undergo myofibroblast differentiation (10). In addition, stiffening of the extracellular matrix itself, in the range noted in fibrotic lung tissue, for example, is capable of providing a myofibroblast-inducing mechanical signal (11–14). The exuberant extracellular matrix deposition and the resultant stiffening of the tissue are thus recognized as critical inducers of myofibroblast differentiation. However, the precise mechanisms, or pathway(s), by which these physical properties are transduced into the biological response remain elusive.
Ca2+ second messenger signals are critical for many essential cellular functions, including differentiation, gene expression, cell proliferation, growth, and death (15). In an in vivo study, mibefradil, a Ca2+ channel blocker, significantly reduced collagen production and fibroblast differentiation in rats treated with angiotensin II or aldosterone (16). Recently, it has been demonstrated that transient receptor potential (TRP) family member–mediated (i.e., mediation by TRPM7 and TRPC6) Ca2+ influx plays an essential role in atrial and dermal fibroblast differentiation (17, 18). These studies demonstrate that intracellular Ca2+ influx via calcium permeable channels can play an important role during fibroblast differentiation. Although Ca2+-initiated signals can regulate actomyosin assembly/dissolution and protein function, the mechanism by which intracellular calcium signals are mediated in response to changes in intracellular tension or matrix stiffness is unknown.
TGF-β1 is the best-studied mediator of both myofibroblast differentiation in vitro and in fibrosis of many organs (19–21). TGF-β1 signals through a plasma membrane receptor complex of TGF-β1 receptor type I and II, which phosphorylates the transcription factors SMAD2/3 to activate canonical (SMAD-dependent) pathways (19–21). TGF-β1 can also signal through noncanonical (SMAD-independent) pathways that include c-JUN N-terminal kinase, p38 kinases, focal adhesion kinases, phosphatidylinositol 3-kinase, and Rho GTPases (19–21). Both canonical and noncanonical pathways have been linked to myofibroblast differentiation and fibrosis (19, 20). However, TGF-β1 also has beneficial roles in regulating immune suppression, inflammation, cell growth, and tissue homeostasis in various normal and pathologic tissues (19–21). Thus, any molecular strategy that targets the profibrotic signals of the TGF-β1 pathway without disrupting its beneficial/homeostatic roles will have great therapeutic potential. The integration of the matrix stiffness signal with that of soluble TGF-β in fibroblasts may provide such a selective target.
We reasoned that a protein that was able to respond to either intracellular tension or matrix stiffness with a calcium signal might be a good candidate to function as a stiffness sensor and myofibroblast differentiation signal initiator. While functional voltage-gated calcium channels in fibroblasts have not been described, the TRP channel family of ion channels are gated by a variety of physical and chemical stimuli, including growth factor receptor ligation, hypotonicity-induced cell swelling, membrane stretching, lipid mediators, and thermal stimuli (22–25). TRP vanilloid 4 (TRPV4) is a nonselective cation channel that was originally identified as an osmosensor in C. elegans (22–25). TRPV4 is ubiquitously expressed in various cell types (such as neurons, endothelial cells, and epithelial cells) and tissues (such as lung, heart, and kidney). Furthermore, mutations in TRPV4 have been linked to diseases including skeletal dysplasia and sensory and motor neuropathies (26–28). TRPV4 has been implicated recently in the differentiation of chondrocytes, osteoclasts, and cardiac myofibroblasts via Ca2+ influx (29–31). It has been reported that TRPV4 initiates the acute calcium-dependent vascular permeability in pulmonary edema that develops upon ventilator-induced lung injury or by elevated pulmonary venous pressure associated with heart failure in mice (32, 33). However, TRPV4 has not been previously shown to be involved in lung myofibroblast differentiation or fibrosis, and the molecular mechanism of its involvement in this process has not been determined. In the present study, we tested the hypothesis that the TRPV4 channel plays a role in lung myofibroblast differentiation and in the generation of lung fibrosis in mice. Our work reveals a novel role for TRPV4 in pulmonary fibrogenesis and for TRPV4-mediated Ca2+ influx in myofibroblast differentiation.
Results
Trpv4 KO mice are protected from the profibrotic effects of bleomycin.
To investigate the role of TRPV4 on pulmonary fibrogenesis in vivo, the effect of bleomycin was studied in Trpv4 KO mice. The Trpv4 KO mice were significantly protected from bleomycin-induced lung fibrosis. Trpv4 KO mice had less lung collagen accumulation after bleomycin (by 75% in mice given 4 U/kg bleomycin, day 14, n ≥ 5 per group, P < 0.05), as quantitated biochemically by measuring hydroxyproline levels (Figure 1A), by measuring collagen-1 in the lung tissue extracts by immunoblotting (n > 5 per group, P < 0.05) (Figure 1B), or by trichrome staining of lung tissue sections (P < 0.01 for WT vs. Trpv4 KO; n = 5 per group) (Figure 1C), as compared with the congenic WT mice. Further, the Trpv4 KO mice had a smaller decrease in lung compliance (by 57% in mice given 4 U/kg bleomycin, day 14, P < 0.05; n = 5 per group) after bleomycin instillation, as compared with the WT mice (Figure 1D). Bleomycin-instilled Trpv4 KO mice showed less myofibroblast differentiation compared with the WT mice, as revealed by immunoblotting analysis of lung tissue extracts for α-SMA (n > 5 per group, P < 0.05) (Figure 1E). Importantly, the lung histology and compliance in saline-instilled WT and Trpv4 KO mice were indistinguishable. Furthermore, we saw no difference in the total wbc counts or cell differentials in lung lavages from WT mice, as compared with those from Trpv4 KO mice that received bleomycin 7 days previously (WT vs. KO, 1,122 ± 258 × 103 total cells/ml vs. 1,000 ± 322 × 103 total cells/ml, n ≥ 5 per group), suggesting that differences in early inflammation do not account for the differences in the later fibrosis (Figure 1F). Given the high mortality (WT, 60% vs. KO, 15%; P < 0.05; number of mice at the start of the experiment: 17 WT and 13 Trpv4 KO; number of mice analyzed: 5 WT and 9 Trpv4 KO) at the 4 U/kg dose and the possibility of a confounding survivor effect, experiments were repeated using a 1 U/kg dose of bleomycin. As with the high dose (4 U/kg) of bleomycin, the Trpv4 KO mice receiving the lower dose had a smaller increase in lung hydroxyproline content (WT, 203% ± 67% vs. KO, 143% ± 44% of WT intratracheal [IT] saline; P < 0.004; n = 5 per group; Figure 1G) and a 2.5-fold less impairment in lung compliance (WT, 69% ± 7% vs. KO, 88% ± 4% of IT saline, which was set as 100%; P = 0.031; n = 5 per group; Figure 1H), with no mortality. Collectively, these data clearly demonstrate that the Trpv4 KO mice are protected from the profibrotic effects of bleomycin.
Figure 1. Trpv4 KO mice are protected from the profibrotic effects of bleomycin.

WT or Trpv4 KO mice were instilled with (A–F) 4 U/kg or (G and H) 1 U/kg bleomycin or saline. (A) Hydroxyproline content in the lungs of WT and Trpv4 KO mice (day 14; n ≥ 5 per group, *P < 0.05). (B) Representative immunoblots of total lung protein lysates show reduced expression of collagen-1. Bar graphs show collagen-1 band density normalized to GAPDH. *P < 0.05, WT vs. Trpv4 KO; n > 5 per group. (C) Representative photomicrographs of trichrome-stained lung tissue (original magnification, ×10). The bar graph shows the percentage of fibrotic area. **P < 0.01, WT vs. Trpv4 KO; n = 5 per group. (D) Static lung compliance (Cst) was measured using the FlexiVent (day 14, static P-V loop; *P < 0.05, WT vs. Trpv4 KO; n = 5 per group). (E) Representative immunoblots of total lung protein lysates show reduced expression of α-SMA protein. The bar graph shows α-SMA band density normalized to GAPDH. *P < 0.05; n > 5 per group. (F) Cells counts and differentials from lung lavage from mice given bleomycin (4 U/kg, day 7, n ≥ 5 per group). Macs, macrophages; PMNs, polymorphonuclear leucocytes; Lymphs, lymphocytes. (G) Hydroxyproline as in A. WT, 203% ± 67% vs. KO, 143% ± 44% of WT IT saline; *P < 0.004; n = 5 per group. (H) Lung compliance as in D. *P < 0.05, WT vs. Trpv4 KO mice; n = 5 per group. (A, F, and G) Results are expressed as mean ± SD. (B–E and H) Results are expressed as mean ± SEM.
TRPV4 is expressed and functional in both primary human and murine lung fibroblasts.
To determine the potential role of TRPV4 in lung myofibroblast differentiation, we first examined its expression in normal lung fibroblasts and those derived from patients with IPF. Immunoblotting analysis detected expression of TRPV4 protein in the lung fibroblasts from both normal control subjects and patients with IPF (Figure 2A). However, there were no differences in total cellular TRPV4 protein expression among normal lung fibroblasts and those derived from patients with IPF, stimulated or unstimulated with TGF-β1. To test whether TRPV4 channel activation induces Ca2+ influx (cytosolic calcium increases) in human lung fibroblasts (HLFs; 19Lu cells) we measured its specific agonist-induced Ca2+ influx. We noted a rapid (within 1 to 2 minutes) increase in intracellular Ca2+ influx in HLFs in response to the selective TRPV4 agonist, GSK1016790A (GSK; ref. 34), and its concentration-dependent inhibition by pretreatment with the selective TRPV4 antagonist, RN-1734 (AB1; ref. 35 and Figure 2, B and C). To further confirm the existence of functional TRPV4 channel in lung fibroblasts, we compared Ca2+ influx in primary lung fibroblasts from Trpv4 KO and congenic WT mice, using a range of concentrations of GSK. TRPV4 agonist–induced (GSK-induced) Ca2+ influx was completely absent in Trpv4 KO fibroblasts, even in the presence of 30-fold excess of GSK (EC50 = 10 nM) (Figure 2D), demonstrating the absence of redundant GSK-responsive Ca2+ influx channels in lung fibroblasts. To substantiate the IPF disease relevance of TRPV4, we showed that TRPV4-mediated Ca2+ influx is 2-fold greater in fibroblasts derived from patients with IPF compared with that in normal lung fibroblasts (Figure 2E; P < 0.05 for IPF vs. normal lung fibroblasts; n = 5 per group). Altogether, these results demonstrate that both HLFs and murine lung fibroblasts express functional TRPV4 channels capable of inducing a rise in intracellular calcium.
Figure 2. TRPV4 calcium channel is expressed and functional in HLFs and murine lung fibroblasts.

(A) Representative immunoblots of TRPV4 protein (~88 kDa) and GAPDH loading control in normal and IPF lung fibroblast lysates with or without TGF-β1 (0, 2, and 10 ng/ml, 48 hours). n = 6 per group. (B) Single cell recording of fura-2 dye–loaded normal HLFs shows that TRPV4 agonist GSK induces Ca2+ influx, which is abrogate d by its antagonist, AB1. (C) Concentration-dependent inhibition of TRPV4 activity (Ca2+ influx) by AB1. Ca2+ influx is shown by relative fluorescence units (RFUs) measuring ΔF/F (max-min), using Calcium 5 dye on intact fibroblast monolayers (FlexStation 3, Molecular Devices). Results are expressed as mean ± SEM. *P < 0.05 compared with no AB1 conditions by ANOVA. (D) Loss of GSK-inducible Ca2+ influx in Trpv4 KO mouse lung fibroblasts. Fibroblast monolayers were incubated with the indicated concentration of GSK, and Ca2+ influx was measured as in C. All experiments were repeated ≥3 times in quadruplicate. Results are expressed as mean ± SEM. (E) Augmentation of GSK-inducible Ca2+ influx in lung fibroblasts from patients with IPF (IPF) compared with normal lung fibroblasts (NL). Fibroblast monolayers were incubated with GSK (10 nM), and Ca2+ influx was measured as in C. Results are expressed as mean ± SEM. *P < 0.05 for IPF vs. NL; n = 5 per group.
TRPV4 is required for TGF-β1–induced lung myofibroblast differentiation.
TRPV4 has recently been linked to the differentiation of chondrocytes, osteoclasts, and cardiac myofibroblasts (29–31). To determine whether TRPV4 contributes to lung myofibroblast differentiation, we performed knockdown of TRPV4 in HLFs with TRPV4 siRNA before induction of myofibroblast differentiation by treatment with TGF-β1. TRPV4 siRNA reduced TRPV4 protein level by >70% (20 and 50 nM TRPV4 siRNA; n = 3, P < 0.05) in HLFs compared with that in scrambled siRNA-treated cells (Figure 3, A and B). This knockdown of TRPV4 protein attenuated TGF-β1–induced myofibroblast differentiation by greater than 50% (50 nM TRPV4 siRNA; n = 3, P < 0.05) compared with scrambled siRNA (Figure 3C), as measured by total cell α-SMA protein levels. To further confirm the requirement of TRPV4 in lung myofibroblast differentiation by a second independent method, we compared TGF-β1–induced myofibroblast differentiation in primary lung fibroblasts from Trpv4 KO and congenic WT mice. TGF-β1–induced lung myofibroblast differentiation was reduced by 60% (n > 18 cells per group; P < 0.01 by ANOVA for WT vs. Trpv4 KO cells treated with TGF-β1) in Trpv4 KO fibroblasts compared with those from WT mice, as examined by α-SMA incorporation into stress fibers (Figure 3, D and E). Furthermore, reconstitution of TRPV4 in Trpv4 KO cells, using a lentivirus expression system, restored the myofibroblast differentiation defect seen in the Trpv4 KO fibroblasts (80% vs. <4%; P < 0.01; n = 20 cells per condition) (Figure 3F). To investigate the role of TRPV4 in regulating IPF fibroblast function, we determined the relative importance of TRPV4 to normal and IPF patient–derived myofibroblast differentiation. Upon using the TRPV4 antagonist, AB1, we noted that TRPV4 activity was a greater determinant of myofibroblast differentiation in fibroblasts derived from patients with IPF than in those from normal lung fibroblasts (Figure 3, G and H; n = 5 per group, P < 0.05). Taken together, these results confirm that TGF-β1–induced lung myofibroblast differentiation is dependent on TRPV4 and that fibroblasts derived from patients with IPF exhibit a greater dependency of myofibroblast differentiation on TRPV4 than those from normal lung fibroblasts.
Figure 3. TRPV4 is required for TGF-β1–induced lung myofibroblast differentiation.

HLFs were plated on fibronectin-coated (10 μg/ml) plastic wells and incubated with or without TGF-β1 (2 ng/ml, 24 hours), TRPV4 siRNA, or scrambled siRNA. (A) Representative immunoblots show knockdown of TRPV4 proteins by TRPV4-specific siRNA and blocking of TGF-β1–induced α-SMA expression under conditions of TRPV4 knockdown. (B and C) Quantification of (B) TRPV4/GAPDH and (C) α-SMA/GAPDH protein bands from A. *P < 0.05 scrambled vs. TRPV4 siRNA-treated cells, #P < 0.05 TGF-β1–treated cells treated with scrambled siRNA vs. TRPV4 siRNA; n = 3. (D) Representative fluorescence micrographs (original magnification, ×20). Myofibroblast differentiation is reduced in fibroblasts from Trpv4 KO mice (colocalization of α-SMA and F-actin, orange). (E) Quantification of results from D by Pearson’s coefficient analysis. **P < 0.01; TGF-β1–treated WT vs. Trpv4 KO cells; n > 18 cells per group. UT, untreated. (F) Reconstitution of TRPV4 into Trpv4 KO mouse lung fibroblasts (MLFs) using a lentivirus expression system (lenti-TRPV4-GFP) restores myofibroblast differentiation in response to TGF-β1. Lenti-GFP–infected Trpv4 KO mouse lung fibroblasts were used as negative control; uninfected WT mouse lung fibroblasts were used as positive control. Original magnification, ×20. (G) TRPV4 blockade has a greater inhibitory effect on myofibroblast differentiation (α-SMA/GAPDH band density in immunoblots) in fibroblasts from patients with IPF than in normal fibroblasts. (H) Quantitation of results from G. *P < 0.05; n = 5 per group. Results are expressed as mean ± SEM.
Blocking of TRPV4 channel activity by selective small-molecule antagonists abrogates Ca2+ influx and TGF-β1–induced myofibroblast differentiation.
To investigate the effect of pharmacologic inhibition of TRPV4 channel activity (Ca2+ influx) on TGF-β1–induced lung myofibroblast differentiation, we first examined whether the presence of extracellular calcium is required for the TRPV4-dependent rise in intracellular calcium. As expected from work in other cell types, the TRPV4-mediated rise in intracellular calcium was absolutely dependent on the presence of external calcium in HLFs (Figure 4A). Recently, it has been reported that TGF-β1 induces a calcium wave in HLFs that is dependent on both an external influx of calcium and the release of calcium from internal pools (36). However, blocking of known regulators of intracellular calcium stores, including the inositol 1,4,5-trisphosphate receptor, ryanodine receptors, and the sarco/endoplasmic reticulum calcium transport ATPase, with selective small-molecule inhibitors (xestospongin C for inositol 1,4,5-trisphosphate receptor, ryanodine for ryanodine receptors, and cyclopiazonic acid for sarco/endoplasmic reticulum calcium transport ATPase) had no effect on the TRPV4-dependent (GSK-induced) intracellular Ca2+ rise (Figure 4B). These results indicate that the mobilization of internally sequestered Ca2+ is not necessary for TRPV4-dependent Ca2+ influx in HLFs. Similarly, in order to test the requirement of extracellular calcium in the TGF-β1–induced differentiation of fibroblasts, we quantified myofibroblast generation under conditions of the presence or absence of extracellular calcium. HLFs showed an absolute requirement for extracellular calcium to differentiate into myofibroblasts, as assessed by multiple measures of myofibroblast cytoskeletal remodeling (Figure 4, C and D). Concordantly, blockade of TRPV4 channel activity by its selective antagonist, AB1, abrogated TGF-β1–induced myofibroblast differentiation by approximately 80% compared with untreated controls in a concentration-dependent manner, as revealed by α-SMA protein expression (Figure 5A), and by incorporation of α-SMA in stress fibers (Figure 5B), as quantified using Pearson’s coefficient (Figure 5C; n > 18 cells per condition, P < 0.01 for TGF-β1–treated cells with vs. without AB1). We also found similar concentration-dependent results using a second structurally distinct, highly selective small-molecule TRPV4 antagonist, HC-067047 (HC) (37). HC suppressed both TRPV4 agonist-induced Ca2+ influx (IC50 = 300 nM) (Figure 5D) and TGF-β1–induced myofibroblast differentiation in a concentration-dependent manner (IC50 = 30 nM; Figure 5E), as quantified using Pearson’s coefficient (Figure 5F; n > 18 cells per condition, P < 0.01 for TGF-β1–treated cells with vs. without HC). We also assessed other key functional properties of myofibroblasts by examining TGF-β1–induced collagen-1 production. Results show that collagen-1 production in lung fibroblasts was abrogated by greater than 50% (P < 0.05 for TGF-β1–treated cells with vs. without AB1) upon blockade of TRPV4 with AB1 (Figure 5, G and H). Taken together (Figures 4 and 5), these results suggest that TRPV4-dependent Ca2+ influx potentiates the TGF-β1 signal and thereby modulates TGF-β1–induced myofibroblast differentiation.
Figure 4. TRPV4-dependent Ca2+ influx from the extracellular space mediates TGF-β1–induced myofibroblast differentiation.

(A) Extracellular calcium is required for TRPV4-mediated intracellular calcium rise. HLF monolayers were compared for their Ca2+ influx in response to the indicated concentrations of GSK, as in Figure 2C, with or without extracellular calcium. A23187 (2 μM), a calcium ionophore, was used as a positive control. Ca2+ influx for each condition is shown by the respective red arrow. (B) TRPV4-dependent calcium rise is independent of intracellular pools and/or regulators of calcium. HLF monolayers were compared for their intracellular calcium rise after TRPV4 activation (GSK, 10 nM) with or without selective inhibitors (30 minutes of pretreatment before addition of GSK) to the inositol 1,4,5-trisphosphate receptor (xestospongin C [Xesto], 10 μM), ryanodine receptors (ryanodine [RyD], 10 μM), or sarco/endoplasmic reticulum calcium transport ATPase (cyclopiazonic acid [CPA], 10 μM). Results are expressed as mean ± SEM. (C) Representative photomicrographs. Extracellular calcium is required for myofibroblast differentiation, as measured by intracellular remodeling of F-actin and α-SMA by immunofluorescence. Original magnification, ×20. (D) Quantification of photomicrographs from C using ImageJ software. Results are expressed as mean ± SEM. *P < 0.001 for TGF-β1–treated cells with vs. without calcium; n > 10 cells per condition, 1-way ANOVA.
Figure 5. TRPV4 small-molecule antagonists abrogate TGF-β1–induced myofibroblast differentiation in a concentration-dependent manner.

(A) TRPV4 blockade reduces TGF-β1–induced expression of α-SMA protein in a concentration-dependent manner, without inhibiting total TRPV4 protein level. n = 3. (B) TRPV4 antagonist, AB1 (50 μM), blocks TGF-β1–induced myofibroblast differentiation. Representative photomicrographic merged images of α-SMA (green) and F-actin (red). UT, no TGF-β1 treatment. Original magnification, ×20. (C) Quantitation of results from B by Pearson’s coefficient. **P < 0.001 for TGF-β1–treated cells with vs. without AB1; n > 18 cells per condition. (D) HC inhibits Ca2+ influx in a concentration-dependent manner. Mouse lung fibroblast monolayers from WT mice were examined for their Ca2+ influx response to GSK (10 nM) with or without selective inhibitor HC at the indicated concentrations, using similar methods as in Figure 2C. All data were repeated more than 3 times in quadruplicate. (E) Representative photomicrographic images as in B, with or without indicated HC concentrations. Original magnification, ×20. (F) Quantitation of results from E by Pearson’s coefficient. **P < 0.01 for TGF-β1–treated cells with vs. without HC; n > 18 cells per condition. (G) AB1 (50 μM) inhibits collagen-1 production in HLFs by collagen/GAPDH protein band density in immunoblots. (H) Quantitation of results from G. *P < 0.05 for TGF-β1–treated cells with vs. without AB1. Results are expressed as mean ± SEM.
Increased matrix stiffness potentiates TRPV4-induced calcium influx and myofibroblast differentiation.
Extracellular matrix deposition, with subsequent stiffening of the tissue microenvironment, is recognized as a critical inducer of myofibroblast differentiation during the fibrotic process (9–14). However, precisely how the matrix stiffness signal is transduced intracellularly and the molecular mechanism by which TRPV4 participates in myofibroblast differentiation has remained elusive. We found that TRPV4 channel activity (as measured by the response in agonist-induced Ca2+ influx) was increased when cells were plated on stiffer matrices within the pathophysiological range (Figure 6A). In addition, TGF-β1 had a greater enhancing effect on myofibroblast differentiation in fibroblasts plated on stiffer matrices, as measured by α-SMA integrated density (Figure 6B). Importantly, the TRPV4 blockade selectively abrogated this stiff matrix response (Figure 6B).
Figure 6. Increased matrix stiffness augments TRPV4-induced Ca2+ influx and myofibroblast differentiation.

(A and B) HLFs were plated (15,000 cells per well) on collagen-coated (100 μg/ml) hydrogels with varying degrees of stiffness (1, 6, 8, and 25 kPa) under vehicle-treated or AB1-treated (50 μM) conditions, with or without TGF-β1 (2 ng/ml, 24 hours). (A) GSK-induced (10 nM) Ca2+ influx, measured as in Figure 2C, is increased by stiffness and abrogated by AB1. (B) Fluorescence microscopy analysis shows that the TGF-β1 response in myofibroblast differentiation seen with increasing stiffness of the matrix is dependent on TRPV4 channel activity. *P < 0.05, **P < 0.01 by ANOVA; n > 12 cells per group. Int., integrated. (C and D) HLFs were plated on normal or stiffer fibrotic lung tissue sections with or without TRPV4 antagonist, AB1 (50 μM). (C) Representative confocal micrographs (original magnification, ×63) of phalloidin-stained F-actin (red). (D) Quantification of actin stress fiber density from C. The results are expressed as mean ± SD. *P < 0.05 by ANOVA for vehicle-treated normal lung vs. fibrotic lung; †P < 0.05 by ANOVA for vehicle-treated vs. AB1-treated fibrotic lung.
To understand the pathophysiological relevance of our observations to fibrotic lung disease, we developed a model system to test the interaction of fibroblasts with actual normal and fibrotic lung tissue (38). The contribution of TRPV4 to myofibroblast differentiation was studied using fibroblasts plated on unfixed normal and fibrotic lung tissue sections from bleomycin-treated WT mice. Myofibroblast differentiation was enhanced in fibroblasts maintained on fibrotic lung tissue compared with those plated on normal lung tissue (Figure 6, C and D). Interestingly, inhibition of TRPV4 function reduced myofibroblast differentiation (as measured by F-actin stress fiber density) selectively on fibrotic lung tissue (Figure 6, C and D). Taken together, these data demonstrate for the first time that TRPV4 mediates the lung myofibroblast differentiation response to matrix stiffness in two pathophysiologically relevant model systems.
TRPV4 activity modulates TGF-β1 actions in a SMAD-independent manner.
Since the profibrotic functions of TGF-β1 are regulated by both SMAD-dependent and SMAD-independent signals, we examined whether TRPV4 activity modulates TGF-β1–induced activation of SMAD2/3 in HLFs. We found that TRPV4-mediated Ca2+ influx in HLFs was insensitive to treatment with SD208, a selective inhibitor of the TGF-βRI kinase (Figure 7A). We also observed that, while TGF-β1 caused a rapid increase in phosphorylation/activation of SMAD2/3 as expected, this response was not inhibited by the TRPV4 antagonist, AB1 (Figure 7B). These results demonstrate that TRPV4 activity regulates TGF-β1–dependent myofibroblast differentiation independent of SMAD2/3 activation.
Figure 7. TRPV4 activity potentiates TGF-β1 actions in a SMAD2/3-independent manner.

(A) TRPV4-mediated Ca2+ influx in HLFs (measured as in Figure 2C) is not blocked by supraphysiologic concentrations of the TGF-βRI kinase inhibitor, SD208. Results are expressed as mean ± SEM. (B) Immunoblots of fibroblast cell lysates, treated as indicated, show no inhibition of TGF-β1–induced phosphorylation of SMAD2/3 or of expression of total TRPV4 proteins by the TRPV4 antagonist, AB1. Total SMAD2/3 was used as loading control. The experiments were repeated 2 times.
TRPV4 activity potentiates TGF-β1–induced actomyosin remodeling.
Recent data demonstrate that the TGF-β1 signal induces the polymerization of actin monomers, whereupon the myocardin-related transcription factor (MRTF-A) is released and then binds to serum response factor coactivator, and the complex translocates to the nucleus to induce α-SMA gene transcription (39, 40). As we showed that actin polymerization (F-actin, stress fiber formation) is dependent on TRPV4, we investigated whether TRPV4 potentiates TGF-β1–induced upregulation of α-SMA expression via modulation of MRTF-A translocation to nucleus. Antagonism of TRPV4 by AB1 lead to an inhibition of TGF-β1–induced actin polymerization (F-actin, ~70% inhibition, compared with untreated controls) (Figure 8, A and B). Furthermore, the downstream MRTF-A translocation to the nucleus was similarly inhibited (~60% of that of the positive control polymerization inhibitor, latrunculin B) (Figure 8, C and D). Similarly, KO of Trpv4 completely abrogated (P < 0.05 for TGF-β1 WT cells vs. Trpv4 KO; n = 3 per condition) the MRTF-A nuclear translocation seen in response to TGF-β1 in the WT fibroblasts (Figure 8, E and F). RhoA/ROCK pathway has been implicated in MRTF-A nuclear translocation and in mechanical stiffness and TGF-β1–induced myofibroblast differentiation (12, 41), thus, we measured TGF-β1–induced RhoA activation under TRPV4-inhibited conditions. TGF-β1–induced activation of RhoA was completely abrogated (n = 3, P < 0.05, Student’s t test) upon pharmacological inhibition of TRPV4 (Figure 8G). TRPV4 inhibition by the antagonist, AB1, similarly attenuated TGF-β1–induced myosin activation, as revealed by inhibition of phosphorylation of myosin regulatory light chain (MLC2) in HLFs (Figure 8H). Altogether, these molecular gain- and loss-of-function data put TRPV4 upstream of RhoA and MRTF-A nuclear translocation in the myofibroblast differentiation pathway.
Figure 8. Inhibition of TRPV4 channel activity abrogates molecular signaling essential for myofibroblast differentiation.

(A–D) HLFs were incubated with or without TGF-β1 (2 ng/ml, 48 hours) as well as with or without indicated TRPV4 inhibitor (AB1) or with or without inhibitor (latrunculin B [Lat-B], 500 nM) or activator (jasplakinolide [Jak], 100 nM) of actin polymerization, as indicated. n = 3. Results are expressed as mean ± SEM. (A) Immunoblots of cell lysates were separated into F-actin (filamentous) and G-actin (monomeric) fractions, which show abrogation of F-actin formation upon TRPV4 inhibition by AB1. (B) Quantification of results in A. *P < 0.05 for TGF-β1–treated cells with or without AB1. (C) MRTF-A nuclear translocation is abrogated by TRPV4 inhibition. Nuclear and cytoplasmic fractions were separated, and an equal amount of protein per lane was immunoblotted for MRTF-A, a nuclear marker (lamin A/C), and a cytoplasmic marker (14-3-3B). (D) Quantification of results from C. *P < 0.05 for TGF-β1–treated cells with vs. without AB1. (E) As in C, immunoblots show MRTF-A nuclear translocation is abrogated in Trpv4 KO mouse lung fibroblasts. (F) Quantification of results from E. Results are expressed as mean ± SEM. *P < 0.05 for TGF-β1 WT cells vs. Trpv4 KO cells (n = 3). (G) G-LISA RhoA activation assay shows inhibition of TGF-β1–induced activation of RhoA by TRPV4 antagonist, AB1. *P < 0.05 for TGF-β1–treated cells with or without AB1 at 10 minutes; n = 3. Results are expressed as mean ± SEM. (H) HLFs were pretreated with or without AB1 (20 μM) followed by TGF-β1 (2 ng/ml) for indicated times. Sonicated cell lysates (n = 2) were immunoblotted for total MLC2 or activated MLC2 (p-MLC2).
Discussion
The major findings of this study are as follows: (a) Trpv4 KO mice are protected from lung fibrosis, (b) functional TRPV4 channel is expressed in primary human and murine lung fibroblasts, (c) TRPV4-dependent Ca2+ influx potentiates TGF-β1–induced lung myofibroblast differentiation under conditions of pathophysiological range stiffness seen in fibrotic lung tissue, and (d) TRPV4 activity is upregulated in IPF fibroblasts and is more of a determinant of myofibroblast differentiation in IPF fibroblasts than it is in normal lung fibroblasts. Absolute TRPV4 dependence of myofibroblast differentiation was shown by both genetic ablation and pharmacological inhibition of TRPV4 function. Furthermore, pathophysiologically relevant matrix stiffness sensitizes both TRPV4 function and myofibroblast differentiation. We provide evidence that cytoskeletal remodeling and consequent release and nuclear translocation of the α-SMA transcription factor, MRTF-A, is downstream of TRPV4 action.
Given its ubiquitous cell type and tissue expression, its sensitivity to multiple stimuli, and the existence of other family members, it is surprising that data are emerging that mice lacking only TRPV4 have altered pressure sensation, vasodilatory responses, osmosensing, neurological function, and bone development and are more susceptible to either high vascular pressure or high volume ventilation–induced acute lung edema (22–29, 32, 33). Furthermore, human diseases, including skeletal and nerve disorders, have been ascribed to mutations in TRPV4 (26–28). A recently described SNP near the TRPV4 gene has been implicated in chronic obstructive pulmonary disease (42). However, the role of TRPV4 in normal fibroblast homeostasis or lung fibrosis has not been explored until now. Using a bleomycin mouse model of human IPF, we demonstrate that TRPV4 deficiency protects mice from fibrosis at the biochemical, histological and physiological levels. These results implicate TRPV4 in the development of lung fibrosis for the first time. Furthermore, the lack of difference in the early inflammatory response, as measured by cell differentials, suggests that the fibrosis-protecting effect of TRPV4 loss is a function of tissue repair.
There are emerging data supporting a role for Ca2+-initiated signals in myofibroblast differentiation and function; however, neither the identity of the calcium channel involved nor the mechanisms by which Ca2+ signals promote these effects have been fully elucidated (36, 43). We show that extracellular calcium is specifically required for myofibroblast differentiation and that the intracellular calcium rise upon TRPV4 activation is solely dependent on TRPV4-mediated extracellular Ca2+ influx. We demonstrate these findings by the loss of TRPV4-dependent calcium signal in the absence of extracellular calcium and the lack of effect of ER calcium pumps and receptors on calcium rise noted after TRPV4 activation. Recently, it has been demonstrated that both external influx of Ca2+ and the release of Ca2+ from internal sources following activation of the inositol 1,4,5-trisphosphate receptor, ryanodine receptors, or sarco/endoplasmic reticulum calcium transport ATPase contribute to TGF-β1–induced calcium wave activity in HLFs (36). The reported TGF-β calcium signal is oscillatory and dependent on ER pumps and receptors (36) and on TGF-βR kinase activity. In contrast, in the present study, the observed TRPV4 calcium signal is uniphasic, persistent (>20 minutes), and independent of ER pumps and receptors and of TGF-βR kinase activity. Thus, these data suggest that there is no direct contribution from the TGF-β–generated calcium or kinase signal on the TRPV4-initiated calcium signal.
In the current study, we demonstrate that TRPV4 proteins are expressed in a functional manner and potentiate TGF-β1–induced myofibroblast differentiation in primary human and murine lung fibroblasts. A recent report demonstrated that TRPV4 mediates TGF-β1 matrix stiffness–induced differentiation of cardiac fibroblasts, but the molecular mechanism by which TRPV4 participates in myofibroblast differentiation was not shown (31). Importantly, HLFs with inhibited TRPV4 function (by siRNA and two distinct pharmacological antagonists) or primary lung fibroblasts isolated from Trpv4 null mice fail to undergo TGF-β1–induced myofibroblast differentiation, clearly demonstrating an absolute requirement for TRPV4 action in this process.
Although both a mechanical signal, likely due to the internally generated cell tension pulling on a stiff matrix (44, 45), and a TGF-β1–initiated signal are essential for optimal myofibroblast differentiation, the actual molecular pathway by which the mechanical signal is transduced is not precisely known. Interestingly, it has been reported that TRPV4 is activated by cyclical stretching of endothelial cell surface or by mechanical forces applied to cell surface β1 integrins in endothelial cells (45, 46). Furthermore, TRPV4 mediates the directional migration of endothelial cells noted upon local matrix compression (46). Here, we investigated whether TRPV4 will integrate mechanical (matrix stiffness) and soluble (TGF-β1) signals to mediate lung myofibroblast differentiation, using collagen-coated polyacrylamide gels of varying degrees of stiffness (47), in a model system we developed using actual lung tissue sections (38). We found that the TRPV4 channel activity (agonist-induced Ca2+ influx) is potentiated when cells are plated on stiffer matrices within the pathophysiological range seen in diseased/fibrotic lung tissue. Furthermore, the inhibition of TRPV4 channel activity significantly abrogated TGF-β1–induced enhancement of myofibroblast differentiation selectively under higher stiffness conditions. Concordantly, inhibition of TRPV4 activity reduces myofibroblast differentiation selectively on fibrotic lung. It has been increasingly recognized that the matrix stiffness acts as an important modulator of myofibroblast differentiation, whose molecular pathway has yet to be fully elucidated (11–14). These data demonstrate the sensitivity of lung myofibroblast differentiation to TRPV4 channel activity for the first time. Further, these data show that TRPV4 channel activity in lung fibroblasts is potentiated by pathophysiologic range matrix stiffness as well as by fibrotic lung tissue in a disease-relevant model system.
Recently, it has been reported that TGF-β1–induced actin polymerization facilitates MRTF-A release from actin monomers, thereby allowing for its binding to serum response factor and translocation into the nucleus to induce α-SMA gene transcription during lung myofibroblast differentiation (39, 40). Inhibition of TRPV4 activity abrogated the extensive actin polymerization otherwise noted upon TGF-β1–induced myofibroblast differentiation. This was shown using both qualitative (i.e., assembly of stress fibers) and quantitative (i.e., filamentous [F-actin] and monomeric [G-actin] actin) measures of actin polymerization. Furthermore, inhibition of TRPV4 activity attenuates TGF-β1–induced nuclear translocation of MRTF-A, similar to that seen upon inhibition of actin polymerization with latrunculin B. Inhibition of TRPV4 function also impairs activation of RhoA in response to TGF-β. Since RhoA has been implicated previously in stiffness-induced differentiation of fibroblasts (12), and in nuclear translocation of MRTF-A (41), our results underscore the importance of TRPV4 as a key initiator of a mechanosensitive pathway. Since calcium-regulated signaling plays a critical role during actin remodeling and we show a strict dependence of actin remodeling on extracellular calcium, it is likely that the mechanism whereby TRPV4 action mediates TGF-β1–induced myofibroblast differentiation is through calcium-triggered actin polymerization-dependent nuclear translocation of MRTF-A. As there are a number of actomyosin-remodeling proteins whose function is calcium dependent (e.g., gelsolin, cofilin, and myosin) (43), we speculate that the actomyosin machinery system is the critical downstream calcium response for myofibroblast differentiation.
While we have put forth several complementary approaches that convincingly demonstrate that TRPV4 is a key mechanosensor for myofibroblast differentiation and is altered in IPF fibroblasts, there are limitations. We show that TRPV4 mediates myofibroblast differentiation in vitro and in vivo through a mechanosensing RhoA/MRTF-A axis. However, it remains possible that TRPV4’s critical fibrosis-inducing effect(s) occurs through other cells or other pathways. Although there is reasonable evidence suggesting that resident mesenchymal cells are the predominant source of myofibroblasts in the bleomycin model, cell types, including bone marrow–derived cells, epithelial cells, endothelial cells, and pericytes, may differentiate into myofibroblasts (48–53). As TRPV4 is not constitutively active in our system, its pathogenic functions are demonstrable by its sensitization by TGF-β, matrix stiffness, and in IPF disease and its physiological relevance to myofibroblast differentiation (29). As we show evidence that TRPV4-mediated calcium influx does not affect TGF-β–induced SMAD phosphorylation, the interface by which TGF-β signaling is modified by TRPV4 may be through noncanonical pathways (i.e., PI3K, MAPK) or downstream of SMAD2/3 phosphorylation (i.e., SMAD localization, NFAT activation).
In summary, as shown in the proposed schematic model (Figure 9), our study reveals for the first time a novel role of TRPV4 in lung fibrogenesis in vitro and in vivo. We show that TRPV4 activity modulates TGF-β1 actions in a SMAD-independent manner and mediates enhanced actomyosin remodeling. Our data suggest that TRPV4 is sensitized through cell-matrix interactions with stiff matrices within the pathophysiological range. TRPV4 channel activity and its biological effects are upregulated in lung fibroblasts derived from patients with IPF. This work may lead to a novel therapeutic approach to the treatment of IPF, as TRP channel-targeted small-molecule inhibitors are currently in developmental/preclinical or early-phase clinical trials for other disorders (54). Furthermore, as myofibroblasts are a pathogenic cell in fibrotic disorders of the liver, heart, skin, and kidney, this work will potentially have impact beyond lung fibrotic diseases.
Figure 9. Schematic model showing the mechanistic pathway by which TRPV4 mediates myofibroblast differentiation and pulmonary fibrosis.
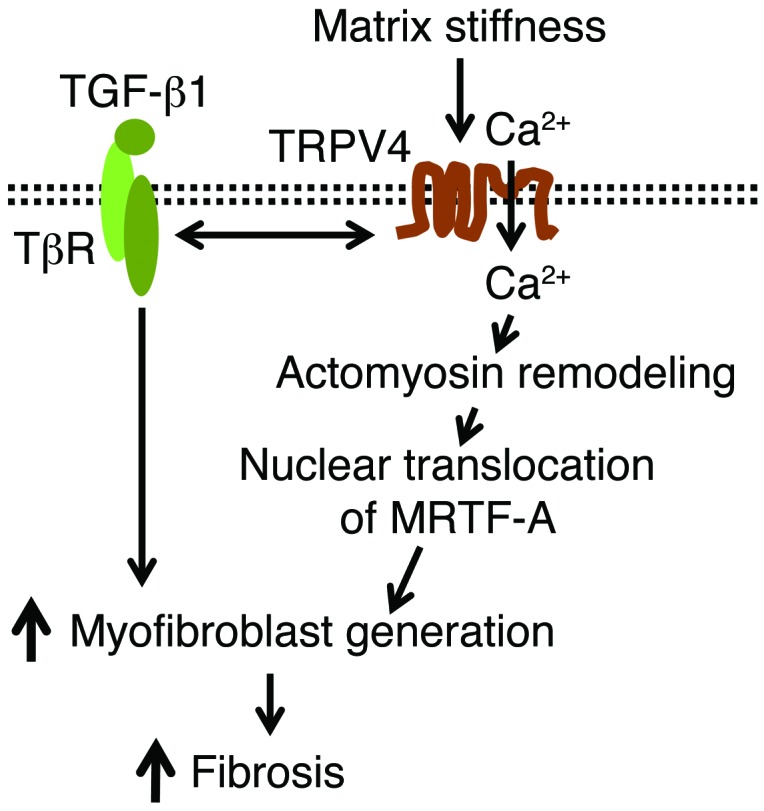
Our data suggest that TRPV4-dependent Ca2+ influx activity is sensitized by stiff matrices within the pathophysiological range. Interaction between TRPV4 activity (Ca2+ influx) and the profibrotic TGF-β1 signals promote nuclear localization of α-SMA transcription factor, MRTF-A, via regulation of actomyosin remodeling to potentiate myofibroblast differentiation during fibrogenesis. TβR, TGF-βR.
Methods
Antibodies and reagents.
The following antibodies were obtained as follows: anti–α-SMA (Sigma-Aldrich); anti-TRPV4 (Alomone Labs); anti–p-MLC2, anti-MLC2, anti-SMAD2/3, and anti–p-SMAD2/3 (Cell Signaling); anti–MRTF-A and anti–14-3-3B (Santa Cruz); anti–collagen-1 and anti-LaminA/C (BD Transduction Laboratories); anti–Collagen-1 (Millipore); and anti-GAPDH (Research Diagnostics). TGF-β1 was purchased from R&D Systems. HC was provided by Hydra Biosciences Inc., RN1734 (AB159908 or AB1) was obtained from ABCR GmbH, and GSK1016790A (GSK101 or GSK) was purchased from Sigma-Aldrich. Alexa Fluor-phalloidin was obtained from Invitrogen and latrunculin B was purchased from EMD Biosciences. SD208 was purchased from Tocris Bioscience. Cyclopiazonic acid, ryanodine, and xestospongin C were purchased from Sigma-Aldrich. The G-LISA RhoA Activation Assay Biochem Kit was purchased from Cytoskeleton Inc. All other chemicals were purchased from Sigma-Aldrich and Fisher Scientific.
Bleomycin-induced pulmonary fibrosis model.
Induction of pulmonary fibrosis in Trpv4 null mice (generated by Mizuno et al.) (55, 56) and age-matched female congenic WT C57BL/6 mice was done by bleomycin (4 U/kg or 1 U/kg) or phosphate-buffered saline (as a control) injection via IT instillation as previously described (57). Fourteen days after bleomycin treatment, the lungs were inflated with OCT (Sakura Finetek) and sectioned for staining with trichrome. Static compliance measurements were done using an animal ventilator (Scireq) equipped with software (FlexiVent, Scireq) to record and analyze the measurements. Anesthetized, tracheostomized, paralyzed, and mechanically ventilated mice were used during all static P-V loop measurements (as a measure of lung compliance). Collagen deposition was quantified biochemically by measuring hydroxyproline levels or detecting collagen-1 in the lung tissue extracts by immunoblotting analysis (58). Fibrosis development was assessed by quantitation of α-SMA expression in the lung tissue extracts by immunoblotting analysis. Lung lavage was performed to determine total wbc counts or cell differentials as described previously (58). All animal protocols were performed according to guidelines approved by the Cleveland Clinic Institutional Review Board.
Cell culture, transfection, and lentiviral transduction.
After informed consent was provided, deidentified explanted lung tissues from patients with IPF and normal controls were provided by the airway tissue procurement program under protocols approved by the University of Alabama at Birmingham for isolation of lung fibroblasts. Adult normal HLFs (19Lu cells) were purchased from ATCC. Primary isolates of HLFs from patients with IPF and normal controls were provided by Patricia Sime (University of Rochester, Rochester, New York, USA) and were obtained with the approval of the University of Rochester Institutional Review Board. Fibroblasts were maintained and propagated in Modified Eagle’s Medium (MEM) supplemented with 10% FBS, and all experiments were performed on early passages of cells. Primary murine lung fibroblasts were derived from 7- to 10-week-old Trpv4 null mice and C57BL/6 mice and propagated as described previously (58). TRPV4 expression was downregulated by transfecting 19Lu cells with TRPV4-specific siRNAs using siLentFect lipid reagent (Bio-Rad) following manufacturer’s protocols. Targeting and control scrambled siRNA duplexes (SMART pool oligos) were synthesized by and purchased from Origene. Lentivirus-mediated expression of TRPV4 was achieved by using a GFP-conjugated TRPV4 fusion protein that has been functionally validated (lenti-TRPV4-GFP) (56). For transduction, primary murine lung fibroblasts were exposed to lentivirus encoding lenti-TRPV4-GFP for 24 hours in MEM supplemented with polybrene (8 μg/ml, Chemicon International). For control experiments, infections with the empty lenti-GFP virus were carried out. Cells were then transferred to 1% BSA-containing serum-free MEM with or without TGF-β1 for 24 hours. The attached cells were fixed, permeabilized, and immunostained for α-SMA to examine the α-SMA–incorporated cytoskeletal fibers.
Nuclear and cytoplasmic fractionation, F-actin and G-actin purification, RhoA activation assay, and Western blot analysis.
Nuclear and cytoplasmic fractions from 19Lu cells or mouse lung fibroblasts (WT and Trpv4 KO) were prepared using the NEPER Nuclear and Cytoplasmic Reagents Kit (Thermo Scientific) following the manufacturer’s protocol. The amount of F-actin and G-actin formation in TGF-β1–treated 19Lu cells was assessed and quantified using the G-Actin/F-Actin In Vivo Assay Biochem Kit from Cytoskeleton. RhoA activation assays were performed using a G-LISA kit from Cytoskeleton according to the manufacturer’s instructions. For the assay, HLFs were serum starved for 24 hours before treatment with TGF-β1 (2 ng/ml or vehicle controls) for the indicated time points with or without AB1 (50 μM). Immunoblotting analysis was performed on 1% NP-40 whole cell lysates, as we described in a previous study (58), and developed with the enhanced chemiluminescence system (Fisher Scientific).
Immunofluorescence staining.
Normal HLFs or primary mouse lung fibroblasts were plated on fibronectin-coated (10 μg/ml) glass coverslips and maintained in MEM supplemented with 1% FBS for 24 hours. Cells were then transferred to 1% BSA-containing serum-free MEM with or without TGF-β1 for 24 to 48 hours. The attached cells were fixed with 3% paraformaldehyde, permeabilized by 0.1% Triton X-100, and blocked with 10% normal goat serum. To examine the α-SMA–incorporated cytoskeletal fibers, the fibroblasts were incubated with primary anti–α-SMA antibodies followed by Alexa Fluor–conjugated secondary antibody. Staining of F-actin was achieved using Alexa Fluor–labeled phalloidin. Digital fluorescence microscopic images were analyzed by ImageJ software. Colocalization of α-SMA with F-actin was compared with colocalization in different conditions using Pearson’s colocalization coefficients.
In vitro myofibroblast differentiation assay on mouse lung tissue and hydrogels with variable stiffness.
Female C57BL/6 mice were injected with 4 U/kg bleomycin or saline via IT instillation, and, after 2 weeks, lungs were inflated with OCT to prepare 10-μm sections containing both fibrotic and normal areas as published previously (38, 59). HLFs were allowed to attach on the 5% BSA-blocked lung sections, and unattached cells were washed off with PBS as published previously (38, 59). After washing, lung section–containing cells were treated with vehicle or AB1 (20 μM) in 1% BSA/serum-free medium (24 hours), and staining for F-actin was performed using Alexa Fluor 594–labeled phalloidin (5 U/ml, 60-minute incubation at room temperature; Molecular Probes). Digital confocal fluorescence images were analyzed by ImageJ software for quantitation of the actin stress fiber density of cells attached to normal and fibrotic areas. Assessment of myofibroblast differentiation as a function of extracellular matrix stiffness was assayed using polyacrylamide hydrogels bound to 96-well glass bottom plates with variable stiffness (1, 8, and 25 kPa) (Matrigen). For the assay, HLFs were plated on collagen-coated (100 μg/ml) hydrogels and maintained in MEM supplemented with 1% FBS for 24 hours. Cells were then transferred to 1% BSA-containing serum-free MEM with or without TGF-β1 under vehicle- or AB1-treated conditions for 24 hours. Staining of α-SMA and digital image analysis was performed as described above.
Measurement of intracellular calcium.
The calcium response in human or mouse lung fibroblasts was measured by either the FlexStation system using the FLIPR Calcium 5 Assay Kit (Molecular Devices) or Fura-2 dye–loaded cells. Cells were plated a day before at 15,000 cells per well (in 1% serum containing MEM) in 96-well plates coated with fibronectin. Cells were then placed in 1% BSA-containing serum-free MEM with or without 2 ng/ml TGF-β1. On the following day, cells were incubated for 45 minutes at 37°C with FLIPR Calcium 5 dye (Molecular Devices) in 1X HBSS solution (pH 7.4) containing 20 mM HEPES (pH 7.4) and 2.5 mM probenecid. Calcium influx was induced by the TRPV4 agonist, GSK, in fibroblasts that were pretreated with vehicle or TRPV4 antagonists (AB1 or HC). Cytosolic calcium increases (Ca2+ influx) were recorded by measuring ΔF/F (max-min), which is shown as relative fluorescence units. For single cell Ca2+ influx measurements, Fura-2 dye–loaded cells were used and the ratio of emitted fluorescence after excitation at 340 nm and 380 nm relative to the ratio measured before cell stimulation (ratio F/F0) was considered. Measurement of Ca2+ influx caused by increasing stiffness was assayed using hydrogels bound to 96-well glass bottom plates with variable stiffness (1, 8, and 25 kPa). HLFs were plated on collagen-coated hydrogels and maintained in MEM supplemented with 1% BSA for 24 hours. The next day, cells were transferred to 1% BSA-containing serum-free MEM with or without TGF-β1 under vehicle- or AB1-treated conditions for 24 hours. Measurement of calcium influx was done by the FlexStation system using FLIPR Calcium 5 Assay Kit (Molecular Devices) as described above.
Statistics.
All data are expressed as mean ± SEM or SD as indicated below. Statistical comparisons between control and experimental groups were performed with the Student’s t tests or 1-way ANOVA using SigmaPlot software. ANOVA followed by the Student-Newman-Keuls test was used for multiple comparisons. P < 0.05 was considered significant.
Acknowledgments
This work was supported by NIH grants (R01HL103553-04 and R01HL085324-07) to M.A. Olman and American Heart Association grant (SDG17310007) to S.O. Rahaman.
Footnotes
Conflict of interest: Magdalene M. Moran is a paid employee of and owns stock options in Hydra Biosciences Inc.
Reference information:J Clin Invest. 2014;124(12):5225–5238. doi:10.1172/JCI75331.
References
- 1.Raghu G, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Noth I, et al. A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012;186(1):88–95. doi: 10.1164/rccm.201202-0314OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Richeldi L, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365(12):1079–1087. doi: 10.1056/NEJMoa1103690. [DOI] [PubMed] [Google Scholar]
- 4.Idiopathic Pulmonary Fibrosis Clinical Research Network, et al. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366(21):1968–1977. doi: 10.1056/NEJMoa1113354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.King TE, Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378(9807):1949–1961. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 6.Chapman HA. Disorders of lung matrix remodeling. J Clin Invest. 2004;113(2):148–157. doi: 10.1172/JCI20729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hinz B, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180(4):1340–1355. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muro AF, et al. An essential role for fibronectin extra type III domain A in pulmonary fibrosis. Am J Respir Crit Care Med. 2008;177(6):638–645. doi: 10.1164/rccm.200708-1291OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marinkovic A, Mih JD, Park JA, Liu F, Tschumperlin DJ. Improved throughput traction microscopy reveals pivotal role for matrix stiffness in fibroblast contractility and TGF-β responsiveness. Am J Physiol Lung Cell Mol Physiol. 2012;303(3):L169–L180. doi: 10.1152/ajplung.00108.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grinnell F, Ho CH. Transforming growth factor beta stimulates fibroblast-collagen matrix contraction by different mechanisms in mechanically loaded and unloaded matrices. Exp Cell Res. 2002;273(2):248–255. doi: 10.1006/excr.2001.5445. [DOI] [PubMed] [Google Scholar]
- 11.Liu F, et al. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol. 2010;190(4):693–706. doi: 10.1083/jcb.201004082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou Y, et al. Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J Clin Invest. 2013;123(3):1096–1108. doi: 10.1172/JCI66700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wells RG. Tissue mechanics and fibrosis. Biochim Biophys Acta. 2013;1832(7):884–890. doi: 10.1016/j.bbadis.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimbori C, Gauldie J, Kolb M. Extracellular matrix microenvironment contributes actively to pulmonary fibrosis. Curr Opin Pulm Med. 2013;19(5):446–452. doi: 10.1097/MCP.0b013e328363f4de. [DOI] [PubMed] [Google Scholar]
- 15.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4(7):517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 16.Ramires FJ, Sun Y, Weber KT. Myocardial fibrosis associated with aldosterone or angiotensin II administration: attenuation by calcium channel blockade. J Mol Cell Cardiol. 1998;30(3):475–483. doi: 10.1006/jmcc.1997.0612. [DOI] [PubMed] [Google Scholar]
- 17.Du J, et al. TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ Res. 2010;106(5):992–1003. doi: 10.1161/CIRCRESAHA.109.206771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davis J, Burr AR, Davis GF, Birnbaumer L, Molkentin JD. A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev Cell. 2012;23(4):705–715. doi: 10.1016/j.devcel.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biernacka A, Dobaczewski M, Frangogiannis NG. TGFβ signaling in fibrosis. Growth Factors. 2011;29(5):196–202. doi: 10.3109/08977194.2011.595714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18(7):1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov. 2012;11(10):790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhu MX, ed. TRP Channels. Boca Raton, Florida, USA: CRC Press; 2011. [Google Scholar]
- 23.Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem. 2007;76:387–417. doi: 10.1146/annurev.biochem.75.103004.142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liedtke W. Molecular mechanisms of TRPV4-mediated neural signaling. Ann N Y Acad Sci. 2008;1144:42–52. doi: 10.1196/annals.1418.012. [DOI] [PubMed] [Google Scholar]
- 25.Vriens J, Watanabe H, Janssens A, Droogmans G, Voets T, Nilius B. Cell swelling, heat, and chemical agonists use distinct pathways for the activation of the cation channel TRPV4. Proc Natl Acad Sci U S A. 2003;101(1):396–401. doi: 10.1073/pnas.0303329101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lamandé SR, et al. Mutations in TRPV4 cause an inherited arthropathy of hands and feet. Nat Genet. 2011;43(11):1142–1146. doi: 10.1038/ng.945. [DOI] [PubMed] [Google Scholar]
- 27.Auer-Grumbach M, et al. Alterations in the ankyrin domain of TRPV4 cause congenital distal SMA, scapuloperoneal SMA and HMSN2C. Nat Genet. 2010;42(2):160–164. doi: 10.1038/ng.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deng HX, et al. Scapuloperoneal spinal muscular atrophy and CMT2C are allelic disorders caused by alterations in TRPV4. Nat Genet. 2009;42(2):165–169. doi: 10.1038/ng.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masuyama R, et al. TRPV4-mediated calcium influx regulates terminal differentiation of osteoclasts. Cell Metab. 2008;8(3):257–265. doi: 10.1016/j.cmet.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 30.Muramatsu S, et al. Functional gene screening system identified TRPV4 as a regulator of chondrogenic differentiation. J Biol Chem. 2007;282(44):32158–32167. doi: 10.1074/jbc.M706158200. [DOI] [PubMed] [Google Scholar]
- 31.Adapala RK, et al. TRPV4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J Mol Cell Cardiol. 2013;54:45–52. doi: 10.1016/j.yjmcc.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hamanaka K, et al. TRPV4 initiates the acute calcium-dependent permeability increase during ventilator-induced lung injury in isolated mouse lungs. Am J Physiol Lung Cell Mol Physiol. 2007;293(4):L923–L932. doi: 10.1152/ajplung.00221.2007. [DOI] [PubMed] [Google Scholar]
- 33.Thorneloe KS, et al. An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Sci Transl Med. 2012;4(159):159ra148. doi: 10.1126/scitranslmed.3004276. [DOI] [PubMed] [Google Scholar]
- 34.Thorneloe KS, et al. N-((1S)-1-{[4-((2S)-2-{[(2,4-dichlorophenyl)sulfonyl]amino}-3-hydroxypropanoyl)-1-piperazinyl]carbonyl}-methylbutyl)-1-benzothiophene-2 carboxamide (GSK1016790A), a novel and potent transient receptor potential vanilloid 4 channel agonist induces urinary bladder contraction and hyperactivity: Part I. J Pharmacol Exp Ther. 2008;326(2):432–442. doi: 10.1124/jpet.108.139295. [DOI] [PubMed] [Google Scholar]
- 35.Vincent F, et al. Identification and characterization of novel TRPV4 modulators. Biochem Biophys Res Commun. 2009;389(3):490–494. doi: 10.1016/j.bbrc.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 36.Mukherjee S, Kolb MR, Duan F, Janssen LJ. Transforming growth factor-β evokes Ca2+ waves and enhances gene expression in human pulmonary fibroblasts. Am J Respir Cell Mol Biol. 2012;46(6):757–764. doi: 10.1165/rcmb.2011-0223OC. [DOI] [PubMed] [Google Scholar]
- 37.Everaerts W, et al. Inhibition of the cation channel TRPV4 improves bladder function in mice and rats with cyclophosphamide-induced cystitis. Proc Natl Acad Sci U S A. 2010;107(44):19084–19089. doi: 10.1073/pnas.1005333107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu S, et al. Urokinase receptor mediates lung fibroblast attachment and migration toward provisional matrix proteins through interaction with multiple integrins. Am J Physiol Lung Cell Mol Physiol. 2009;297(1):L97–L108. doi: 10.1152/ajplung.90283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sandbo N, Lau A, Kach J, Ngam C, Yau D, Dulin NO. Delayed stress formation mediates pulmonary myofibroblast differentiation in response to TGF-β. Am J Physiol Lung Cell Mol Physiol. 2011;301(5):L656–L666. doi: 10.1152/ajplung.00166.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sandbo N, Dulin NO. Actin cytoskeleton in myofibroblast differentiation: ultrastructure defining form and driving function. Transl Res. 2011;158(4):181–196. doi: 10.1016/j.trsl.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cen B, et al. Megakaryoblastic leukemia 1, a potent transcriptional coactivator for serum response factor (SRF), is required for serum induction of SRF target genes. Mol Cell Biol. 2003;3(18):6597–6608. doi: 10.1128/MCB.23.18.6597-6608.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu G, et al. Association of TRPV4 gene polymorphisms with chronic obstructive pulmonary disease. Hum Mol Genet. 2009;18(11):2053–2062. doi: 10.1093/hmg/ddp111. [DOI] [PubMed] [Google Scholar]
- 43.Follonier Castella L, Gabbiani G, McCulloch CA, Hinz B. Regulation of myofibroblast activities: calcium pulls some strings behind the scene. Exp Cell Res. 2010;316(15):2390–2401. doi: 10.1016/j.yexcr.2010.04.033. [DOI] [PubMed] [Google Scholar]
- 44.Schwartz MA. Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb Perspect Biol. 2010;2(12):a005066. doi: 10.1101/cshperspect.a005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thodeti CK, et al. TRPV4 channels mediate cyclic strain-induced endothelial cell reorientation through integrin-to-integrin signaling. Circ Res. 2009;104(9):1123–1130. doi: 10.1161/CIRCRESAHA.108.192930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matthews BD, Thodeti CK, Tytell JD, Mammoto A, Overby DR, Ingber DE. Ultra-rapid activation of TRPV4 ion channels by mechanical forces applied to cell surface beta1 integrins. Integr Biol (Camb). 2010;2(9):435–442. doi: 10.1039/c0ib00034e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mih JD, Sharif AS, Liu F, Marinkovic A, Symer MM, Tschumperlin DJ. A multiwell platform for studying stiffness-dependent cell biology. PLoS One. 2011;6(5):e19929. doi: 10.1371/journal.pone.0019929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barkauskas CE, Noble PW. Cellular mechanisms of tissue fibrosis. 7. New insights into the cellular mechanisms of pulmonary fibrosis. Am J Physiol Cell Physiol. 2014;306(11):C987–C996. doi: 10.1152/ajpcell.00321.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hutchison N, Fligny C, Duffield JS. Resident mesenchymal cells and fibrosis. Biochim Biophys Acta. 2013;1832(7):962–971. doi: 10.1016/j.bbadis.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tanjore H, et al. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med. 2009;180(7):657–665. doi: 10.1164/rccm.200903-0322OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hung C, et al. Role of lung pericytes and resident fibroblasts in the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med. 2013;188(7):820–830. doi: 10.1164/rccm.201212-2297OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Piera-Velazquez S, Li Z, Jimenez SA. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am J Pathol. 2011;179(3):1074–1080. doi: 10.1016/j.ajpath.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hashimoto N, Jin H, Liu T, Chensue SW, Phan SH. Bone marrow-derived progenitor cells in pulmonary fibrosis. J Clin Invest. 2004;113(2):243–252. doi: 10.1172/JCI18847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moran MM, McAlexander MA, Bíró T, Szallasi A. Transient receptor potential channels as therapeutic targets. Nat Rev Drug Discov. 2011;10(8):601–620. doi: 10.1038/nrd3456. [DOI] [PubMed] [Google Scholar]
- 55.Mizuno A, Matsumoto N, Imai M, Suzuki M. Impaired osmotic sensation in mice lacking TRPV4. Am J Physiol Cell Physiol. 2003;285(1):C96–C101. doi: 10.1152/ajpcell.00559.2002. [DOI] [PubMed] [Google Scholar]
- 56.Zheng X, et al. Arachidonic acid-induced dilation in human coronary arterioles: convergence of signaling mechanisms on endothelial TRPV4-mediated Ca2+ entry. J Am Heart Assoc. 2013;2(3):e000080. doi: 10.1161/JAHA.113.000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Olman MA, Mackman N, Gladson CL, Moser KM, Loskutoff DJ. Changes in procoagulant and fibrinolytic gene expression during bleomycin-induced lung injury in the mouse. J Clin Invest. 1995;96(3):1621–1630. doi: 10.1172/JCI118201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ding Q, et al. FAK-related nonkinase is a multifunctional negative regulator of pulmonary fibrosis. Am J Pathol. 2013;182(5):1572–1584. doi: 10.1016/j.ajpath.2013.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grove LM, et al. Urokinase-type plasminogen activator receptor (uPAR) ligation induces a raft-localized integrin signaling switch that mediates the hypermotile phenotype of fibrotic fibroblasts. J Biol Chem. 2014;289(18):12791–12804. doi: 10.1074/jbc.M113.498576. [DOI] [PMC free article] [PubMed] [Google Scholar]