

Abstract
Growing evidence supports a link between inflammation and cancer; however, mediators of the transition between inflammation and carcinogenesis remain incompletely understood. Sphingosine-1-phosphate (S1P) lyase (SPL) irreversibly degrades the bioactive sphingolipid S1P and is highly expressed in enterocytes but downregulated in colon cancer. Here, we investigated the role of SPL in colitis-associated cancer (CAC). We generated mice with intestinal epithelium-specific Sgpl1 deletion and chemically induced colitis and tumor formation in these animals. Compared with control animals, mice lacking intestinal SPL exhibited greater disease activity, colon shortening, cytokine levels, S1P accumulation, tumors, STAT3 activation, STAT3-activated microRNAs (miRNAs), and suppression of miR-targeted anti-oncogene products. This phenotype was attenuated by STAT3 inhibition. In fibroblasts, silencing SPL promoted tumorigenic transformation through a pathway involving extracellular transport of S1P through S1P transporter spinster homolog 2 (SPNS2), S1P receptor activation, JAK2/STAT3-dependent miR-181b-1 induction, and silencing of miR-181b-1 target cylindromatosis (CYLD). Colon biopsies from patients with inflammatory bowel disease revealed enhanced S1P and STAT3 signaling. In mice with chemical-induced CAC, oral administration of plant-type sphingolipids called sphingadienes increased colonic SPL levels and reduced S1P levels, STAT3 signaling, cytokine levels, and tumorigenesis, indicating that SPL prevents transformation and carcinogenesis. Together, our results suggest that dietary sphingolipids can augment or prevent colon cancer, depending upon whether they are metabolized to S1P or promote S1P metabolism through the actions of SPL.
Introduction
A connection between inflammation and cancer has been recognized for over a hundred years, exemplified by Virchow’s observation that cancer behaves like a wound that does not heal (1, 2). This connection is particularly evident in colon carcinogenesis, because patients with inflammatory bowel disease (IBD) have as much as 7-fold higher incidence of colon cancer than the general population (3). Also, the use of NSAIDs reduces the risk of colon cancer in patients with familial adenomatous polyposis coli and patients with sporadic adenomas (4–6). However, chemoprevention with celecoxib was associated with deleterious effects on the cardiovascular system, limiting their utility and revealing the need for safer chemopreventive agents (5). Key inflammatory pathways are constitutively activated in many colorectal cancers, and inflammatory infiltrates and elevated cytokines are often present (7). Although these may represent a defense mechanism, they are also procarcinogenic (7). As developing nations westernize, both IBD and colon cancer incidences rise in association with the adoption of unhealthy diets (8). Together, these findings implicate a link between diet, inflammation and colon cancer. However, the molecular mechanisms underlying these connections remain incompletely understood. It has been suggested that genetic mutations are the “match that lights the fire” of cancer, whereas inflammation is the “fuel that feeds the flame” (2). However, there is increasing evidence that inflammation contributes to the earliest stages of carcinogenesis, namely in the process of cell transformation, in which the cell acquires many aspects of the cancer phenotype (9).
Bioactive sphingolipids play fundamental roles in carcinogenesis via their ability to regulate programmed cell death pathways, stress responses, angiogenesis, innate and adaptive immunity, and inflammation (10). For example, ceramide and its metabolite sphingosine promote apoptosis in colon cancer cells (11). Further, sphingolipids have been shown to exhibit a preventive effect against colon cancer in preclinical models (12–14). In contrast, the phosphorylated form of sphingosine, sphingosine-1-phosphate (S1P), acts through G protein–coupled receptors and through intracellular mechanisms to inhibit apoptosis, promote angiogenesis, and enhance inflammatory signaling through activation of the NF-κB and STAT3 pathways (15, 16). Further, cellular accumulation of S1P due either to its increased biosynthesis or reduced catabolism results in cell transformation (17, 18).
The impact of S1P signaling and metabolism is particularly germane in colon cancer, as gut epithelial cells are exposed to sphingolipid metabolites generated by the breakdown of dietary sphingolipids (19). Enzymes in the brush border generate sphingosine from higher-order mammalian sphingolipids. Sphingosine enters epithelial cells, in which it is phosphorylated by sphingosine kinases, generating S1P. The S1P degradative enzyme S1P lyase (SPL) is highly expressed in differentiated enterocytes, in which it rapidly catabolizes S1P, thereby maintaining low levels of S1P relative to sphingosine in healthy gut mucosa. There is evidence that disruption of normal S1P metabolism occurs during malignant transformation and colon cancer progression. The major sphingosine kinase, sphingosine kinase 1 (SPHK1), is overexpressed in colon cancer (17, 20, 21). Mice lacking SPHK1 are less susceptible to experimentally induced colitis and colon cancer and exhibit less tumor progression in genetic models of colon cancer (20, 22, 23). In contrast to SPHK1, SPL is downregulated in colon cancer cells, leading to accumulation of S1P (22, 24). However, the role of SPL in colon carcinogenesis has not been directly examined. Further, the fact that S1P functions as a proangiogenic and proinflammatory promoter of carcinogenesis, whereas dietary sphingolipids have been shown to exert chemopreventive activity against colon cancer, creates a paradox that, to date, has not been explained.
STAT3 is a key transcription factor implicated in inflammation and carcinogenesis (25), and its phosphorylation, in response to activation of gp130 receptors by cytokines, including its major activator, IL-6, enables its nuclear translocation and function. Crucially, S1P signaling participates in a reciprocal positive feedback loop with STAT3 (15), and cross-talk between S1P and the STAT3 pathway can sustain persistent STAT3 signaling in cancer, thereby promoting tumor progression and metastasis (15). Recent studies have demonstrated that STAT3 regulates the expression of microRNAs (miRNAs) that promote cell transformation (9). Specifically, miRNAs miR-21 and miR-181b-1 were shown to suppress expression of their respective targets — phosphatase and tensin homolog (PTEN; a lipid phosphatase that negatively regulates the PI3K/AKT pathway) and cylindromatosis (CYLD; a deubiquitinating enzyme that negatively regulates NF-κB). In so doing, STAT3 activation mediates the epigenetic switch that underlies the transformed phenotype (9). These findings provide a molecular basis that directly links inflammation to carcinogenesis. How S1P signaling may be involved in these events remains unknown.
In the present study, we have explored the effect of intestinal SPL downregulation on colon carcinogenesis by generating and characterizing a gut-specific SPL1 (Sgpl1) knockout (KO) mouse. Our findings demonstrate that SPL plays a critical role in intestinal tumorigenesis by regulating extracellular levels of S1P formed from mammalian sphingosine, thereby affecting the induction of STAT3-activated miRNAs that contribute to the process of cell transformation. In contrast, we show that plant-type sphingolipids called sphingadienes (SDs) cannot be metabolized to S1P and instead enhance the metabolism of S1P by upregulating SPL, as SPL irreversibly catabolizes S1P. Our findings provide a mechanistic link between diet, inflammation, and cancer and simultaneously provide evidence supporting the further investigation of SDs as colon cancer chemopreventive agents.
Results
Generation of a conditional KO mouse lacking SPL in gut epithelium.
To study the role of SPL in colitis-associated cancer (CAC) in vivo, a conditional Sgpl1 KO targeting vector was generated by bacterial recombineering (outlined in Figure 1A, Supplemental Table 1, and Supplemental Figure 1A; supplemental material available online with this article; doi:10.1172/JCI74188DS1). Targeted embryonic stem cells were isolated and used to create mice heterozygous for the mutant allele. The β-geo gene-trapping cassette was excised by mating these mice with mice expressing a Flp recombinase deleter transgene to generate a conditional KO or “floxed” allele for Sgpl1fl/fl. In the floxed allele, exons 10–12 of Sgpl1, which includes the enzyme cofactor-binding site encoded by exon 11, are flanked by loxP sites. Deletion of exons 10 to 12 by Cre recombinase eliminates SPL expression (a KO phenotype). Mice homozygous for the floxed allele (Sgpl1fl/fl mice) reproduced normally, exhibited normal life span, and showed no evidence of disrupted SPL function or expression (P. Bandhuvula, unpublished observations). In contrast, all Sgpl1fl/fl mice harboring a constitutively expressed Cre recombinase transgene exhibited the same phenotypes as KO mice: runting and death within 1 month of age (P. Bandhuvula, unpublished observations). Examination of these pups at 18 days of age revealed very low or undetectable Sgpl1 expression by quantitative RT-PCR (qRT-PCR) (Figure 1B) or immunoblotting (IB) (Figure 1C).
Figure 1. Global and tissue-specific Sgpl1 KO mouse models.

(A) Conditional KO strategy. A targeting vector introduces a lacZ reporter 5′ to exon 10, resulting in an Sgpl1 truncation. The reporter was deleted by crossing mice with FLPR transgenic mice. The resulting Sgpl1 allele has “floxed” exons 10–12 that can be excised by Cre recombinase. The asterisks indicate that exon 11 contains a critical enzyme cofactor-binding site. (B) mRNA expression of Sgpl1 in tissues of Sgpl1fl/fl (white bars) and Sgpl1fl/fl global Cre+ transgenic (black bars) mice. (C) IB of representative tissues from Sgpl1fl/fl and Sgpl1fl/fl global Cre+ transgenic KO mice. (D) IB of SPLGutKO tissues. Sgpl1fl/fl mice were crossed with mice expressing Cyp1a1-Cre transgene, which was inducible in gut epithelium by oral treatment with NF. After 14 days, WT noninduced (WT), induced (KO), and NF-treated control (C) mice were euthanized. (E) Jejunum SPL activity. (F) Jejunum S1P levels. (G) Plasma S1P levels (no significant difference). (H) SPL IHC (original magnification, ×10) of SPLGutKO mouse colon and thymus tissues. *P < 0.05.
An inducible intestinal-specific KO line was generated by breeding of Sgpl1fl/fl mice with mice harboring a Cre transgene under control of an intestine-specific cytochrome p450 promoter element (Cyp1a1) that is induced in response to lipophilic xenobiotics such as β-napthoflavone (NF) (26). Turning on the expression of the Cyp1a1-Cre transgene with NF leads to the inactivation of Sgpl1 only in the small and large intestinal epithelium (SPLGutKO line). We confirmed the fidelity of the gut-specific Sgpl1 KO line; there was a near complete inactivation of SPL in the jejunum after 5 days of induction with NF, with no effect on SPL expression in thymus, stomach, or esophagus, as judged by IB and immunohistochemistry (IHC) (Figure 1, D and H). Note that relative SPL expression throughout the intestinal tract is variable: it is low in esophagus and stomach, moderate in ileum and colon, and high in duodenum and jejunum. Treatment of Sgpl1fl/fl mice with NF did not affect SPL expression in control mice lacking the Cre transgene, which expressed SPL at levels similar to those in WT mice (Figure 1D). Importantly, SPL enzyme activity was virtually absent in the lower gastrointestinal tract of the SPLGutKO KO (Figure 1E). Consistent with the loss of SPL activity, S1P levels were more than 8-fold higher in jejunum tissues of SPLGutKO mice than in NF-treated Sgpl1fl/fl control mice (hereafter referred to as control mice) (Figure 1F). In contrast, circulating S1P levels were not different from those of control mice (Figure 1G). Histology was performed on SPLGutKO mice and control mice 42 days after induction with NF. Our results showed no appreciable difference in histology or immune cell composition by microscopic pathology, as graded by a pathologist blinded to genotype. We also observed no differences in TNF-α, IL-1β, or IL-6 levels in colon tissue harvested 1 week after induction of SPL recombination. These results are shown in Supplemental Figure 1, B and C. We also measured the levels of sphingosine, dihydrosphingosine, total ceramide, and sphingomyelin in the colons of SPLGutKO and control mice. We found no differences in the abundance of those sphingolipid species in the SPLGutKO and control mice (A. Eltanawy, unpublished observations).
Loss of SPL in gut epithelium promotes susceptibility to azoxymethane/dextran sodium sulfate–induced CAC.
SPLGutKO and control mice exhibited similar adult weights, litter sizes, and life spans. To define the impact of SPL downregulation on CAC, SPLGutKO mice and control mice were treated with 10 mg/kg i.p. azoxymethane (AOM), followed by 3 rounds of 2% dextran sodium sulfate (DSS) in the drinking water for 7 days separated by 2-week intervals. Disease activity index (DAI), a measure of disease-associated symptoms in IBD, was calculated by measurement of body weight, stool consistency, and rectal bleeding, as previously described (27). On this regimen, survival of SPLGutKO mice was lower than that of controls (Figure 2A). DAI of control mice increased modestly with each DSS treatment, and mice exhibited clinical recovery after DSS was removed from the drinking water (Figure 2B). Beginning on day 15 of treatment, the DAI scores of the SPLGutKO mice were uniformly higher than those of the control mice, reaching 3- to 4-fold higher levels than those in control mice during and immediately after DSS treatment and failing to recover completely after cessation of DSS treatment. The levels of the circulating and colon tissue cytokines that are elevated in IBD and other inflammatory conditions (e.g., TNF-α, IL-1β, IL-6, IL-17, IL-21, IL-23) were higher in SPLGutKO mice than in controls (Figure 2, C and D). To further characterize the inflammatory response in SPLGutKO mice, Th1 and Th17 subsets were analyzed by flow cytometry by labeling of splenocytes from control and SPLGutKO mice with subset-specific antibodies. SPLGutKO mice exhibited a shift from the Th1 subset to the Th17 subset, which was not observed in control mice, quantified by the average percentage of Th1 and Th17 cells in all CD4-positive lymphocytes in untreated WT mice, AOM/DSS-treated controls, and AOM/DSS-treated SPLGutKO mice (Figure 2E). (A representative histogram from 1 mouse per group is shown in Figure 2F). In addition, SPLGutKO colon tissues contained higher numbers of macrophages (Figure 2G). These findings demonstrate that SPLGutKO mice exhibited a greater global inflammatory response to AOM/DSS compared with that of controls. Gross and microscopic pathologic analyses were also performed on SPLGutKO and control mouse organs, tissues, and blood. SPLGutKO colon lengths were shorter than those of control mice, indicating greater inflammation-associated stricture (Figure 2H). In SPLGutKO mice, hematocrit levels were lower (Figure 2I) and spleen size and weight were greater than those in control mice (Figure 2J), reflecting more loss of blood in the stool, resulting in greater degree of anemia and more compensatory extramedullary hematopoiesis.
Figure 2. Loss of SPL in gut epithelium promotes susceptibility to AOM/DSS-induced CAC, tumor formation, and crypt cell proliferation.

(A) Kaplan-Meier plot of control (n = 12) and KO (n = 21) survival on AOM/DSS regimen. (B) DAI. Starting on day 16, significance of control (blue line) and KO (red line) mouse DAI was P < 0.05. (C) Plasma and (D) colon inflammatory cytokines. White bars represent controls, and black bars represent KO. (E) Average percentage of Th1 (CD4+IFN-γ+) and Th17 (CD4+IL-17+) cells plotted from 2 independent experiments. No treatment (NT), n = 3; control with AOM/DSS treatment (C), n = 7; SPLGutKO with AOM/DSS treatment (KO), n = 5. (F) Representative dot plots for 1 individual mouse per group from the experiment shown in E. (G) Macrophages (F4/80-positive cells) per ×10 microscopic field. Results represent the average of 3 fields per group. (H) Colon length. (I) Hematocrit. (J) Spleen weight. (K) Average tumor number per mouse. (L) Average tumor size distribution as percentage of total. Green bars represent tumors >16 mm3; red bars represent tumors between 4 and 16 mm3; blue bars represent tumors <4 mm3; no significant difference. (M) Representative colon section Ki-67 IHC (original magnification, ×40). (N) Proliferating cells per crypt (average) determined by counting 50 well-oriented intact crypts with visible lumen per section (n = 5 per group). *P < 0.05.
Loss of SPL in gut epithelium promotes tumorigenicity and crypt cell proliferation in response to AOM/DSS.
To determine whether enhanced inflammation observed in SPLGutKO mice correlated with an increase in cancer development, we compared colon tumor incidence, location, size, and total tumor burden in SPLGutKO and control mice exposed to AOM/DSS and euthanized mice on day 63 of the regimen. SPLGutKO mice exhibited approximately 2-fold more colonic tumors than control mice, as judged by counting all macroscopically detectible tumors in the lower gastrointestinal tract of each mouse (Figure 2K). The majority of colon tumors in both SPLGutKO and control groups occurred in the distal colon and rectum, and the increase in tumor number in SPLGutKO mice was accounted for mainly by an increase in tumors of the distal colon. The size distribution of tumors was not significantly different between the groups (Figure 2L). In addition, the histological tumor grade, percentage of apoptotic cells, and percentage of proliferating cells were not significantly different in tumors of each group (Supplemental Figure 2 and A. Pandurangan, unpublished observations). However, the number of proliferating cells per crypt in the nonneoplastic colons was significantly higher in SPLGutKO mice than in control mice (Figure 2, M and N). Collectively, these results indicate a critical role for intestinal SPL in protection against CAC. Specifically, our finding that intestinal SPL silencing leads to expansion of the proliferating crypt cell population and increases colon tumor incidence without affecting tumor size or grade suggests that SPL downregulation influences an early stage of tumorigenesis.
Loss of SPL in gut epithelium enhances STAT3 activation and induction of STAT3-activated miRNAs.
STAT3 is constitutively activated in most colon cancers, and JAK2/STAT3 signaling influences colon cancer cell proliferation and survival (28). Further, STAT3 was recently found to participate in a positive feedback loop with S1P signaling (15). Therefore, we investigated whether SPL mediates its proinflammatory and protumorigenic effects through STAT3 signaling. Immunofluorescence (IF) microscopy with an antibody to detect STAT3 phosphorylation revealed a higher level of activated STAT3 protein in nonneoplastic colonic tissues of SPLGutKO mice compared with that in controls (Figure 3A). Detection of activated STAT3 by IHC in neoplastic and nonneoplastic colonic tissues showed that STAT3 activation was equally intense in the interstitial infiltrates and tumor tissue of both groups but was more prominent in the differentiated enterocytes of SPLGutKO mice compared with that in control mice (Figure 3B). IB of colon extracts confirmed higher levels of phosphorylated STAT3 in the colons of SPLGutKO mice (Figure 3C and Supplemental Figure 3, A and B). JAK2 autophosphorylation on Tyr1007/1008 in its putative activation loop was also increased in the colon tissues of SPLGutKO mice compared with that in controls (Figure 3C and Supplemental Figure 3, A and B). MCL-1, a BCL-2 family member and downstream STAT3 target, and c-MYC, which is also a STAT3 target, were also more highly expressed in the colons of SPLGutKO mice (Figure 3C and Supplemental Figure 3A). Total STAT3 levels were also elevated, presumably due to the well-documented autoregulation of Stat3 transcription (29).
Figure 3. Loss of SPL in gut epithelium enhances STAT3 activation and induction of STAT3-activated miRNAs.

(A) IF of activated STAT3 phosphorylated on Tyr705 (STAT3-P, green) and counterstained with DAPI (blue) in control and KO mouse colon tissues, following administration of AOM/DSS. Scale bar: 50 μm. (B) Activated STAT3 phosphorylated on Tyr705 IHC in representative colon sections (original magnification, ×10). (C) Protein expression of phosphorylated STAT3 (STAT3-P), total STAT3 (STAT3-T), phosphorylated JAK2 (JAK2-P), total JAK2 (JAK2-T), C-MYC, and MCL-1 in colon tissues of control and KO mice. Note that in the last lane of JAK2-T IB there is a nonspecific band. (D) mRNA expression of murine S1pr1 in nonneoplastic colon tissues of control, KO, and KO mice treated with NSC 74859. (E) mRNA expression of human S1PR1 in SPL knockdown (SPL-KD) and vector control DLD1 cells. (F) miR-181b-1 and (G) miR-21 expression in nonneoplastic colon tissues of control and KO mice. (H) Protein expression of PTEN and CYLD in nonneoplastic colon tissues of control and KO mice. *P < 0.05.
The major S1P receptor S1PR1 has been shown to be a downstream transcriptional target of STAT3 (15). To determine whether S1PR1 is upregulated in response to colonic STAT3 activation, we performed a comparative analysis of murine S1pr1 gene expression by qRT-PCR in the gut epithelia of WT and SPLGutKO mice after AOM/DSS treatment. As a control for the interaction between STAT3 and S1pr1, we also analyzed gut epithelia of SPLGutKO mice that had been treated with STAT3 inhibitor NSC 74859 in addition to AOM/DSS. We observed a higher level of S1pr1 expression in SPLGutKO mouse colonic tissues compared with that in control mouse tissues (Figure 3D). In contrast, SPLGutKO mice treated with STAT3 inhibitor showed a significant reduction of S1pr1 expression, well below that of either control or SPLGutKO mice. To avoid potential paracrine effects associated with inflammation, we also compared human S1PR1 expression in DLD1 colon cancer cells in which SPL was silenced by a lentiviral shRNA with that in lentiviral vector control cells. We observed that SPL silencing was associated with significant S1PR1 upregulation in DLD1 cells (Figure 3E). These findings confirm the interaction between STAT3 and human S1PR1 in colonic epithelium.
STAT3 was recently shown to play a direct and crucial role in mediating cell transformation through the induction of 2 miRNAs, miR-21 and miR-181b-1 (9). These miRNAs are STAT3 targets that repress the expression of the well-established tumor suppressor PTEN and the lesser known tumor suppressor CYLD, respectively. Therefore, we used qRT-PCR to address whether SPL disruption in gut epithelium altered the expression of miR-21 and miR-181b-1 and their respective targets in colonic tissues of AOM/DSS-treated mice. We observed a 25-fold increase in miR-21 expression levels and a 12-fold increase in miR-181b-1 expression levels in nonneoplastic colonic tissues of SPLGutKO mice compared with those in control mice (Figure 3, F and G). Further, induction of miR-21 and miR-181b-1 correlated with repression of PTEN and CYLD in nonneoplastic colon tissues of SPLGutKO mice compared with that in control mice (Figure 3H and Supplemental Figure 3C). These findings indicate that SPL disruption enhances JAK2/STAT3 signaling and promotes induction of STAT3-activated miRNAs that mediate cell transformation through suppression of PTEN and CYLD.
Inhibition of STAT3 ameliorates the pathological effects of gut SPL knockdown.
To determine whether STAT3 activation plays a critical role in mediating the proinflammatory and protumorigenic effects of gut SPL disruption, we administered the STAT3 inhibitor NSC 74859 or vehicle to SPLGutKO mice throughout the course of AOM/DSS treatment. STAT3 inhibition significantly reduced inflammatory cytokines, including the levels of TNF-α, IL-1β, and IL-6, in SPLGutKO mouse colons compared with that in vehicle-treated SPLGutKO mouse colons (Figure 4A). Pathological shortening of the colon was also reduced in SPLGutKO mice treated with NSC 74859 (Figure 4B). Importantly, STAT3 inhibition reduced the frequency of colon tumors in SPLGutKO mice (Figure 4C). Treatment with NSC 74859 also suppressed expression of miR-181b-1 and miR-21 (Figure 4, D and E) and normalized PTEN and CYLD expression in nonneoplastic tissues of SPLGutKO mice (Figure 4F and Supplemental Figure 4). These findings suggest that loss of SPL in the gut epithelium promotes CAC through a STAT3-dependent mechanism that results in induction of STAT3-activated miRNAs and suppression of their target tumor suppressor proteins.
Figure 4. Loss of SPL in gut epithelium promotes cytokine production, PTEN and CYLD repression, and colon tumorigenesis in a STAT3-dependent manner.

KO mice were treated with AOM/DSS with or without NSC 74859 (10 mg/kg i.p.) or saline every other day on days 30 to 60. White bars represent KO plus saline, and black bars represent KO plus NSC 74859. (A) Colon inflammatory cytokines. (B) Average colon length. (C) Average tumor number per mouse. (D) miR-181b-1 expression. (E) miR-21 expression. (F) PTEN and CYLD protein expression. n = 6 per group. *P < 0.05.
Loss of SPL in gut epithelium promotes STAT3 activation, induction of STAT3-activated miRNAs, and inflammatory signaling after DSS treatment.
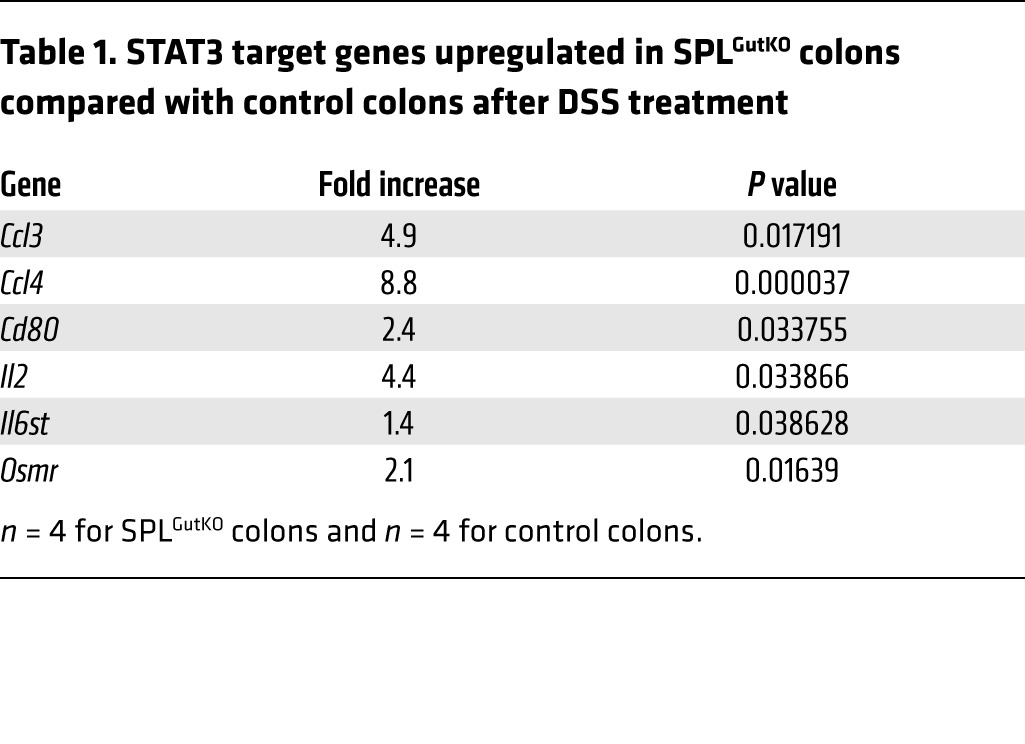
To characterize the timeline by which SPL inactivation in gut epithelium promotes colonic inflammation, we compared the effects of 2 rounds of treatment with 3% DSS in the drinking water on SPLGutKO mice and controls. DAI scores were significantly higher in SPLGutKO mice than in control mice after short-term DSS treatment (Figure 5A). Also, colon lengths were shorter in SPLGutKO mice than in control mice (Figure 5B). Treatment of control mice with a DSS regimen increased colon S1P levels, and DSS-associated S1P accumulation in the colon was higher in DSS-treated SPLGutKO mice than in DSS-treated controls (Figure 5C). In contrast, we did not observe changes in plasma S1P levels after DSS treatment or the complete AOM/DSS regimen (Supplemental Figure 5A). STAT3 activation was higher in SPLGutKO mouse colons (Figure 5D). This finding was corroborated by higher STAT3 transcriptional target gene expression levels in SPLGutKO mice, as judged by qRT-PCR (Table 1, Supplemental Table 2, and Supplemental Figure 5B). Specifically, of 86 STAT3 transcriptional targets queried, 28 were upregulated by 2-fold or greater (and 6 target genes were highly upregulated) in SPLGutKO mice. No STAT3 target genes were downregulated in SPLGutKO mice compared with control mice. The most significantly upregulated genes in SPLGutKO mice included macrophage inflammatory protein 1α (Ccl3), macrophage inflammatory protein 1β (Ccl4), Cd80, Il2, oncostatin M receptor (Osmr), and interleukin 6 signal transducer (Il6st) (significant but not 2-fold upregulated) (all of the significantly upregulated genes are involved in gp130 signaling, proinflammatory, and chemokinetic functions). In addition to STAT3 gene targets, the STAT3-activated miRNAs miR-21 and miR-181b-1 were higher in colons of SPLGutKO mice than in control mouse colons (Figure 5, E and F). Thus, gut SPL downregulation promotes early inflammatory responses in murine gut mucosa, including STAT3 activation, STAT3-mediated target gene induction, and induction of STAT3-activated miRNAs.
Figure 5. The S1P/SPL/STAT3 axis in experimental murine colitis and human IBD.

Control and KO mice were treated for 2 cycles of 3% DSS. (A) DAI. (B) Colon length. (C) Colon S1P levels in WT mice treated with vehicle or DSS and control and KO mice treated with DSS. (D) IHC (original magnification, ×10) of activated STAT3 phosphorylated on Tyr705 in representative colon sections of DSS-treated control and KO mice. (E) Colon miR-181b-1 expression. (F) Colon miR-21 expression. n = 4 per group. (G–K) Colon tissues from patients with IBD and controls evaluated for expression of S1P-related genes. (G) SGPL1. Control, n = 6; IBD, n = 8. (H) SPHK1. Control, n = 6; IBD, n = 8. (I) S1PR1. Control, n = 4; IBD, n = 7. (J) miR-181b-1 expression in control (n = 4) and IBD (n = 5) colons. (K) miR-21 in control (n = 5) compared with IBD (n = 6) colons. *P < 0.05.
Table 1.
STAT3 target genes upregulated in SPLGutKO colons compared with control colons after DSS treatment
Colitis in human patients with IBD is associated with enhanced S1P and STAT3 signaling.
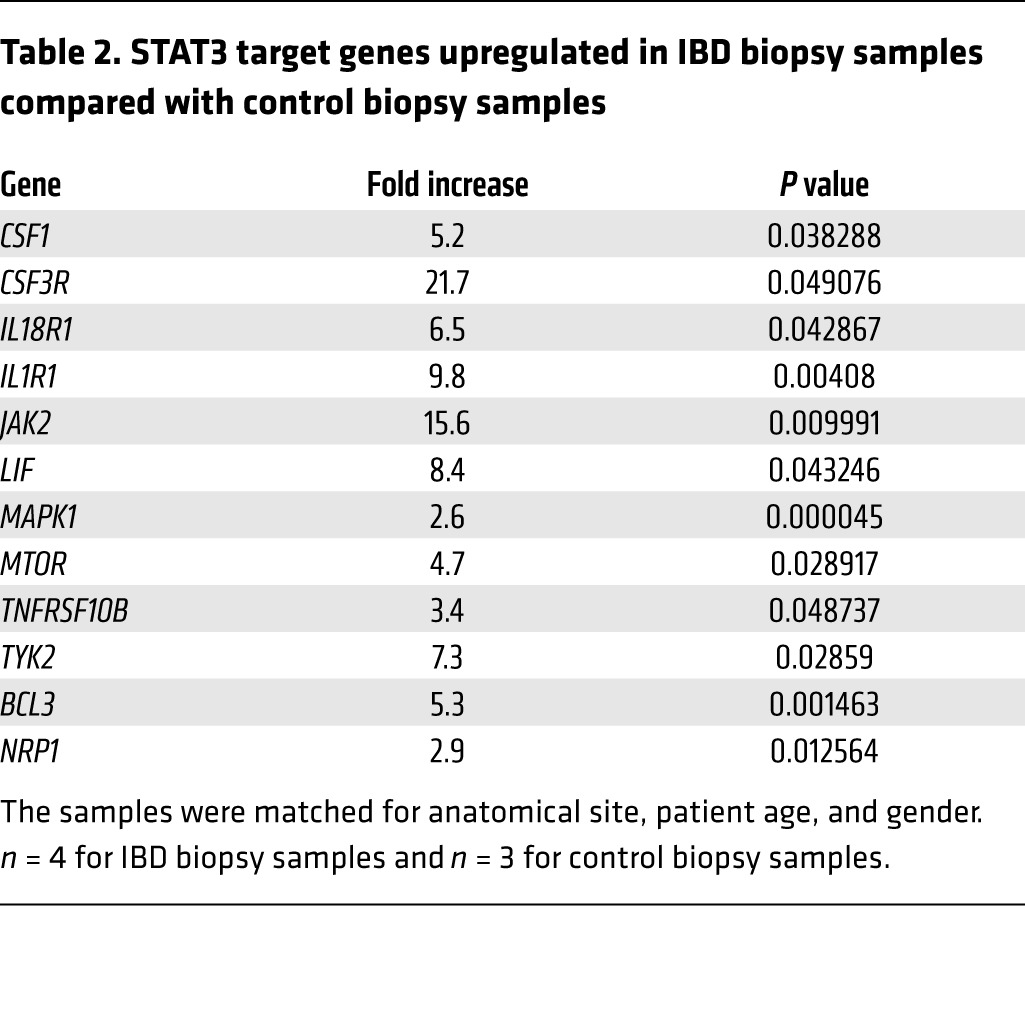
To investigate the relevance of our findings to the pathophysiology of IBD, we first measured the expression of the two key genes of S1P metabolism, SPHK1 and SGPL1, in colon tissues of patients with IBD and controls. We observed significant upregulation of SPHK1 in IBD colons compared with control colons, whereas SGPL1 expression was reduced (Figure 5, G and H). The finding of increased SPHK1 is consistent with the findings of Snider and colleagues, who have demonstrated elevated SPHK1 protein expression in human IBD samples (30). Expression of S1PR1, the major S1P receptor and a target of STAT3 activation, was also elevated in IBD colons (Figure 5I). Comparison of STAT3 target gene expression in IBD and control colons revealed a consistent pattern of enhanced STAT3 signaling in IBD colons (Table 2, Supplemental Table 3, and Supplemental Figure 5C). The average expression levels of 66 of 86 STAT3 target genes examined were upregulated more than 2-fold in IBD colon tissue compared with that in control colon tissue, with 12 of these exhibiting a highly significant increase, whereas no STAT3 target genes were downregulated in IBD. The genes in this list include JAK family members JAK2 and TYK2, proinflammatory cytokines, and receptors (e.g., TNFRSF10B, IL1R1, IL18R1, macrophage colony-stimulating factor 1 [CSF1] and its receptor CSF3R, and the antiinflammatory cytokine leukemia inhibitory factor [LIF]). Interestingly, the upregulation of CSF3R and CSF1 suggests that macrophage recruitment is a hallmark of IBD, consistent with our finding of elevated macrophages in the colon tissues of SPLGutKO mice. Several members of the list of upregulated genes are components of signaling pathways in which S1P plays a role, including TNFRSF10B, MAPK, MTOR, JAK2, and IL1R1. Other targets included the proto-oncogene and NF-κB–interacting protein BCL3 and NRP1 (a signaling protein involved in cell migration and regulation of Treg lymphocytes). In addition, the STAT3-activated miRNA miR-181b-1 was significantly elevated in IBD colons (Figure 5J), and miR-21 showed a trend toward upregulation in IBD colons (Figure 5K).
Table 2.
STAT3 target genes upregulated in IBD biopsy samples compared with control biopsy samples
SPL downregulation promotes cell transformation through a molecular pathway that involves activation of S1PR1, JAK2, and STAT3 and miR-dependent silencing of CYLD.
Forced overexpression of SPHK1 or molecular downregulation of SPL in embryonic fibroblasts result in accumulation of S1P and are associated with the acquisition of the transformed phenotype (17, 18). Based on our in vivo findings, we hypothesized that SPL downregulation might mediate cell transformation in a STAT3-dependent manner. To address this question, we used a lentiviral system to silence SPL in WT murine embryonic fibroblasts (MEFs) as well as in MEFs in which the Stat3 gene was inactivated by Cre-lox technology (ref. 31, Figure 6A, and Supplemental Figure 6A). Silencing of SPL in Stat3+/+ MEFs did not affect intracellular S1P levels (Figure 6B). However, SPL silencing resulted in an increase in extracellular S1P levels and a corresponding increase in expression of the S1P transporter spinster homolog 2 (Spns2) (Figure 6, C and D). When Spns2 was silenced using siRNA, the increase in extracellular S1P levels observed in response to SPL knockdown was attenuated (Figure 6E). Importantly, SPL silencing in Stat3+/+ MEFs led to an increase in proliferation rate compared with vector control cells, and this growth advantage was reversed when Spns2 was silenced using siRNA (Figure 6F). SPL knockdown additionally resulted in elevated Il6 expression (Figure 6G). In contrast, Stat3–/– MEFs exhibited low intracellular and extracellular S1P levels, low Il6 expression, and no change in Spns2 expression regardless of SPL status (Figure 6, B–D and G). Treatment of Stat3+/+ MEFs with 125 nM S1P led to the activation of both JAK2 and STAT3. This was shown by an increase in the phosphorylated forms of both proteins, as detected by IB of whole cell extracts (Figure 6H), as well as the ratio of phosphorylated to total protein levels obtained by image quantification and normalization (Supplemental Figure 6, B and C). Further, MEFs in which SPL was silenced exhibited higher baseline activation of JAK2 and STAT3 compared with control MEFs, consistent with the increase in S1P exported from these cells (Figure 6H). Whereas silencing of SPL in Stat3+/+ MEFs increased cell proliferation compared with untreated control cells, silencing of SPL in Stat3–/– MEFs did not affect proliferation (Figure 6I). More importantly, SPL silencing in Stat3+/+ MEFs resulted in cell transformation, as determined by the ability of MEFs to form tumors in NOD/SCID mice, whereas SPL silencing in Stat3–/– MEFs failed to induce transformation/tumor formation (Figure 6J). Stat3+/+ MEFs in which SPL was silenced exhibited higher levels of the STAT3-activated mRNA miR-181b-1, whereas Stat3–/– MEFs in which SPL was silenced did not (Figure 6K). In contrast, levels of miR-21 were similar in all cell lines (E. Degagné, unpublished observations), which suggests that this gene is not activated by STAT3 in MEF cells. Consistent with the induction of miR-181b-1, we found that expression of its target gene, Cyld, was repressed in tumors from Stat3+/+ MEFs in which SPL was silenced compared with control Stat3+/+ MEFs and compared with Stat3–/– MEFs (either containing or lacking SPL) (Figure 6L). We were interested in exploring the potential role of S1PR1 in mediating the effects of SPL silencing on transformation. Therefore, we evaluated the effects of treating MEFs with W123 (5 μM), a competitive antagonist of S1PR1, or with FTY720 (100 nM), a nonselective antagonist of S1PR1, on the proliferative capacity of the SPL knockdown MEF line compared with the control MEF line. We also tested the impact of inhibiting STAT3 pharmacologically using NSC 74859 (25 μM) or the JAK2/STAT3 inhibitor, WP1066 (1 μM). In addition, we tested the effects of treatment with rolipram (10 μM), an inducer of the CYLD anti-oncogene targeted by miR-181b-1. We found that the growth advantage displayed by SPL knockdown MEFs was reversed by inhibiting S1PR1 as well as by inhibiting STAT3 (Figure 6M). Importantly, rolipram also reversed the growth-activating effects of SPL knockdown (Figure 6M). These cumulative findings implicate a pathway to cell transformation that is initiated by SPL silencing and involves extracellular transport of S1P via SPNS2; activation of S1PR1, JAK2, and STAT3; induction of STAT3-activated miR-181b-1; and suppression of the miR-181b-1 target, the anti-oncogene CYLD.
Figure 6. SPL downregulation promotes cell transformation in a STAT3-dependent manner.

(A) IB of SPL and STAT3 expression in Stat3+/+ and Stat3–/– MEFs in which SPL was silenced (SPL knockdown) compared with vector-only control (C) cell lines. (B) Intracellular and (C) extracellular S1P levels in MEF lines. (D) Spns2 mRNA expression levels in MEF lines. (E) Extracellular S1P levels in Stat3+/+ SPL knockdown MEFs with or without SPNS2 siRNA and MEF Stat3+/+ control cells. (F) Cell proliferation at day 3 of MEFs grown in 0% FBS in Stat3+/+ SPL knockdown MEFs with and without SPNS2 siRNA and MEF Stat3+/+ control cells. Representative bar graph from 1 individual experiment is shown. Statistical analysis was performed with data from 3 independent experiments. (G) Il6 mRNA expression levels in MEF lines. (H) Protein expression of activated JAK-2 (JAK2-P), total JAK-2 (JAK2-T), activated STAT3 (STAT3-P), and total STAT3 (STAT3-T) in control and SPL knockdown Stat3+/+ MEFs, following treatment with 125 nM S1P. (I) Cell proliferation of MEFs grown in 0% FBS. (J) Time course of tumor growth, following s.c. injection of MEF cell lines into NOD/SCID mice. Blots shown are from different gels. (K) miR-181b-1 expression and (L) Cyld expression in tumors or injection sites of NOD/SCID mice. (M) Cell proliferation at day 3 of MEFs grown in 0% FBS with or without W123 (10 μM), FTY720 (100 nm), rolipram (10 μM), NSC 74859 (25 μM), and WP1066 (1 μM) added at day 0. *P < 0.05 compared with control Stat3+/+ MEFs. #P < 0.05 compared with control SPL knockdown Stat3+/+ MEFs.
Oral SDs induce SPL expression, reduce colon S1P levels, prevent STAT3 activation, and reduce cytokine levels and CAC.
Our results indicate that SPL downregulation is a key step in the development of CAC. We next asked whether it was possible to reverse SPL downregulation in the gut, thus making it a potential target for chemoprevention. We recently reported the chemopreventive activity of SDs, compounds derived from the metabolism of sphingolipids in soy and other nonmammalian food sources (32, 33). SDs are cytotoxic to colon cancer cells and reduce adenoma formation when administered orally to ApcMin/+ mice (32, 33). To address the effects of SDs in colitis, we evaluated colon S1P levels in WT mice treated with 25 mg/kg SD (or vehicle alone) by gavage for 6 days. SD treatment significantly reduced colon S1P levels (Figure 7A). When WT mice were treated with DSS with or without the addition of oral SDs, the SD-treated mice exhibited a slight reduction in symptoms, as determined by DAI (Figure 7B). Further, loss of body weight was lower in SD-treated mice (Figure 7C). To investigate the mechanism by which SDs reduce S1P levels, we next measured the effect of SDs on SPL expression in human colon cancer cells. Importantly, SPL protein expression was induced in DLD1 colon cancer cells after a 24-hour incubation with SDs (applied at concentrations as low as 10 μM) (Figure 7D and Supplemental Figure 7A). To test whether SDs attenuate CAC, SDs were administered by gavage to WT mice receiving the AOM/DSS regimen. SPL expression levels were higher, and activated STAT3 levels were lower, in colon tissues of mice treated with AOM/DSS plus SDs compared with mice treated with AOM/DSS plus vehicle (Figure 7E and Supplemental Figure 7B). These results establish that SPL upregulation can be achieved in vivo by oral SD treatment. We next evaluated the impact of SDs on cytokines and CAC. SD treatment suppressed cytokine levels (including TNF-α, IL-6, IL-21, and IL-23) in CAC in response to AOM/DSS (Figure 7F). Importantly, SDs reduced colon tumor incidence in response to AOM/DSS (Figure 7G). In contrast to the effects of SDs on tumorigenicity, the mammalian sphingoid base sphingosine had no affect on tumor number (Figure 7G). Finally, STAT3 activation and downstream tumor-promoting signals were reduced in response to SDs, as shown by reduced STAT3 activation in nonneoplastic colon tissue (as shown by IF microscopy) (Figure 7H) and higher expression of tumor suppressor proteins PTEN and CYLD in colons of SD-treated mice (Figure 7I and Supplemental Figure 7C). These findings demonstrate that SDs induce SPL, lower colonic S1P levels, and inhibit cytokines, STAT3 activation, protumorigenic signaling, and tumor formation in CAC.
Figure 7. Oral SD induces SPL expression, reduces colon S1P levels, prevents STAT3 activation, and reduces cytokines and CAC.

(A) Colon S1P levels in WT mice treated with 25 mg/kg SD (n = 4) or vehicle (n = 8) for 1 week. (B) DAI of SD-treated mice compared with vehicle-treated mice. (C) Percentage of weight loss in SD-treated and vehicle-treated mice. (D) SPL expression in SD-treated DLD1 cells for 24 hours. (E) SPL, phosphorylated STAT3 (STAT3-P), and total STAT3 (STAT3-T) expression in WT mice treated with AOM/DSS plus 25 mg/kg SD or vehicle. Blots shown are from different gels. (F) Colon inflammatory cytokines in vehicle-treated (white bars) and SD-treated (black bars) mice. (G) Average tumor number per mouse in vehicle, SD-treated, or sphingosine-treated (25 mg/kg) mice. (H) IF of activated STAT3 phosphorylated on Tyr705 (STAT3-P, green) in colon tissues of SD-treated and vehicle-treated mice. Nuclei were counterstained with DAPI. Scale bar: 50 μm. (I) PTEN and CYLD protein expression in colons of SD-treated and vehicle-treated mice. *P < 0.05.
Discussion
Although SPL controls tissue levels of the immunoregulatory and mitogenic lipid S1P and is enriched in gut epithelium and downregulated in intestinal tumors of mice and humans, its impact on colitis and colon carcinogenesis has hitherto remained unknown. The strikingly proinflammatory and protumorigenic phenotype of SPLGutKO mice demonstrates an important role for SPL in the regulation of global inflammatory responses, innate immune functions of the gut epithelium, and CAC.
Sgpl1 deletion in the SPLGutKO mouse is highly specific and nearly complete. Thus, this mouse model is an ideal system for studying interactions among S1P, SPL, and dietary sphingolipids and their influence on colitis and CAC. Deletion of Sgpl1 in gut tissues does not result in early lethality (unlike the global Sgpl1 KO), which suggests that SPL expressed in other tissues can compensate and maintain circulating S1P levels at or near the physiologic state. Nevertheless, local tissue S1P levels are highly influenced by SPL, because SPLGutKO mice exhibit an accumulation of S1P in the gut. However, under basal conditions Sgpl1 deletion in intestinal epithelium does not affect levels of other sphingolipids, including sphingosine, dihydrosphingosine, total ceramide and sphingomyelin. SPLGutKO mice showed no abnormalities in growth, breeding, weight gain, or survival and no changes in colon inflammatory cytokines under routine laboratory conditions. However, following oral exposure to DSS (with or without the addition of the carcinogen AOM), SPLGutKO mice exhibited exacerbated symptoms compared with control mice. This phenotype was associated with pathological evidence of inflammation — increased proinflammatory cytokines along with increased numbers of tissue macrophages. In response to AOM/DSS treatment, SPL disruption was also associated with a higher tumor incidence and an increase in crypt cell proliferation in nonneoplastic colon tissues. SPL has been shown to regulate lymphocyte trafficking (34, 35). In addition, global Sgpl1 KO mice exhibit high levels of circulating inflammatory cytokines and increased liver expression of acute phase response genes (36). These findings occurred in association with an impairment of neutrophil recruitment into inflamed tissues, leading to disruption of the IL-23/IL-17/G-CSF cytokine signaling loop. Our study demonstrates that gut SPL plays a major role in regulating inflammatory signaling, particularly in response to stimuli from the gut lumen, independently of SPL activity in other tissues. These findings suggest that, by promoting local S1P accumulation, STAT3 signaling, and inflammatory responses, tissue-specific Sgpl1 KO mice may serve as a unique model of inflammatory disease, including but not necessarily limited to the gut.
Intestinal epithelial cells are an integral part of the innate immune system. They represent the first line of defense against pathogens and other harmful agents (37). They are also known to secrete multiple cytokines and chemokines that lead to chemoattraction of leukocytes, monocytes/macrophages, and lymphocytes (37). This can result in a counterproductive inflammatory response in some disease states, such as IBD, in which inflammation can lead to tissue destruction. STAT3 pathway activation has been observed in many autoimmune and inflammatory-related diseases. Two members of the S1P receptor family, S1PR1 and S1PR2, have been identified as regulators of the STAT3 pathway (15, 38). S1PR1 signaling activates the JAK2/STAT3 pathway by increasing production of IL-6 (15). S1PR2-related STAT3 pathway activation requires activation of Src through phosphorylation (38). A recent study demonstrated that S1P activation links persistent STAT3 activation to CAC (23). In that study, compensatory upregulation of SPHK1 in a global Sphk2 KO mouse was shown to promote CAC. Further, the authors demonstrated that disruption of SPHK2 in hematopoietic cells recapitulated the effects of the global Sphk2 KO, indicating that S1P in hematopoietic cells contributed to STAT3 activation and CAC. Our findings also demonstrate that S1P signaling contributes to STAT3 activation, colitis, and CAC. In contrast to the previous study, however, our findings demonstrate that SPL activity within the gut epithelium and its impact on local S1P levels and epithelial cell STAT3 activation serve to organize and promote innate immune functions of the gut epithelium — independently of S1P metabolism in hematopoietic cells and independently of circulating S1P levels. Our findings suggest that SPL downregulation in adenomas may affect the surrounding colon niche in ways that exacerbate carcinogenesis. Specifically, by increasing local S1P levels, SPL downregulation in adenomas may promote STAT3-mediated inflammatory signaling within colonic epithelial cells, cytokine release from these cells, recruitment of macrophages and other inflammatory cells, and STAT3-dependent neoplastic transformation and tumorigenesis. Consistent with this notion, others have observed that, in more than half of specimens examined from patients with colon cancer, nonneoplastic tissues adjacent to colon cancers exhibited significant STAT3 activity, suggesting that STAT3 activation may precede pathologic alterations in intestinal tissue (39).
We found evidence of STAT3 activation in SPLGutKO colon epithelium based on IF and IHC staining of colon tissues and elevation of phosphorylated JAK2, STAT3, and STAT3 target expression, including the STAT3 target S1PR1. This finding is consistent with the observation of others that S1P and STAT3 signaling is linked in a coactivating pathway, in which S1P acts through S1PR1 to activate JAK2/STAT3. STAT3 is a transcriptional activator of IL-6 and S1PR1 (15, 40). Whereas the prior studies used artificial expression systems to demonstrate this interaction, we have found that treatment of WT MEFs promotes JAK2 activation in a manner that requires S1P export via SPNS2 and extracellular S1PR1 signaling.
STAT3 controls the expression of miR-21 and miR-181b (9). miR-21 has been shown to be upregulated in patients with Crohn’s disease and ulcerative colitis (41). Moreover, miR-21 and miR-181b expression levels are increased in colorectal cancer, and expression levels have been linked to a poor prognosis in an American cohort (42). In this study, we demonstrated that these 2 STAT3-activated miRNAs were significantly increased in nonneoplastic colon tissues lacking SPL. Further, we showed that the respective targets of miR-21 and miR-181b-1, PTEN, and CYLD, were suppressed in nonneoplastic SPLGutKO tissues. Our finding that STAT3-activated miRNAs are induced in nonneoplastic tissue adjacent to tumors and in DSS-treated mice prior to the onset of tumor formation supports the notion that SPL downregulation and consequent S1P accumulation may initiate a process of STAT3-dependent signaling that contributes to cell transformation and CAC.
We observed that colon S1P levels rose in WT mice treated with a regimen of oral DSS without AOM and that the accumulation of colonic S1P was exacerbated in SPLGutKO mice. The higher S1P levels in SPLGutKO mouse colons correlated with worse DAI scores, worse inflammation (as evidenced by shorter colon length), and a rise in STAT3 activation and induction of STAT3-activated miRNAs. The significance of the S1P/STAT3 signaling hub in colitis was further demonstrated by the elevated levels of SPHK1, S1PR1, and STAT3-related gene expression as well as induction of STAT3-activated miRNAs in colitic bowel compared with normal control bowel. CAC is linked with increased expression of proinflammatory cytokines and chemokines that results in the sustained activation of NF-κB and STAT3 pathways (7, 43). The elevated STAT3 signaling observed in IBD colon tissue suggests that STAT3-dependent inflammatory signaling may initiate the process of carcinogenesis by promoting cell proliferation and onco-miRNA expression within the colon niche.
SPL disruption in gut epithelium promoted CAC tumor incidence without affecting tumor volume. This suggests that the effect of SPL silencing on tumorigenesis occurs at an early step. Consistent with this notion, silencing SPL in MEFs resulted in increased proliferation and enabled tumor formation in immunodeficient mice. These effects correlated with elevated extracellular S1P levels and elevated expression of Il6 and the S1P transporter Spns2, each of which were dependent upon the presence of STAT3, as they were not observed in Stat3-null MEFs. Extracellular S1P accumulation and the growth advantage conferred by SPL knockdown in MEFs were dependent on export of S1P into the extracellular environment via SPNS2. The growth advantage also depended upon the activity of S1PR1. Further, miR-181b-1 was significantly elevated in tumors that formed from xenografted SPL-silenced Stat3+/+ MEFs compared with that in tissues from mice xenografted with MEFs harboring WT SPL levels. These findings confirm that SPL downregulation promotes tumorigenesis through STAT3-mediated cell transformation and suggest the specific involvement of miR-181b-1 and CYLD. This notion is further supported by our observation that the increased rate of cell proliferation exhibited by MEFs lacking SPL was reversed when the cells were treated with an inducer of CYLD. We do not exclude the possibility that SPL may influence later stages in the complex process of tumorigenesis, for example, angiogenesis or inflammatory effects on tumor progression. However, our study demonstrates that the link between SPL/S1P and STAT3 is critical for transformation and thereby establishes their role in promoting the translation of inflammatory signals into the earliest steps of carcinogenesis. Further, the induction of miR-181b-1 caused by SPL silencing suggests that, in addition to the putative role of S1P as a cofactor in the NF-κB activation pathway, S1P may promote NF-κB signaling indirectly via suppression of the NF-κB repressor, CYLD, itself a target of miR-181b-1. Our findings are consistent with several recent reports demonstrating that NF-κB can be activated independently of intracellular S1P synthesis via SPHK1 and that S1P can have proinflammatory effects that are independent of direct NF-κB activation (44, 45).
SDs are sphingolipids found in soy, spinach, eggplant, and other natural products. We have shown previously that SDs are cytotoxic to colon cancer cells but not normal intestinal epithelial cells and reduce tumorigenicity in the ApcMin/+ mouse model of colon cancer (24, 33). Dietary SDs are antiinflammatory lipids that are taken up by the gut but are poorly absorbed, making them well suited as colon cancer chemoprevention agents (46, 47). SDs derived from soy and other nonmammalian dietary sources cannot be metabolized to S1P and are not readily incorporated into mammalian sphingolipids (46). This fate of SDs stands in contrast to mammalian sphingolipids, which are readily metabolized to sphingosine, the substrate for sphingosine kinase, which is present in the colon and generates S1P from sphingosine. Importantly, whereas SDs inhibited the development of CAC, the mammalian sphingoid base sphingosine had no effect. Although we did not observe enhancement of tumorigenesis by sphingosine, we suspect that longer exposure to sphingosine might potentiate tumor formation via S1P generation. The critical distinction in the metabolic fate of dietary sphingolipids from different food sources, combined with our findings and those of others implicating S1P in IBD and CAC (23), provide a biochemical link between diet, inflammation, and cancer. Our findings suggest that dietary sphingolipids may augment or prevent colon cancer, depending on their ability to be converted to S1P. In addition, our finding that oral SDs induce SPL expression in colon epithelial cells in vitro as well as in vivo demonstrates that SPL downregulation during intestinal tumorigenesis is a reversible step that can be targeted through dietary or nutraceutical strategies to enhance S1P metabolism and inhibit the S1P/STAT3 signaling pathway.
In summary, our study demonstrates that gut epithelial SPL, which is highly expressed in healthy gut mucosa but downregulated in intestinal adenomas, plays a critical role as a gatekeeper of intestinal carcinogenesis that acts by regulating extracellular S1P levels and a STAT3-activated miRNA-mediated process of cell transformation. Further, our study solves the conundrum that dietary sphingolipids exhibit chemopreventive activity against colon cancer whereas the sphingolipid metabolite S1P is proinflammatory and serves to facilitate carcinogenesis by demonstrating that sphingolipids from different sources may promote or prevent cancer, depending upon their ability to either be metabolized to S1P or, alternatively, to promote S1P metabolism.
Methods
Study design.
We used animal models of colitis or CAC in mice in which the expression of SPL was disrupted in colonic epithelial cells. This enabled us to determine the role of SPL in these diseases by looking at proinflammatory cytokines, S1P levels, miR-181b-1 and mir-21 expression, and their related target proteins. We also used MEF cells to determine whether the deletion of SPL would affect S1P metabolism and promote tumorigenesis in a xenograft model of NOD/SCID mice. All animal used were randomly assigned to experimental groups, and all experiments were repeated at least 3 times.
Animals.
The Sgpl1fl/fl AhCre transgene (SPLGutKO) and Sgpl1fl/fl (control) lines were generated with a sequence-replacement targeting vector designed to insert a gene-trapping cassette containing a lacZ reporter in intron 8 of Sgpl1 (48). This strategy was based on a report showing that deletion of exons 9 to 11 results in SPL-null embryonic stem cells (49). The gene-trapping vector, which was flanked by Frt sites, consisted of a splice acceptor site (flanked by a loxP and a lox71 site), followed by a promoter-less β-geo cassette. This is the same gene-trapping cassette used by the Knockout Mouse Project (50). The gene-trapping cassette was designed to yield an in-frame fusion with exon 9 of Sgpl1. The gene-targeting vector also inserted a loxP site into intron 12.
To create a targeting vector, a mouse genomic C57BL/6 BAC library (library ID: CHORI-39) was constructed with over 200,000 clones (160-kb average insert size). The library is “recombineering-ready” by using retrofitted BAC cloning vector with integrated λ-Red–inducible recombineering system into pBACe3.6. Arrayed 528- × 384-well BACs were gridded on eleven 22- × 22-cm nylon high-density filters for screening by probe hybridization (51). The filters were screened by 2 “overgo” probes (52) for the Sgpl1 gene: GACAGTCTACCAAAGCCAGCTCTTCCCCTCGAACAATGTA and CACCACTGATGAACCTTTAGGAGCATAGCCATACTGAGGG. Three BAC vectors (clone ID: CH39-118K18, CH39-138B21, and CH39-276E19) were confirmed to include the Sgpl1 gene, and one BAC vector (BAC CH39-118K18) was used to construct a targeting vector through 3 recombineering and 2 site-specific recombination and L/R Gateway reactions. This BAC vector–based λ-Red recombineering system was the same system described for the prophage λ-Red recombineering system (53). Briefly, λ-Red was under the control of temperature-sensitive cI-repressor (allele cI857), and the recombination was suppressed at 32°C. Incubation at 42°C for 15 minutes inactivates the repressor and induces λ-Red exo, bet, gam genes. The recombinant cassettes were PCR amplified with chimeric oligos consisting of the 5′ 50-nt primer site homologous to the genomic target and the 3′ 20-nt to 22-nt primer sites to amplify the recombinant cassette with positive (and negative) selection. Two sequential recombineering steps placed Gateway attR1 and attR2 sites flanking a positive/negative region (zeocin/PheS) (54) and selection markers upstream of exon 10 between AAACCAGTGATTACATTGTCCTCACAGCAAAAGGCATAAGCAGCTAGGAC and CTCTCTTCATTGCCTAGGGATTTTAAAGTTTAAGAGAAGTAAGACTCTTA sites and floxed kanamycin marker downstream of exon 12 between TGATCTCCATAAAACCCACCTGTCCCAGCCTAGCCTCTGTAGTGTAAGAG and TCCTTATCTCAGTTGATGATCAGAGCCTACTCTCCTGGGACTTGACTTGT. An approximately 9-kb fragment between ATAGTTGAGATTAAGTCTACAAATCAGCAACAACACCCTGTGGTCTAGCC and TGTGTATATGTCCACTTCCCTCCTGTATGCTCTACAGTGCATTATGAGG, including the 2 modified sites, was retrieved by recombineering into a p15A-origin plasmid. Clones with the retrieved genomic fragment were obtained following transformation into the EL350 E. coli strain (55) and selection using zeocin (Life Technologies) and ampicillin (Sigma-Aldrich). The EL350 E. coli strain includes an arabinose-inducible Cre-recombinase (55) permitting the removal of the floxed neomycin phosphotransferase II (nptII), confirmed by comparing growth of candidate clones on Petri dishes containing or lacking kanamycin (Sigma-Aldrich). The intermediate constructs were also checked by PCR for the presence of all new recombination junctions. The final construct was prepared by an in vitro Gateway reaction to replace the attR1/attR2 flanked fragment with the attL1/attL2 flanked mammalian selection/reporter cassette. This cassette includes a β-galactosidase reporter and neomycin (G418) markers (flanked by FRT sites) and a 5′ lox71 site. The FRT sites permit removal of the reporter/selection cassette following proper targeting in ES cells, such that the knockin allele is converted into a conditional allele.
The gene-targeting vector was electroporated into 129/OlaHsd strain embryonic stem cells, which were then subjected to G418 selection. G418-resistant embryonic stem cell clones that produced a fusion transcript extending from exon 9 of Sgpl1 to β-geo were identified by 5′ rapid amplification of 5′ complementary DNA ends. PCR was used to identify clones that also retained the distant loxP site (56).
A targeted clone was injected into C57BL/6 blastocysts to generate chimeric mice. Male chimeras carrying the reporter transgene were crossed with female C57BL/6 mice. Germ line transmission of the reporter was confirmed by PCR, RT-PCR, and IHC. The lacZ reporter was removed by breeding the mice with FLPR transgenic mice, generating mice with a floxed Sgpl1 allele (Sgpl1fl). To test the fidelity of that allele, Sgpl1fl/fl mice were bred to mice harboring a constitutively expressed Cre transgene. To assess recombination, we performed PCR on tail DNA with the forward and reverse primer pair “exon 8–9,” which yielded a 123-bp band in WT mice but no product in KO mice that had undergone complete recombination (Supplemental Table 1). Genomic PCR was also performed using the forward and reverse primer pair “exon 8–12,” which yielded a 612-bp band in WT mice and a 128-bp product in KO mice that had undergone recombination (Supplemental Table 1). All the pups examined exhibited the characteristic SPL KO phenotype (perinatal death). Also, circulating S1P levels were 7- to 10-fold higher in the Sgpl1fl/fl Cre transgene mice than in Sgpl1fl/fl mice. Western blotting, IHC, and enzyme assays confirmed complete loss of SPL expression and activity.
The Sgpl1fl/fl line was used to generate a second line in which intestinal epithelium-specific SPL inactivation could be induced with a Cre transgene that is driven by a cytochrome p450 promoter element that is upregulated in response to lipophilic xenobiotics such as NF (26). WT C57BL/6 mice were obtained from Taconic. Animals were housed under pathogen-free conditions in the CHORI AAALAC-accredited animal facility under 12-hour-light/dark cycles and received food and water ad libitum.
IF.
Colons were fixed in 4% paraformaldehyde and washed in PBS containing 0.01% Tween 20 (PBST). Sections were blocked with PBST containing 5% goat serum and 0.3% Triton X-100. After washing, sections were incubated with mouse anti-phospo-STAT3 Tyr705 (Cell Signaling), followed by incubation with secondary Alexa Fluor 488 goat anti-mouse IgG (Life Technologies) in PBST containing 5% goat serum. Sections were then counterstained with Hoechst after washing and mounted in Vectashield mounting medium (Vector Labs). Fluorescence was observed with a Carl Zeiss Axioskop 50 fluorescent microscope, and images were captured using Nikon DS-Ri1 camera.
IHC.
Formalin-fixed sections were processed and embedded in paraffin. They were deparaffinized and incubated for 30 minutes in 3% hydrogen peroxide/methanol to quench endogenous peroxidases. For SPL detection, sections were rinsed in PBS and immunostained with anti–murine SPL antisera at 1:200 dilution in 0.5% PBS/Ova albumin at 37°C for 1 hour after antigen retrieval with citrate buffer, pH 6.0, in a small autoclave set for 125°C for 2 minutes; slides were cooled for 1 hour at room temperature before adding secondary antibody. The secondary antibody was biotinylated anti-rabbit (Vector laboratories) diluted 1:1,000 in 0.5% PBS/Ova albumin and incubated for 30 minutes at room temperature. Sections were incubated with the Elite ABC Kit (Vector laboratories) for 30 minutes and rinsed in PBS. Signal was detected with DAB (Vector laboratories) for 2 minutes, and the slides were counterstained in hematoxylin. For phospho-STAT3 staining, a 1:50 dilution of anti-rabbit phospho-STAT3 from Santa Cruz Biotechnologies was used and protocol followed as written above for SPL staining. To measure tumor and crypt proliferation, Ki-67 staining was performed with Ki-67 rabbit monoclonal antibody (Vector Laboratories). Numbers of proliferating cells per crypt were determined by counting 50 well-oriented crypts per section.
IB.
After AOM/DSS treatment or DSS treatment, colon tissues were harvested, homogenized, and lysed, and samples were processed for protein IB, as described previously (57). IB was performed using a 1:1,000 dilution of rabbit anti-phospho-STAT3 (Tyr705), anti-phospho-JAK2, anti-c-MYC, anti-MCL1, anti-PTEN, anti-CYLD, and anti-SPL antibodies. Specific protein bands on membranes were detected with a 1:10,000 dilution of HRP-conjugated anti-rabbit as the secondary antibody and visualized on autoradiographic films with the SuperSignal West-Pico Kit (Pierce) and quantified by densitometry with ImageJ software (NIH). For normalization of signals, membranes were stripped of antibodies and reprobed with a 1:5,000 dilution of mouse anti-STAT3, a 1:1,000 dilution of rabbit anti-JAK2, a 1:1,000 dilution of rabbit anti-GAPDH, or a 1:10,000 dilution of mouse monoclonal anti-β-actin, followed by a 1:10,000 dilution of HRP-conjugated anti-rabbit IgG or HRP-conjugated anti-mouse IgG, respectively (57).
SPL activity.
SPL activity was measured in intestinal tissues by HPLC using a 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene–labeled (BODIPY-labeled) fluorescent product detection system as described previously (58).
S1P quantitation.
S1P was extracted from 10 μl mouse plasma by adding 0.4 ml methanol, followed by vortexing and 30-minute incubation at 30°C. The sample was spun down at 14,000 g, and the supernatant was recovered. C17-S1P was used as an internal standard. Lipids were separated on a C18 column (2.1 × 50 mm; Kinetex) (Phenomenex) at a flow rate of 0.25 ml per minute. The gradient used was from 45% to 99% methanol, containing 1% acetic acid and 5 mM ammonium acetate. The data were acquired with electrospray ionization in the positive mode on a Micromass Quattro LC mass spectrometer (Waters Corp.). Lipids were identified based on their specific precursor and product ion pair and quantified with multiple reaction monitoring as described previously (59). For intestinal S1P measurements, extraction was performed by homogenizing the tissue in 1 ml methanol using a homogenizer (Omni TH). Homogenates were combined with 100 pmol C17-S1P internal standard and 2 ml chloroform/methanol (1:2). Samples were incubated overnight at 48°C. The samples were spun down to collect the supernatant and dried under nitrogen. S1P extraction was accomplished by 2-phase separation. Samples were resuspended in 1.5 ml chloroform/methanol (2:1). The extract was made basic by adding 50 μl 1 M KOH in methanol. To accomplish 2-phase separation, 0.25 ml water was added. The aqueous phase was transferred to a new tube and made acidic by adding 100 μl glacial acetic acid. A second aqueous phase was created by adding 0.5 ml chloroform/methanol (2:1) and 0.1 ml water. The organic phase was recovered, dried down under nitrogen, resuspended in 50 μl methanol containing 5 mM ammonium acetate, mixed by vortexing, and placed in a water bath sonication device for 5 minutes, before S1P measurement by mass spectrometry. All other steps were the same as for plasma S1P quantitation but with some modifications. A 10-μl sample was injected into an HPLC, and lipids were separated on a Luna-RP column (3-μm particle size C18(2) 100A pore size [50 mm l × 2.00 mm i.d.]) (Phenomenex) at a flow rate of 0.3 ml per minute. The mobile phase consists of eluant A (methanol/water/acetic acid in 5 mM ammonium acetate, 50:49:1 [vol/vol]) and eluant B (methanol/acetic acid in 5 mM ammonium acetate, 99:1 [vol/vol]). The gradient flow is as follows: t = 0 to 0.5 minutes 60% eluent A and 40% eluent B, followed by a linear gradient change to 100% eluent B until t = 2 minutes. Gradient was kept at 100% eluent B for 4.5 minutes, before a linear gradient change from 100% eluent B to 60% eluent A and 40% eluent B until t = 7.5 minutes. Finally, gradient was kept at 60% eluent A and 40% eluent B for 4.5 minutes. Quantitative analysis was based on the following equation: [(C18-S1P peak area) / (C17-S1P peak area)] × concentration of C17-S1P internal standard. The resulting concentrations were further normalized against total phosphatidylcholine using the Phosphatidylcholine Assay Kit from Abcam according to the manufacturer’s instructions.
CAC murine model.
6- to 8-week-old SPLGutKO and control mice were given NF (80 mg/kg initial body weight i.p.) for 5 days. One week later, mice were given AOM (10 mg/kg initial body weight i.p.) (Sigma Aldrich). After 7 days, mice were given 3 cycles of 2% DSS (MP Biomedicals) in the drinking water for 7 days, followed by 2 weeks of consumption of water ad libitum. Mice were euthanized 7 days after cessation of 2% DSS administration. In addition, two groups of SPLGutKO mice received either 5 mg/kg NSC 74859 i.p. every other day or vehicle alone, starting the first day of the second DSS cycle until the end of the AOM/DSS treatment. For SD treatment, C57BL/6 mice received the AOM/DSS regimen described above plus SD (25 mg/kg body weight in 0.5% methylcellulose in sterile water, pH 5–6, by gavage) every other day, starting from the first day of the second DSS cycle until the end of the AOM/DSS treatment. DAI was monitored every day and calculated by scoring mice for weight loss, stool consistency, and blood in the stool as described previously (60). Survival was plotted by product-limit Kaplan-Meier method. P values were evaluated by the Mantel log-rank test with GraphPad Prism software. Mice were euthanized, colons were harvested, and colon lengths were measured. Colons were divided longitudinally; visible tumors were counted, and the tumor size was measured. Colons were fixed flat in 10% buffered formalin for 24 hours and paraffin embedded as Swiss rolls. Sections were stained with hematoxylin and eosin. Sections of colons in which there were no visible tumors were flash frozen in liquid nitrogen for miRNA and protein expression studies as described herein. S1pr1 mRNA expression was measured as described below. Primers used can be found in Supplemental Table 1.
Model of chronic colitis.
Mice received two cycles consisting of 3% DSS in drinking water for 7 days, followed by 14 days of normal water. DAI was monitored as described previously (61). Pathological analysis of the colon was performed as described earlier.
Cytokine measurements.
IL-1β, TNF-α, IL-6, IL-17, IL-21, and IL-23 cytokine levels were measured by ELISA (Affymetrix eBioscience) on 5 μl plasma or 100 mg colon tissue according to the manufacturer’s instructions.
Measurement of miRNAs.
Total RNA was extracted using the miRNeasy Mini Kit (Qiagen). Complementary DNA was synthesized from total RNA using gene-specific primers from the TaqMan Small RNA Assays (Applied Biosystems) in conjunction with the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems). Real-time PCR reaction was carried out with an ABI 7900HT thermocycler (Applied Biosystems) using the TaqMan Universal Master Mix II (Applied Biosystems) and the supplied miRNA-specific RT primer/probe sets from the TaqMan Small RNA Assays. The threshold cycle (Ct) data were calculated by SDS 2.4 software (Applied Biosystems) with the default threshold settings. All real-time PCR reactions were run in triplicate. The average expression levels of miR-21 and miR-181b-1 in control and SPLGutKO tissues were normalized with U6 small nuclear ribonucleic acid (snRNA) as a reference gene, and all data were subsequently analyzed by the 2–ΔΔCt method. Statistical differences between the levels of miR-21 and 181b-1 in control and SPLGutKO tissues were evaluated. P values of less than 0.05 were considered significant.
Gene expression in human IBD and control samples.
Human tissues were frozen in liquid nitrogen and stored at –80°C. Total RNA was extracted using the miRNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. For the measurement of miRNA, refer to methods described earlier. To measure SGPL1, S1PR1, and SPHK1 gene expression, we synthesized complementary DNA with the Real Masterscript Super Mix Kit from 5 PRIME (Fischer Scientific) according to the manufacturer’s instructions. Real-time PCR reaction was carried out in a 384-well optical plate on an ABI 7900HT thermocycler (Applied Biosystems) in conjunction with the Real Master Mix Fast SYBR ROX from 5 PRIME and specific primers listed in Supplemental Table 1. All real-time PCR reactions were run in triplicate. The average expression levels of SGPL1, S1PR1, and SPHK1 in tissues were normalized with HPRT1 or B2M as a reference gene, and all data were subsequently analyzed by the 2–ΔΔCt method.
MEF cell culture and SPL silencing.
Stat3 KO MEFs and corresponding control MEFs were a gift from Valeria Poli (University of Dundee, Dundee, United Kingdom) (31). Cells were maintained in DMEM supplemented with 10% (vol/vol) heat-inactivated FCS, 2 mM l-glutamine, 50 units/ml penicillin and 50 μg/ml streptomycin. To downregulate SPL, MEFs were stably transfected with a lentiviral shRNA construct specific for murine SPL as described previously (62). Intracellular and extracellular S1P levels in MEF lines were measured as described above. Il6 and Spns2 mRNA expression was measured as described above. Primers used are listed in Supplemental Table 1. IB of protein lysates from MEF cells treated for 0, 35, or 70 minutes with 125 nM S1P (Huzzah S1P, Avanti Polar Lipids) was performed as described above. MEF cells were transfected with an siRNA against Spns2 from Origene using Lipofectamine RNAiMAX from Life Technologies according to the manufacturer’s instructions for reverse transfection protocol. MEF S1P was measured at the indicated time per the method described above.
DLD1 cell culture and SPL silencing.
DLD1 cells were obtained from ATCC and maintained in DMEM supplemented with 10% (vol/vol) heat-inactivated FBS, 2 mM l-glutamine, 50 units/ml penicillin, and 50 μg/ml streptomycin. To downregulate SPL, DLD1 cells were stably transfected with a lentiviral shRNA construct specific for human SPL as described previously (63). S1PR1 mRNA expression was measured as described above. Primers used are listed in Supplemental Table 1.
Mouse tumor model.
6-week-old female NOD/SCID mice were injected s.c. with one of the MEF cell lines in both flanks. Briefly, 2 × 106 cells were suspended in PBS and mixed in a 1:1 ratio with Matrigel (BD Biosciences) and injected s.c. in the mouse flanks. Tumor size was measured using a digital caliper once the tumors were visible. Tumor volume was measured using the equation V = π/6 × LW2, as described previously (64), where V stands for tumor volume, L stands for tumor length, and W stands for tumor width. Growth curves were plotted with tumor volume ± SEM from 4 animals per group. Mice were euthanized on day 45. Tumors were harvested and flash frozen in liquid nitrogen for miRNA and RNA expression studies.
Flow cytometry.
Spleen cells were isolated from uninduced NF control mice or from AOM/DSS-treated NF-induced control mice and AOM/DSS-treated SPLGutKO mice. Spleens were harvested and dispersed into single-cell suspensions by forcing the tissue through a 70-μm cell strainer (Thermo Fisher Scientific). Red blood cells were lysed by hypotonic shock, and after washing cells were suspended in FACS buffer (PBS containing 0.1% BSA and 0.01% sodium azide). Cells were counted using a Beckman Coulter counter (Beckman). 1 × 106 to 2 × 106 cells were stimulated for 4 hours with PMA (50 ng/ml) and ionomycin (1 μg/ml) with monensin (2 mM) to increase the amount of cytokines available for detection. Cells were then stained with anti-mouse CD4 eFluor 450 antibody. Cells were incubated in Intracellular Fixation & Permeabilization Buffer (eBioscience) according to the manufacturer’s instructions, followed by intracellular staining of IL-17 and IFN-γ using anti-mouse IL-17A FITC and anti-mouse IFN-γ PE-Cyanine7 antibodies. All antibodies were from eBioscience. Data were acquired with the BD LSRFortessa flow cytometer. For each sample, 100,000 events were recorded. Subsequent analysis and flow cytometry plots were generated using FlowJo v.7.2.5 (TreeStar Inc.).
IL-6/STAT3 PCR array.
RNA extraction from control or KO mice or from human colon tissue was performed as described earlier. Complementary DNA was synthesized using the RT2 First Strand Kit from Qiagen according to the manufacturer’s instructions. Mouse and human IL-6/STAT3 RT2 Profiler PCR Array and RT2 Real-Time SYBR Green/ROX PCR Mix were purchased from SABiosciences, and real-time PCR reactions were performed with an ABI 7900HT thermocycler (Applied Biosystems). All data were subsequently analyzed by the 2–ΔΔCt method with RT2 Profiler PCR Array Data Analysis software version 3.5 from SABiosciences.
Proliferation assay.
750 MEF cells were plated in 96-well plates in 10% FBS DMEM. 18 hours later (t = 0) the medium was replaced by DMEM without FBS and, when indicated, 10 μM W123 and 100 nM FTY720 (both from Cayman Chemicals), 10 μM rolipram and 25 μM NSC 74859 (both from Tocris Bioscience), and 1 μM WP1066 (EMD Millipore) were added. 3,000 MEF cells were also transfected with siRNA against SPNS2 as mentioned above. At the indicated times, cell proliferation was measured using the CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay from Promega according to the manufacturer’s instructions.
Statistics.
Two-tailed unpaired Student’s t test was used to compare two data sets. Comparisons among more than two groups were tested by 1-way ANOVA using Prism GraphPad (GraphPad Software). Data are expressed as mean ± SD. When the P value obtained from ANOVA was significant, Tukey’s test was applied to test for differences among groups. Significance was considered to be P < 0.05.
Study approval.
All animal experiments were conducted in accordance with the CHORI Institutional Animal Care and Use Committee–approved protocol and the NIH guidelines for use of live animals. For human studies, deidentified frozen Crohn’s disease or ulcerative colitis colon tissues (n = 8–12) and age/gender-matched control colon tissue samples were provided by the Cooperative Human Tissue Network (funded by the National Cancer Institute). Other investigators may have received specimens from the same subjects. Additional IBD colon samples were obtained from IBD and age/gender-matched controls with a protocol approved by the CHORI Institutional Review Board. All experiments on human tissue were conducted in accordance with the CHORI Institutional Review Board–approved protocol. Informed consent from patients or their guardians was obtained.
Supplementary Material
Acknowledgments
We would like to thank Ervin Epstein for critical review of the manuscript and Nelle Cronen for expert administrative support. This work was supported by Broad Medical Research Program grant IBD-0353; Crohn’s and Colitis Foundation of America Senior Research Award 277014; NIH grants CA129438 and R21AT005336; American Institute for Cancer Research grant 09A041; Swim Across America Foundation support (to J.D. Saba); Crohn’s and Colitis Canada and Fonds de recherche du Québec-Santé fellowship grant 28137 (to E. Degagné); NIH grants HL090553, HL76839, HL86683, and GM66152; March of Dimes grant 6-FY2007-2012; Ellison Medical Foundation Senior Scholar Award (S.G. Young); and the National Center for Advancing Translational Sciences, NIH grant UCSF-CTSI UL1 TR000004. We dedicate this report to our good friend and colleague Robert Bittman who passed away while this manuscript was in press. Dr. Bittman synthesized novel, clinically relevant small molecules with ingenuity and elegance. His creativity, dedication and attention to detail were essential to the outcome and significance of this study and to the advancement of the field of sphingolipid biochemistry.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2014;124(12):5368–5384. doi:10.1172/JCI74188.
References
- 1.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357(9255):539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 3.Beaugerie L, et al. Risk of colorectal high-grade dysplasia and cancer in a prospective observational cohort of patients with inflammatory bowel disease. Gastroenterology. 2013;145(1):166–175. doi: 10.1053/j.gastro.2013.03.044. [DOI] [PubMed] [Google Scholar]
- 4.Steinbach G, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med. 2000;342(26):1946–1952. doi: 10.1056/NEJM200006293422603. [DOI] [PubMed] [Google Scholar]
- 5.Solomon SD, et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005;352(11):1071–1080. doi: 10.1056/NEJMoa050405. [DOI] [PubMed] [Google Scholar]
- 6.Arber N, et al. Celecoxib for the prevention of colorectal adenomatous polyps. N Engl J Med. 2006;355(9):885–895. doi: 10.1056/NEJMoa061652. [DOI] [PubMed] [Google Scholar]
- 7.Terzic J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology. 2010;138(6):2101–2114 e2105. doi: 10.1053/j.gastro.2010.01.058. [DOI] [PubMed] [Google Scholar]
- 8.McCormack VA, Boffetta P. Today’s lifestyles, tomorrow’s cancers: trends in lifestyle risk factors for cancer in low- and middle-income countries. Ann Oncol. 2011;22(11):2349–2357. doi: 10.1093/annonc/mdq763. [DOI] [PubMed] [Google Scholar]
- 9.Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell. 2010;39(4):493–506. doi: 10.1016/j.molcel.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maceyka M, Harikumar KB, Milstien S, Spiegel S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012;22(1):50–60. doi: 10.1016/j.tcb.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahn EH, Schroeder JJ. Sphingoid bases and ceramide induce apoptosis in HT-29 and HCT-116 human colon cancer cells. Exp Biol Med (Maywood). 2002;227(5):345–353. doi: 10.1177/153537020222700507. [DOI] [PubMed] [Google Scholar]
- 12.Lemonnier LA, et al. Sphingomyelin in the suppression of colon tumors: prevention versus intervention. Arch Biochem Biophys. 2003;419(2):129–138. doi: 10.1016/j.abb.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 13.Duan RD. Anticancer compounds and sphingolipid metabolism in the colon. In Vivo. 2005;19(1):293–300. [PubMed] [Google Scholar]
- 14.Schmelz EM, et al. Modulation of intracellular beta-catenin localization and intestinal tumorigenesis in vivo and in vitro by sphingolipids. Cancer Res. 2001;61(18):6723–6729. [PubMed] [Google Scholar]
- 15.Lee H, et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat Med. 2010;16(12):1421–1428. doi: 10.1038/nm.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alvarez SE, et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature. 2010;465(7301):1084–1088. doi: 10.1038/nature09128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xia P, et al. An oncogenic role of sphingosine kinase. Curr Biol. 2000;10(23):1527–1530. doi: 10.1016/S0960-9822(00)00834-4. [DOI] [PubMed] [Google Scholar]
- 18.Colie S, et al. Disruption of sphingosine 1-phosphate lyase confers resistance to chemotherapy and promotes oncogenesis through Bcl-2/Bcl-xL upregulation. Cancer Res. 2009;69(24):9346–9353. doi: 10.1158/0008-5472.CAN-09-2198. [DOI] [PubMed] [Google Scholar]
- 19.Duan RD, Nilsson A. Sphingolipid hydrolyzing enzymes in the gastrointestinal tract. Methods Enzymol. 2000;311:276–286. doi: 10.1016/s0076-6879(00)11089-4. [DOI] [PubMed] [Google Scholar]
- 20.Kawamori T, et al. Role for sphingosine kinase 1 in colon carcinogenesis. FASEB J. 2009;23(2):405–414. doi: 10.1096/fj.08-117572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Snider AJ, Orr Gandy KA, Obeid LM. Sphingosine kinase: Role in regulation of bioactive sphingolipid mediators in inflammation. Biochimie. 2010;92(6):707–715. doi: 10.1016/j.biochi.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kohno M, et al. Intracellular role for sphingosine kinase 1 in intestinal adenoma cell proliferation. Mol Cell Biol. 2006;26(19):7211–7223. doi: 10.1128/MCB.02341-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang J, et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic Intestinal Inflammation, and development of colitis-associated cancer. Cancer Cell. 2013;23(1):107–120. doi: 10.1016/j.ccr.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oskouian B, et al. Sphingosine-1-phosphate lyase potentiates apoptosis via p53- and p38-dependent pathways and is downregulated in colon cancer. Proc Natl Acad Sci U S A. 2006;103(46):17384–17389. doi: 10.1073/pnas.0600050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sansom OJ, et al. Myc deletion rescues Apc deficiency in the small intestine. Nature. 2007;446(7136):676–679. doi: 10.1038/nature05674. [DOI] [PubMed] [Google Scholar]
- 27.Hayakawa Y, et al. Effectiveness of IkappaB kinase inhibitors in murine colitis-associated tumorigenesis. J Gastroenterol. 2009;44(9):935–943. doi: 10.1007/s00535-009-0098-7. [DOI] [PubMed] [Google Scholar]
- 28.Bromberg J, Wang TC. Inflammation and cancer: IL-6 and STAT3 complete the link. Cancer Cell. 2009;15(2):79–80. doi: 10.1016/j.ccr.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ichiba M, Nakajima K, Yamanaka Y, Kiuchi N, Hirano T. Autoregulation of the Stat3 gene through cooperation with a cAMP-responsive element-binding protein. J Biol Chem. 1998;273(11):6132–6138. doi: 10.1074/jbc.273.11.6132. [DOI] [PubMed] [Google Scholar]
- 30.Snider AJ, et al. A role for sphingosine kinase 1 in dextran sulfate sodium-induced colitis. FASEB J. 2009;23(1):143–152. doi: 10.1096/fj.08-118109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Costa-Pereira AP, et al. Mutational switch of an IL-6 response to an interferon-gamma-like response. Proc Natl Acad Sci U S A. 2002;99(12):8043–8047. doi: 10.1073/pnas.122236099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fyrst H, et al. Natural sphingadienes inhibit Akt-dependent signaling and prevent intestinal tumorigenesis. Cancer Res. 2009;69(24):9457–9464. doi: 10.1158/0008-5472.CAN-09-2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar A, et al. Chemopreventive sphingadienes downregulate Wnt signaling via a PP2A/Akt/GSK3β pathway in colon cancer. Carcinogenesis. 2012;33(9):1726–1735. doi: 10.1093/carcin/bgs174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwab S, et al. Lymphocyte sequestration through S1P lyase inhibition an disruption of S1P gradients. Science. 2005;309(5741):1735–1739. doi: 10.1126/science.1113640. [DOI] [PubMed] [Google Scholar]
- 35.Vogel P, et al. Incomplete inhibition of sphingosine 1-phosphate lyase modulates immune system function yet prevents early lethality and non-lymphoid lesions. PLoS One. 2009;4(1):e4112. doi: 10.1371/journal.pone.0004112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Allende M, et al. Sphingosine-1-phosphate lyase deficiency produces a pro-inflammatory response while impairing neutrophil trafficking. J Biol Chem. 2011;286(9):7348–7358. doi: 10.1074/jbc.M110.171819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pott J, Hornef M. Innate immune signalling at the intestinal epithelium in homeostasis and disease. EMBO Rep. 2012;13(8):684–698. doi: 10.1038/embor.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frias MA, James RW, Gerber-Wicht C, Lang U. Native and reconstituted HDL activate Stat3 in ventricular cardiomyocytes via ERK1/2: role of sphingosine-1-phosphate. Cardiovasc Res. 2009;82(2):313–323. doi: 10.1093/cvr/cvp024. [DOI] [PubMed] [Google Scholar]
- 39.Corvinus FM, et al. Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia. 2005;7(6):545–555. doi: 10.1593/neo.04571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Y, et al. S1PR1 is an effective target to block STAT3 signaling in activated B cell-like diffuse large B-cell lymphoma. Blood. 2012;120(7):1458–1465. doi: 10.1182/blood-2011-12-399030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ludwig K, et al. PDCD4/miR-21 dysregulation in inflammatory bowel disease-associated carcinogenesis. Virchows Archiv. 2013;462(1):57–63. doi: 10.1007/s00428-012-1345-5. [DOI] [PubMed] [Google Scholar]
- 42.Schetter AJ, et al. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA. 2008;299(4):425–436. doi: 10.1001/jama.299.4.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ullman TA, Itzkowitz SH. Intestinal inflammation and cancer. Gastroenterology. 2011;140(6):1807–1816. doi: 10.1053/j.gastro.2011.01.057. [DOI] [PubMed] [Google Scholar]
- 44.Xiong Y, et al. Sphingosine kinases are not required for inflammatory responses in macrophages. J Biol Chem. 2013;288(45):32563–32573. doi: 10.1074/jbc.M113.483750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adada MM, et al. Sphingosine kinase 1 regulates tumor necrosis factor-mediated RANTES induction through p38 mitogen-activated protein kinase but independently of nuclear factor kappaB activation. J Biol Chem. 2013;288(38):27667–27679. doi: 10.1074/jbc.M113.489443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sugawara T, et al. Intestinal absorption of dietary maize glucosylceramide in lymphatic duct cannulated rats. J Lipid Res. 2010;51(7):1761–1769. doi: 10.1194/jlr.M002204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rozema E, et al. Effects on inflammatory responses by the sphingoid base 4,8-sphingadienine. Int J Mol Med. 2012;30(3):703–707. doi: 10.3892/ijmm.2012.1035. [DOI] [PubMed] [Google Scholar]
- 48.Ohtsuka M, Kimura M, Tanaka M, Inoko H. Recombinant DNA technologies for construction of precisely designed transgene constructs. Curr Pharm Biotechnol. 2009;10(2):244–251. doi: 10.2174/138920109787315033. [DOI] [PubMed] [Google Scholar]
- 49.Kihara A, et al. Sphingosine-1-phosphate lyase is involved in the differentiation of F9 embryonal carcinoma cells to primitive endoderm. J Biol Chem. 2003;278(16):14578–14585. doi: 10.1074/jbc.M211416200. [DOI] [PubMed] [Google Scholar]
- 50.Skarnes WC, et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 2011;474(7351):337–342. doi: 10.1038/nature10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Osoegawa K, et al. An improved approach for construction of bacterial artificial chromosome libraries. Genomics. 1998;52(1):1–8. doi: 10.1006/geno.1998.5423. [DOI] [PubMed] [Google Scholar]
- 52. Ross M, LaBrie S, McPherson J, Stanton V Jr. Screening large-insert libraries by hybridization. In: Boyl A, ed. Current Protocols in Human Genetics. New York, New York, USA: Wiley; 1999:5.6.1–5.6.5. [Google Scholar]
- 53.Yu D, et al. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A. 2000;97(11):5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kast P. pKSS — a second-generation general purpose cloning vector for efficient positive selection of recombinant clones. Gene. 1994;138(1–2):109–114. doi: 10.1016/0378-1119(94)90790-0. [DOI] [PubMed] [Google Scholar]
- 55.Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13(3):476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blake J, Salinas P, Hughes S. n beta geo, a combined selection and reporter gene for retroviral and transgenic studies. Biotechniques. 1997;23(4):690–695. [PubMed] [Google Scholar]
- 57.Grbic DM, Degagne E, Langlois C, Dupuis AA, Gendron FP. Intestinal inflammation increases the expression of the P2Y6 receptor on epithelial cells and the release of CXC chemokine ligand 8 by UDP. J Immunol. 2008;180(4):2659–2668. doi: 10.4049/jimmunol.180.4.2659. [DOI] [PubMed] [Google Scholar]
- 58.Bandhuvula P, Li Z, Bittman R, Saba JD. Sphingosine 1-phosphate lyase enzyme assay using a BODIPY-labeled substrate. Biochem Biophys Res Commun. 2009;380(2):366–370. doi: 10.1016/j.bbrc.2009.01.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sullards MC, Merrill AH., Jr Analysis of sphingosine 1-phosphate, ceramides, and other bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Sci STKE. 2001;2001(67):pl1. doi: 10.1126/stke.2001.67.pl1. [DOI] [PubMed] [Google Scholar]
- 60.Fitzpatrick LR, Wang J, Le T. In vitro and in vivo effects of gliotoxin, a fungal metabolite: efficacy against dextran sodium sulfate-induced colitis in rats. Dig Dis Sci. 2000;45(12):2327–2336. doi: 10.1023/A:1005630723111. [DOI] [PubMed] [Google Scholar]
- 61.Lowe EL, et al. Toll-like receptor 2 signaling protects mice from tumor development in a mouse model of colitis-induced cancer. PLoS One. 2010;5(9):e13027. doi: 10.1371/journal.pone.0013027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smith G, Kumar A, Saba J. Sphingosine phosphate lyase regulates murine embryonic stem cell proliferation and pluripotency through an S1P2/STAT3 signaling pathway. Biomolecules. 2013;3(3):351–368. doi: 10.3390/biom3030351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kumar A, et al. S1P lyase regulates DNA damage responses through a novel sphingolipid feedback mechanism. Cell Death Dis. 2011;2(1):e119. doi: 10.1038/cddis.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Soubeyran P, et al. Homeobox gene Cdx1 regulates Ras, Rho and PI3 kinase pathways leading to transformation and tumorigenesis of intestinal epithelial cells. Oncogene. 2001;20(31):4180–4187. doi: 10.1038/sj.onc.1204551. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.