

Abstract
Angioplasty and stenting is the primary treatment for flow-limiting atherosclerosis; however, this strategy is limited by pathological vascular remodeling. Using a systems approach, we identified a role for the network hub gene glutathione peroxidase-1 (GPX1) in pathological remodeling following human blood vessel stenting. Constitutive deletion of Gpx1 in atherosclerotic mice recapitulated this phenotype of increased vascular smooth muscle cell (VSMC) proliferation and plaque formation. In an independent patient cohort, gene variant pair analysis identified an interaction of GPX1 with the orphan protooncogene receptor tyrosine kinase ROS1. A meta-analysis of the only genome-wide association studies of human neointima-induced in-stent stenosis confirmed the association of the ROS1 variant with pathological remodeling. Decreased GPX1 expression in atherosclerotic mice led to reductive stress via a time-dependent increase in glutathione, corresponding to phosphorylation of the ROS1 kinase activation site Y2274. Loss of GPX1 function was associated with both oxidative and reductive stress, the latter driving ROS1 activity via s-glutathiolation of critical residues of the ROS1 tyrosine phosphatase SHP-2. ROS1 inhibition with crizotinib and deglutathiolation of SHP-2 abolished GPX1-mediated increases in VSMC proliferation while leaving endothelialization intact. Our results indicate that GPX1-dependent alterations in oxido-reductive stress promote ROS1 activation and mediate vascular remodeling.
Introduction
Balloon angioplasty and stenting (BAS) is the most commonly performed method of revascularization for flow-limiting atherosclerosis, with over 1.5 million procedures performed annually in the United States (1). The immediate vascular injury following BAS predisposes to acute thrombosis and initiates vascular smooth muscle cell (VSMC) proliferation and neointimal hyperplasia, leading to a phenotype significantly different from that of de novo atherosclerosis (2). This reaction to vascular injury activates a homeostatic healing response that can lead to neointima-induced in-stent stenosis, the major limitation to the most widely used method for treatment of coronary and peripheral arterial disease. The use of drug-eluting stents (DESs), which target excessive VSMC proliferation and neointimal hyperplasia, has greatly reduced clinical restenosis rates (3). However, current agents used in DESs inhibit reendothelialization, potentially increasing late thrombosis risk within the stented vessel (4). Considering that over 1 million patients undergo DES implantation each year within the United States (1), there remains a pressing need to identify more specific and effective pharmacological agents targeting neointimal hyperplasia without detrimental effects on reendothelialization. Accordingly, we developed and validated a mouse model of BAS that allows for interrogation of relevant molecular pathways by high-throughput genomic approaches not readily feasible using larger animal models (5).
Glutathione peroxidase-1 (GPX1), the most abundant intracellular isoform of the GPX family of selenoprotein antioxidant enzymes, diminishes oxidant stress by utilizing reduced glutathione (GSH) to reduce hydrogen peroxide and lipid peroxides to alcohols. In conditions in which GPX1 becomes deficient, electron transfer is no longer coupled to GSH reduction, leading to increased bioavailability of ROS. In humans with coronary artery disease, GPX1 is independently associated with cardiovascular events (6). GPX1 activity is decreased in atherosclerotic plaque excised from carotid artery (7). Mice with GPX1 deficiency have endothelial dysfunction, impaired capacity for angiogenesis due to endothelial progenitor cell dysfunction (8), and significant structural vascular and cardiac abnormalities (9). Correspondingly, when GPX1 is deficient in animal models of vascular disease, such as hypercholesterolemia (10) or diabetes (11), atherosclerosis is increased. Despite such evidence, large-scale clinical trials using nonspecific antioxidant drugs to prevent atherosclerosis (12) or in-stent stenosis (13) have not been successful. Such lack of success speaks to a need for a more advanced understanding of redox signaling mechanisms as mediators of vascular processes.
In studies reported here, we identify GPX1 as a determinant of vascular remodeling and identify the orphan RTK ROS1 as a mediator of these effects. We find that loss of GPX1 leads to increased VSMC proliferation, migration, and apoptosis, in vitro and in vivo, and that this is attenuated by inhibition of ROS1 through cell-fate regulation. Further, in contrast to the oxidative stress induced by GPX1 deficiency, we find ROS1 activation coincides with reductive stress induced by GSH. We show that this reductive stress has no direct effect on ROS1, but rather that it leads to inhibition of the regulatory phosphatase SHP-2 by s-glutathiolation of 2 critical phosphatase active-site cysteine residues. Inhibition of SHP-2 prevents dephosphorylation of ROS1, resulting in sustained ROS1 kinase activation, triggering VSMC processes that manifest as neointimal hyperplasia and abnormal vascular remodeling. Further, we determined that pharmacological inhibition of ROS1 attenuates these processes without affecting reendothelialization. Finally, in humans, we show that genetic variation within ROS1 is associated with risk for abnormal vascular remodeling leading to in-stent stenosis, confirming ROS1 as a pivotal regulator of the response to vascular injury in humans and a therapeutic target with substantial clinical impact.
Results
Network analysis of human coronary neointima prioritizes the GPX1 subnetwork for investigation.
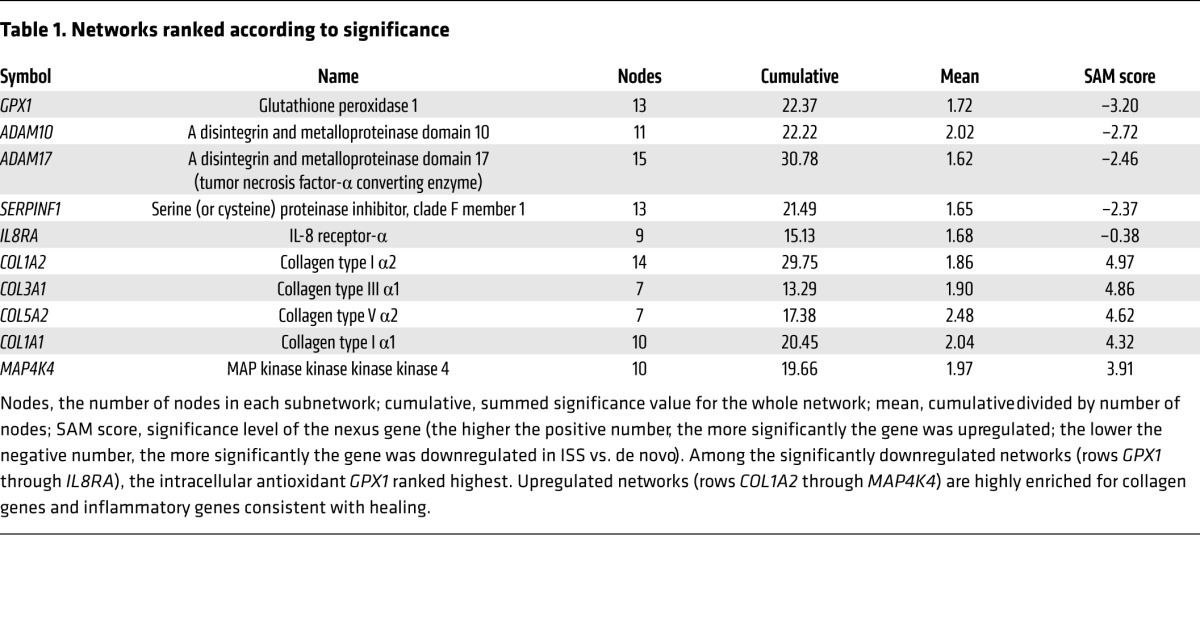
We analyzed coronary vascular gene expression from 89 patients who underwent coronary atherectomy. RNA samples derived from de novo atherosclerosis (n = 55) and in-stent stenosis (n = 34) were hybridized to microarrays with genome-wide representation of coding genes (14). Independently, networks of gene-gene interactions were generated using text mining of the entire abstracted academic literature. Generated networks exhibited scale-free topology. Gene expression from atherectomy tissue was overlaid on these networks and individual networks were scored according to the average differential significance of network members within the experimental data set. While upregulated networks were highly enriched for collagen and inflammatory genes involved in the healing response, we found the network with GPX1 as its hub to be the most significantly downregulated of all gene networks (nodes, 13; cumulative D statistic for the network, 22.4; mean statistic for the network, 1.72; SAM statistic for GPX1, –3.2; Table 1). In contrast, the significance analysis of microarrays alone ranked GPX1 only 247th in a list of most differentially regulated genes.
Table 1.
Networks ranked according to significance
Constitutive deletion of Gpx1 in atherosclerotic mice increases aortic smooth muscle–rich plaque.
Human coronary artery atherectomy specimens demonstrated higher GPX1 gene expression in de novo atherosclerosis than in in-stent neointima (Figure 1A). We used mice with ubiquitous homozygous deletion of Gpx1 crossed with hypercholesterolemic atherosclerotic Apoe–/– (Gpx1–/– Apoe–/–) mice as our primary model. As expected, GPX1 activity was decreased in Gpx1–/– Apoe–/– mice without differences in body weight or cholesterol (Supplemental Results, Supplemental Figure 1, and Supplemental Table 1; supplemental material available online with this article; doi:10.1172/JCI77484DS1). Atherosclerotic plaque burden in the aortic root (42%, P = 0.02; Figure 1B) and the thoracoabdominal aorta (41%, P = 0.01; Figure 1B) of Gpx1–/– Apoe–/– mice was increased compared with that in Gpx1+/+ Apoe–/– controls. Plaque composition analysis revealed a significant increase in the percentage area occupied by α-actin–positive smooth muscle cells (SMCs) (42%, P = 0.02), but not macrophages, lipid, or collagen in Gpx1–/– Apoe–/– mice (Supplemental Table 2 and Supplemental Figure 2). Thus, targeted deletion of Gpx1 leads to increased VSMC-rich plaque formation in atherosclerotic mice.
Figure 1. Increased atherosclerosis and in-stent neointima in Gpx1–/– Apoe–/– mice.

(A) Transcriptional profiling of human coronary atherectomy specimens of control (AHA class I lesions), atherosclerosis (AHA class III–V lesions), or in-stent stenosis identified that GPX1 expression was incrementally lower in atherosclerosis and in-stent stenosis. *P < 0.01 vs. control; #P < 0.05 vs. atherosclerosis. n = 55 atherosclerosis/34 in-stent stenosis. (B) Relative area of atherosclerosis in the aortic root and lesion burden in aorta were increased in Gpx1–/– Apoe–/– mice compared with Gpx1+/+ Apoe–/– mice (*P < 0.02, n = 8–15/group). Black scale bar: 50 μm. White scale bar: 0.5 mm. (C) mRNA levels of Gpx1 in Gpx1+/+ Apoe–/– mice were lower in aorta that had undergone BAS compared with uninjured aorta harvested at 28 days. *P = 0.05. n = 4/group. (D) Gpx1–/– Apoe–/– mice 4 weeks following BAS developed significantly greater neointimal hyperplasia compared with control Gpx1+/+ Apoe–/–. *P < 0.05. Although overall neointimal cell density was higher in Gpx1–/– Apoe–/– mice, there was no difference in cell density when expressed per unit area between groups. Scale bar: 50 μm. Black arrowheads denote neointima.
In-stent neointima is increased in mice with constitutive deletion of Gpx1.
We developed a mouse model of BAS analogous to human coronary stenting, allowing us to mimic human vascular disease states in a manner not possible in larger animal models (5). In our model, balloon deployment of a custom stent in the descending aorta of hypercholesterolemic atherosclerotic mice was performed in a donor mouse just prior to euthanasia and the stented segment explanted. The stented segment was then secured microsurgically as a vascular graft using end-to-end anastomoses (Supplemental Figure 3, A and B). We performed BAS in Gpx1–/– Apoe–/– and control Gpx1+/+ Apoe–/– mice. In keeping with our transcriptomic findings from human coronary artery in-stent neointima (Figure 1A), we found that Gpx1 mRNA was significantly decreased in Gpx1+/+ Apoe–/– vessels that had undergone BAS compared with uninjured aorta (P = 0.05; Figure 1C). Correspondingly, we found an increase in neointima (50%, P = 0.006; Figure 1D) in Gpx1–/– Apoe–/– stented vessels compared with control Gpx1+/+ Apoe–/–. There was no difference in total vessel wall area or lumen area between groups (Supplemental Figure 3C). The extent of vascular injury caused by stent deployment is an important determinant of neointimal hyperplasia (15). As stents were deployed using an identical angioplasty balloon inflated to a consistent pressure in all animals, there were no differences between groups in either stent expansion (0.82 ± 0.11 vs. 0.89 ± 0.13 mm2, P = 0.67) or mean Schwartz vessel injury score (1.4 ± 0.4 vs. 1.4 ± 0.3, P = 0.50) (Supplemental Figure 3C). Total neointimal cell count was greater in the Gpx1–/– Apoe–/– stented vessels; however, there was no difference in cell count between groups when expressed as a proportion of the neointimal area (Figure 1D). Taken together, these findings confirm the hypothesis generated by network analysis of human tissue that GPX1 has an important role in the process of vascular remodeling following injury.
Stent endothelialization is impaired in GPX1-deficient mice.
Previous studies have shown that acceleration of reendothelialization following vascular injury leads to attenuation of the neointimal response and provides protection from stent thrombosis (16). To directly investigate the role of GPX1 deficiency in regulating endothelial cell regeneration following BAS, we generated Gpx1–/– Apoe–/– and Gpx1+/+ Apoe–/– knockout mice with an additional transgene to allow endothelium-targeted LacZ expression. Endothelial cells in freshly harvested aorta from both Gpx1–/– Apoe–/– Tie2-LacZ+ and Gpx1+/+ Apoe–/– Tie2-LacZ+ control mice demonstrated β-gal staining (Figure 2, A and B, and Supplemental Figure 3D), whereas no β-gal staining was evident in animals without LacZ. Twenty-eight days following BAS, stented vessels from Gpx1–/– Apoe–/– Tie2-LacZ+ mice had over 50% reduction in endothelial coverage compared with Gpx1+/+ Apoe–/– Tie2-LacZ+ (P = 0.03; Figure 2C), suggesting that GPX1 is required for endothelial repopulation.
Figure 2. Stent endothelialization is impaired in Gpx1-deficient mice.

In Gpx1–/– Apoe–/– and Gpx1+/+ Apoe–/– mice additionally expressing β-gal (LacZ) targeted to endothelial cells (Tie2), en face macroscopic vessel images identify (A) stents struts (black arrowheads), endothelium (blue stain) on the luminal surface, and (B) areas with no endothelium (gray arrowheads). Scale bar: 0.5 mm. (C) Reendothelialization was impaired in Gpx1–/– Apoe–/– Tie2-LacZ+ mice 4 weeks following BAS. Left panel shows high-power en face microscopy images of the luminal surface of the stented vessel. Right panel shows cross-sectional histomorphometry. *P < 0.05. n = 6–10 mice/group. White arrowheads denote stent struts; black arrowheads denote endothelial cells. Scale bar: 50 μm.
Increased bioavailability of ROS in Gpx1–/– Apoe–/– mice.
Given the central role of GPX1 in protecting cells from ROS and the impairment in endothelialization shown above, we next investigated redox state in different layers of the aortic wall from experimental animals using dihydroethidium (DHE) oxidative fluorescence microtopography. We observed striking differences in superoxide production, finding vessel wall fluorescence 3-fold higher in Gpx1–/– Apoe–/– mice compared with Gpx1+/+ Apoe–/–. Surprisingly, superoxide production was markedly increased only in the media (73%, P < 0.01), but not the endothelium or adventitia (Figure 3A). Similarly, total vascular superoxide production, measured by lucigenin-enhanced chemiluminescence, was 67% higher in Gpx1–/– Apoe–/– aortas (P < 0.01) (Figure 3B). When aortas were incubated with the endothelial NOS (eNOS) inhibitor L-NAME, total superoxide levels were not significantly different, suggesting that the endothelial contribution to superoxide generation from eNOS was minimal (Figure 3B).
Figure 3. Increased bioavailability of ROS in Gpx1–/– Apoe–/– mice.

(A) Vascular superoxide production, measured by DHE fluorescence, in Gpx1–/– Apoe–/– mice was higher than in the Gpx1+/+ Apoe–/– mouse medial layer (red fluorescence), but not the endothelium or adventitia. Elastic laminae exhibit green autofluorescence. Scale bar: 5 μm. *P < 0.01. n = 6/group. (B) Aortic superoxide production using lucigenin chemiluminescence was higher in Gpx1–/– Apoe–/– compared with Gpx1+/+ Apoe–/– mice without a contribution by the eNOS inhibitor L-NAME, suggesting minimal contribution from endothelium. *P < 0.01. n = 7/group. (C) Net NO levels were significantly decreased in Gpx1–/– Apoe–/– compared with Gpx1+/+ Apoe–/– mice that underwent BAS using Fe-DETC EPR. L-NAME did not contribute to net NO levels. n = 6/group. (D) Immunoblotting for eNOS protein in aortic lysates revealed no difference in eNOS band intensity normalized to GAPDH control between groups. n = 6–8.
We measured NO synthesis in intact aortas harvested from Gpx1–/– Apoe–/– and Gpx1+/+ Apoe–/– animals that had undergone BAS and found significantly decreased NO levels in Gpx1–/– Apoe–/– (Figure 3C) without change following addition of L-NAME. These findings suggest that loss of NO by inhibition of eNOS provides only a small contribution to the redox balance of the vessel wall in Gpx1–/– Apoe–/– animals. Further, we found no difference in eNOS protein levels in aortas from Gpx1–/– Apoe–/– versus Gpx1+/+ Apoe–/– mice (Figure 3D).
Together, these findings show that regulation of the redox state is a critical component of GPX1-mediated vascular remodeling. Multiple approaches indicate that the endothelial contribution to redox balance in the aortas of Gpx1–/– Apoe–/– mice is small, while the majority of ROS are generated in the medial layer, rich in VSMCs.
GPX1-ROS1 interaction may mediate pathological vascular remodeling and in-stent neointima.
As the prevailing view has been that the vascular phenotype in mice deficient in Gpx1 was dependent on the endothelium (8, 9, 17, 18), we searched for alternative mechanisms that could implicate the VSMC as the putative mediator of the phenotype in Gpx1–/– Apoe–/– mice. Through our network analysis, we identified a previous report that demonstrated common polymorphisms in the genes encoding GPX1 and one of the last remaining orphan RTKs, ROS1, were associated with in-stent stenosis (19). ROS1 encodes an orphan type I integral membrane protein with RTK activity and is structurally similar to cell-adhesion molecules (20). The physiological function of ROS1 in humans has not been fully elucidated, and mechanisms by which ROS1 might mediate cardiovascular disease have not previously been explored. Given the role of RTKs as important regulators of cellular signal transduction pathways that play crucial roles in the development of vascular disease by regulating cellular proliferation, differentiation, migration, and death, we hypothesized that the polymorphisms in the genes encoding GPX1 and ROS1 might interact.
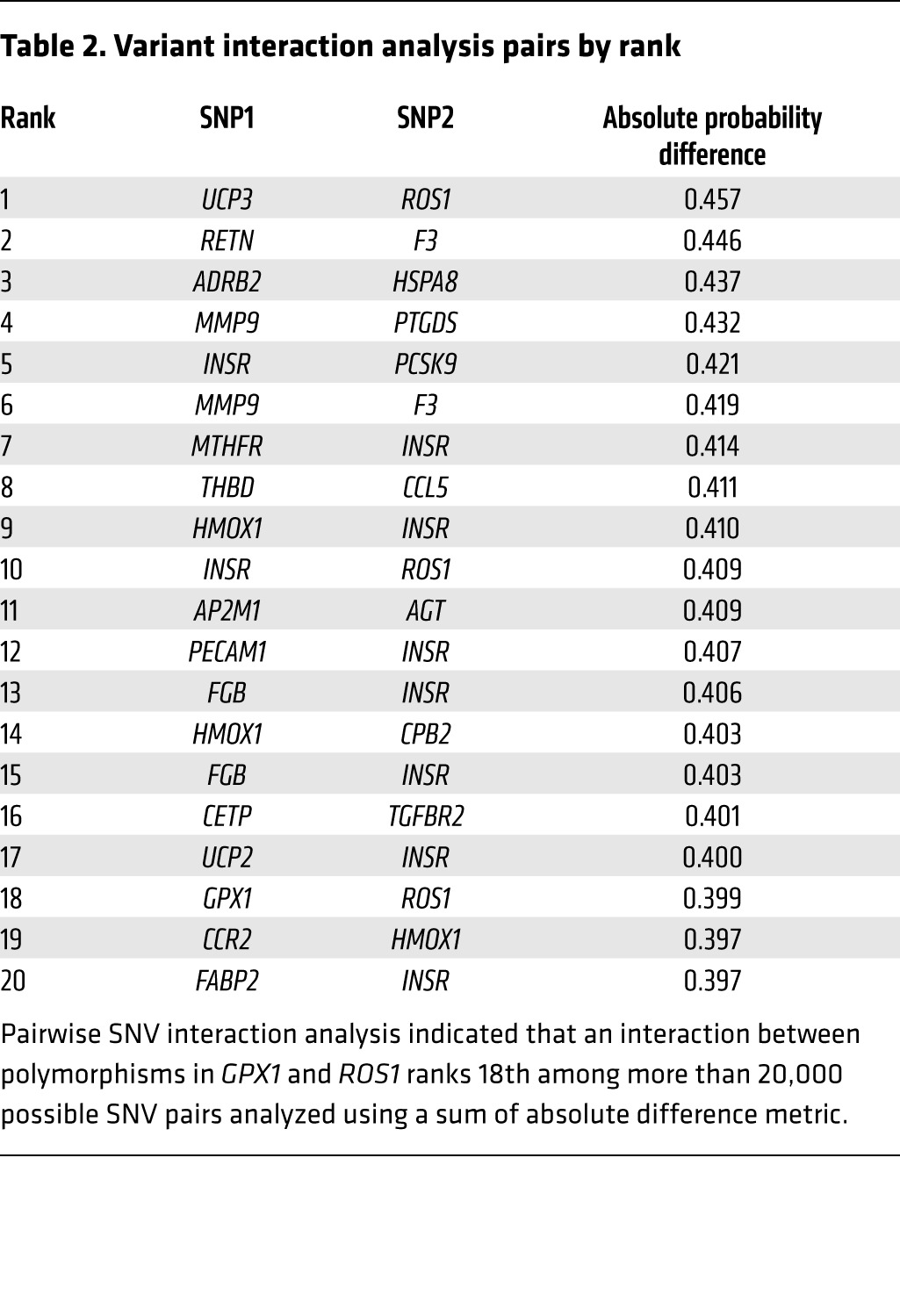
In 461 patients who underwent BAS, 107 of whom developed in-stent stenosis, variants in GPX1 Pro198Leu (rs1050450) and ROS1 Asp2213Asn (rs529038) ranked fifth and sixth, respectively, in terms of informative single-nucleotide variants for the prediction of in-stent stenosis (Supplemental Results and Supplemental Table 3), while gene-pair interaction analysis using an absolute probability difference metric showed that the variant interaction pair ranked 18th out of more than 20,000 possible pairs evaluated (P < 0.002, Table 2). Finally, a linear regression model conditioned on the presence of these variants demonstrated an increasing risk of in-stent stenosis with each additional risk allele, consistent with an additive risk model (Supplemental Figure 4).
Table 2.
Variant interaction analysis pairs by rank
The ROS1 variant associates with in-stent stenosis in GWAS.
To replicate the association of rs529038 ROS1 with in-stent stenosis in humans, we performed an independent meta-analysis of the only GWAS of in-stent stenosis (CardioGene, GeneBank, deCODE, and GENDER). Participants in these studies comprise a total of 1,033 cases of in-stent stenosis and 2,669 controls. In this, the largest genotyped cohort of patients after BAS, we found an association between rs529038 and the development of in-stent stenosis (P = 0.056) to be of borderline significance.
ROS1 is a mediator of atherosclerosis and in-stent stenosis.
To investigate the mechanistic basis of the GPX1-ROS1 interaction, we first measured basal levels of expression of ROS1 in tissues from experimental animals and in cell lines relevant to atherosclerosis and in-stent neointima and found that ROS1 is not expressed in vascular tissue and overall is present at very low levels in tissues and cells (Supplemental Figure 5A). In contrast, we found dramatic differences in ROS1 gene expression in human coronary artery tissue (expression was increased in de novo atherosclerosis, but was significantly higher in in-stent neointima compared with control; Figure 4A). This pattern of expression is in contrast to that of GPX1 expression (Figure 1A), suggesting a counterregulatory role. We thus hypothesized that ROS1 would be upregulated following vascular injury. In keeping with human coronary artery gene expression (Figure 1A), we found Ros1 mRNA was increased nearly 150-fold in Gpx1+/+ Apoe–/– vessels that had undergone stenting compared with uninjured aorta (P < 0.001; Figure 4B).
Figure 4. ROS1 is upregulated in in-stent neointima and involved in basic VSMC fates.

(A) Transcriptional profiling of human coronary artery atherectomy specimens of control (AHA class I lesions), de novo atherosclerosis (AHA class III–V lesions), or in-stent stenosis identified that ROS1 expression was increased in atherosclerosis and furthermore in in-stent stenosis. *P < 0.01 vs. control; #P < 0.05 vs. atherosclerosis. n = 55 atherosclerosis/34 in-stent stenosis. (B) mRNA levels of Ros1 in Gpx1+/+ Apoe–/– mice were dramatically higher in aorta that had undergone BAS compared with uninjured aorta harvested at 28 days. *P < 0.001. n = 4/group. (C) HCASMCs were grown under both serum-fed proliferative and serum-starved nonproliferative phenotypes. ROS1 expression was minimal, but was upregulated as these cells differentiated into “contractile” phenotype and further by confluence, returning to contractile phenotype levels following passage. ROS1 expression was significantly decreased by siROS1. *P < 0.05 vs. (+) serum; **P < 0.05 vs. confluent; †P < 0.05 vs. (–) serum. (D) In primary aortic VSMCs from experimental animals that underwent angioplasty, proliferation was higher in Gpx1–/– Apoe–/– VSMCs compared with control Gpx1+/+ Apoe–/– VSMCs. siROS1, genistein, and crizotinib decreased proliferation in Gpx1–/– Apoe–/– mice, but not in Gpx1+/+ Apoe–/– controls, indicating that the phenotype was dependent on GPX1 deficiency. Daidzein, the structural analogue of genistein with similar antioxidant effects but no effect on tyrosine kinase inhibition, did not inhibit proliferation in Gpx1–/– Apoe–/– cells. Addition of siROS1 to Daidzein showed a reduction in proliferation similar to that of siROS1 alone. *P < 0.05 vs. Gpx1+/+ Apoe–/–; †P < 0.05 vs. baseline Gpx1–/– Apoe–/–.
Given that we previously demonstrated reduced reendothelialization, but increased medial layer superoxide generation, in Gpx1–/– Apoe–/– vessels, we next aimed to determine the vascular cell type in which this putative interaction was constitutively active. We first measured mRNA levels of ROS1 in human coronary artery endothelial cells grown under both serum-fed proliferative and serum-starved nonproliferative conditions. We found that ROS1 mRNA levels were not detectable in endothelium under either condition (Supplemental Figure 5B). Conversely, in VSMCs, we found that ROS1 was expressed at low levels in serum-fed proliferative cells, but was dramatically upregulated as these cells differentiated into a serum-starved contractile phenotype (5-fold, P < 0.05 vs. serum-fed), furthered by confluence (15-fold, P < 0.05 vs. serum-fed), and then decreased to baseline following passage. Addition of siRNA specific for ROS1 (siROS1) to serum-starved VSMCs successfully decreased mRNA levels to very low levels (P < 0.05 vs. serum-fed; Figure 4C).
ROS1 regulates VSMC migration, proliferation, and survival.
We hypothesized that ROS1 might directly affect vascular remodeling via proliferation, migration, and survival of VSMCs. We derived primary aortic VSMCs from Gpx1–/– Apoe–/– mice following balloon angioplasty and found significantly higher proliferation than in Gpx1+/+ Apoe–/– controls (Figure 4D). Interestingly, siROS1, the broad-spectrum RTK antagonist genistein, and the more specific ALK-MET-ROS1 inhibitor crizotinib significantly decreased proliferation in Gpx1–/– Apoe–/–, but not Gpx1+/+ Apoe–/– cells (Figure 4D), indicating a dependency on perturbation of the network hub. When siROS1 and genistein or siROS1 and crizotinib were used in combination, there was no further reduction in proliferation (Figure 4D), suggesting these agents share a common pathway. To clarify that tyrosine kinase inhibition rather than oxidative quenching was the mechanism, we used a structural analogue of genistein, which has no effect on tyrosine kinase inhibition, but similar antioxidant effects. Daidzein did not inhibit proliferation in Gpx1–/– Apoe–/– VSMCs (Figure 4D). Furthermore, addition of siROS1 to daidzein-treated cells showed a reduction in proliferation similar to that in siROS1 alone (Figure 4D), further clarifying that the principal mode of action was through RTK inhibition. Inhibition of proliferation by crizotinib indicated this RTK was likely ROS1.
Migration is an important parallel process to proliferation in VSMCs. We found that migration was regulated in a manner similar to that of proliferation (Supplemental Figure 5C). Apoptosis was lower in Gpx1–/– Apoe–/– VSMCs at baseline compared with Gpx1+/+ Apoe–/– (Supplemental Figure 5D). siROS1, genistein, or crizotinib with or without siROS1 in Gpx1+/+ Apoe–/– VSMCs decreased apoptosis to Gpx1–/– Apoe–/– levels (Supplemental Figure 5D). These findings confirm that the proproliferative and antiapoptotic functions of ROS1 as a protooncogene are active in VSMCs.
Reductive stress from intracellular accumulation of GSH activates ROS1.
To explore more directly the mechanism of action linking GPX1 to ROS1, we assessed the ratio of reduced (GSH) to oxidized GSH (GSSG) (GPX oxidizes GSH to GSSG). We found GSH levels to be mildly elevated in Gpx1–/– Apoe–/– aortas compared with Gpx1+/+ Apoe–/– at baseline (Figure 5A). Although we could not detect ROS1 phosphorylation in Gpx1+/+ Apoe–/– mice, phosphorylated ROS1 protein was constitutively present in Gpx1–/– Apoe–/– animals (Figure 5A). When we performed balloon angioplasty in Gpx1–/– Apoe–/– mice, we found a time-dependent increase in GSH reaching nearly 10-fold higher than baseline (Figure 5B) and a corresponding increase in GSH/GSSG ratio (Figure 5C) compared with that of uninjured controls. We also found a corresponding time-dependent increase in phosphorylation of ROS1 at Y2274 (Figure 5D), a phosphorylation site known to activate the kinase (21), which was in line with the time course of neointima development following vascular injury (5). Taken together, these findings suggest GSH reductive stress as a mediator of ROS1 phosphorylation and activation.
Figure 5. GPX1 deletion–dependent reductive stress activates ROS1.

(A) ROS1 activity, assessed semiquantitatively, expressed as phosphorylated ROS1, was detectable in aortic lysates of Gpx1–/– Apoe–/– mice. *P < 0.05 versus Gpx1+/+ Apoe–/– P-ROS1; †P < 0.05 versus Gpx1+/+ Apoe–/– GSH. (B and C) Deficiency of GPX1 in Gpx1–/– Apoe–/– mice that underwent balloon angioplasty led to a time-dependent increase in intracellular GSH levels and GSH/GSSG ratios with a (D) corresponding increase in ROS1 phosphorylation indicative of increasing ROS1 activity. *P < 0.05 compared with same time point; **P < 0.05 across time points. n = 3–6/group.
We next aimed to determine the cause of increased GSH levels in Gpx1–/– Apoe–/– mice, particularly following vascular injury. We measured expression levels of key enzymes in the GSH metabolic pathway, including those that synthesize GSH and those that utilize GSH as a substrate. We found significant increases in expression levels of the enzymes involved in GSH synthesis. GCS–heavy subunit and GCS–light subunit as well as GSH synthetase were increased in both Gpx1–/– Apoe–/– vessels that had undergone balloon angioplasty (Figure 6A) and human neointima from in-stent stenosis (Figure 6B). These findings are in keeping with the inflammation, oxidative stress, and cell proliferation that occur as a response to vascular injury known to promote the synthesis of GSH through these enzymes (22). When we measured expression levels of enzymes involved in GSH utilization, we found a significant increase in the deglutathiolating enzyme glutaredoxin 1 (GRX1) in Gpx1–/– Apoe–/– mice (Figure 6C).
Figure 6. Vascular injury leads to increased expression of GSH-synthesizing and -mobilizing enzymes.

(A) Vascular injury led to a significant increase in the expression of enzymes involved in biosynthesis of GSH, suggesting vascular injury as a major determinant of GSH reductive stress in Gpx1–/– Apoe–/– vessels following angioplasty. *P < 0.05. (B) These findings were confirmed in human in-stent neointima. The expression of enzymes involved in biosynthesis of GSH was significantly increased compared with human internal mammary artery harvested at the time of coronary artery bypass surgery. *P < 0.05. Fold change in mRNA versus GAPDH. (C) mRNA levels of enzymes involved in the metabolic fate of GSH showed absence of expression of Gpx1 in Gpx1–/– Apoe–/– vessels and an increase in expression of the deglutathiolating enzyme Grx1. *P < 0.05. Fold change in mRNA versus GAPDH. Gsr, glutathione reductase; G6pd, glucose6-phosphate dehydrogenase; Gst, glutathione S-transferase; Ggt, glutathione hydrolase.
We performed further in vitro studies examining proliferation, migration, and survival in primary aortic VSMCs from experimental animals that had undergone angioplasty, additionally administered GSH in vitro. Proliferation in Gpx1+/+ Apoe–/– VSMCs treated with high concentrations of GSH monoethyl ester could be inhibited by siROS1, suggesting a phenotype switch toward Gpx1–/– Apoe–/– VSMCs (Figure 7). The addition of GSH to Gpx1–/– Apoe–/– cells did not affect proliferation patterns, suggesting preexisting saturation of intracellular GSH in these cells. Having found a physiological upregulation of the deglutathiolating enzyme GRX1 (Figure 6B) and complementing the effects of GSH augmentation, adenoviral overexpression of GRX1 (AdGRX1) decreased proliferation to levels similar to those in siROS1, while siRNA directed toward GRX1 (siGRX1) led to increased proliferation in VSMCs from both groups of mice (Figure 7). These findings not only support GSH reductive stress as a mediator of ROS1 activation, but also suggest protein s-glutathiolation as a potential mechanism.
Figure 7. Gpx1 deletion–dependent reductive stress activates ROS1 and regulates proliferation.

Following balloon angioplasty and treatment with high concentrations of GSH, primary aortic VSMC proliferation was inhibited by siROS1 in Gpx1+/+ Apoe–/– mice, while Gpx1–/– Apoe–/– cells were not affected. Adenoviral overexpression of GRX1 inhibited proliferation in both groups, while siGRX1 increased proliferation in Gpx1+/+ Apoe–/– VSMCs, suggesting proliferation was mediated by protein s-glutathiolation. Representative images of cell proliferation (green fluorescence) in Gpx1+/+ Apoe–/– mice treated with GSH and siROS1 or AdGRX1. *P < 0.05 vs. control; †P < 0.05 vs. AdGFP. Scale bar: 20 μm.
Migration of VSMCs was regulated in a manner similar to proliferation (Supplemental Figure 6A). In cell apoptosis assays, addition of GSH to Gpx1+/+ Apoe–/– VSMCs led to a decrease in apoptosis that could be partially reversed by the addition of siROS1 or AdGRX1 (Supplemental Figure 6B), suggesting that GSH sensitizes Gpx1+/+ Apoe–/– VSMCs to ROS1 activation by s-glutathiolation, producing a phenotype similar to that of Gpx1–/– Apoe–/– cells (Supplemental Figure 6B).
s-Glutathiolation of critical SHP-2 cysteines inhibits ROS1 deactivation.
We next sought to determine the mechanism of ROS1 as a redox sensor in VSMCs. Disulfide bonds in proteins are formed by oxidation of the thiol groups of cysteine residues. Specifically, GSH, which is composed of glutamate-cysteine-glycine, can react with oxidatively modified cysteine on other proteins to form a GSH conjugate: s-glutathiolation. To investigate the role of s-glutathiolation on ROS1 function, HEK293T cells were transfected with myc-tagged ROS1 plasmid. s-Glutathiolation was induced by treating cells with the oxidizing agent diamide and incubating cells with biotinylated GSH-ethyl-ester (Figure 8A). s-Glutathiolation could be inhibited by the reducing agent DTT, indicating the need for oxidative modification of Cys for s-glutathiolation to occur (Figure 8A). IP for ROS1 following s-glutathiolation did not identify ROS1 as a glutathiolated protein (Figure 8B); however, other GSS-protein conjugates between the molecular weights 55 and 90 kDa were detected (Figure 8B). To identify these proteins, we performed MS peptide mass fingerprinting of the IP and screened proteins at 55–90 kDa (Supplemental Table 4). Among the potential candidates, we identified the 68-kDa protein tyrosine phosphatases SHP-1 and SHP-2, which we and others have previously shown regulate ROS1 function (23, 24). When s-glutathiolation was induced in HEK293T cells transfected with myc-tagged SHP-1 or SHP-2 plasmid, we noted s-glutathiolation of SHP-2 (Figure 8C) following IP.
Figure 8. s-Glutathiolation of SHP-2 catalytic and backdoor cysteines inhibits ROS1 deactivation.

(A) HEK293 cells transfected with myc-tagged ROS1 and incubated with biotinylated GSH-ethyl-ester induced cysteine activation and s-glutathiolation of many proteins inhibitable by the reducing agent DTT. (B) s-Glutathiolation of ROS1 was not detected following immunoprecipitation; however, other GSS-protein conjugates were detected. Peptide mass fingerprinting showed that SHP-1 and SHP-2 were coimmunoprecipitated with ROS1, and (C) immunoprecipitation identified s-glutathiolation of SHP-2. LC-ESI-MS/MS identified the sites of glutathiolation. Extracted ion chromatograms of the SHP-2 peptides (D) Q450-R469 and (E) S326-R343 in the native form (top panel, control), s-glutathiolated form (middle panel), and reduced form (bottom panel) identify the catalytic Cys 463 and backdoor Cys 333 as the sites of s-glutathiolation. Insets are the representative MS/MS spectra of the Q450-R469 and S326-R343 peptide, providing site-specific localization of s-glutathiolation in the case of the modified forms. Upper insets represent the specific B and y ions as detected in the MS/MS spectra, showing inter-residue bond breakage generating b13 and y7 ions for Cys 463 and b8 and y11 for Cys 333, providing absolute confirmation. (F) (Upper panel and inset on left) Schematic representation of SHP-2 showing catalytic Cys 463 and backdoor Cys 333 and Cys 367 in the active PTP domain. (Inset on right) Modeling of s-glutathiolation of catalytic Cys 463 depicted by the spheres, demonstrating a physical presence within the active phosphatase domain of the enzyme.
To confirm our findings and determine the Cys sites of s-glutathiolation on SHP-2, we performed liquid chromatograph–electron spray ionization–mass spectrometry/mass spectrometry (LC-ESI-MS/MS). To maintain the physical relationship between ROS1 and SHP-2, we induced s-glutathiolation in HEK 293T cells overexpressing myc-ROS1 instead of myc–SHP-2. MS/MS confirmed s-glutathiolation of SHP-2, specifically the catalytic Cys 463 (Figure 8D) and backdoor Cys 333 (Figure 8E). Interestingly, unlike s-glutathiolation of Cys 463, which could be reversed by the addition of the potent reducing agent DTT (total ion chromatogram [TIC]; 6.0 × 106 → 0 with DTT), s-glutathiolation of Cys 333 could not be reduced using even 0.1 M DTT (TIC 6.0 × 104 → 1.3 × 104 with DTT), suggesting an irreversible ionic interaction. Computational protein structure modeling identified s-glutathiolation of the catalytic Cys 463 as physically blocking the phosphatase active site of the enzyme (Figure 8F).
To test the functional significance of s-glutathiolation on critical cysteines in SHP-2, we performed site-directed mutagenesis of catalytic Cys 463 and the 2 backdoor cysteines, Cys 333 and Cys 367, which protect catalytic Cys 463 from irreversible oxidation (25). When Cys 367 and Cys 333 are oxidized, their physical distance favors the generation of a disulfide bridge between them, thus protecting the catalytic domain Cys 463 from oxidation. In conditions in which 1 backdoor cysteine is irreversibly oxidized or in a mixed disulfide with another protein or molecule, it is then possible for the free backdoor cysteine to interact directly with the catalytic cysteine, rendering the phosphatase inactive (26). When phosphatase activity of SHP-2 mutants was assessed, with or without the SH2 domain activator bisphosphoryl tyrosine-based activation motif (BTAM), we found that mutation of the C367S was not different from that of wild-type SHP-2. However, the C333S mutant had less than 10% phosphatase activity, and C463S was completely inactive (Figure 9A).
Figure 9. Catalytic and backdoor cysteines inhibit ROS1 deactivation in vitro and in vivo.

(A) Cys/Ser site-directed mutagenesis of catalytic Cys 463 and backdoor Cys 333 and Cys 367 were performed and phosphatase activity was assessed with and without stimulation. C367S did not affect phosphatase activity, whereas C333S led to a 90% reduction in phosphatase activity, and C463S abolished all activity compared with wild-type. *P < 0.05 compared with wild-type; **P < 0.05 compared with C333. (B) SHP-2 time-dependent inactivation by H2O2 and reactivation by DTT. Full-length SHP-2 was oxidized or glutathiolated and phosphatase activities measured in a continuous assay, after which DTT was added. Both oxidation and glutathiolation could induce deactivation of SHP-2; however, recovery following addition of the reducing agent DTT was significantly impaired in the glutathiolation group. *P < 0.05 compared with H2O2 + DTT. (C) SHP-2 is glutathiolated in vivo in experimental animals. Western blotting for glutathiolated proteins in vessel homogenate identified increased band intensity in Gpx1–/– Apoe–/– mice compared with Gpx1+/+ Apoe–/–. Balloon angioplastied vessels were homogenized and immunoprecipitated for ROS1 or SHP-2 under nonreducing conditions. The immunoprecipitated protein was immunoblotted with anti-GSH antibody. Blotting of immunoprecipitate identified the presence of glutathiolated SHP-2, but also ROS1, suggesting a physical interaction in vivo. (D) Increased ROS1 activity in Gpx1–/– Apoe–/– mice in vivo. Balloon angioplastied vessels harvested 28 days postprocedurally were assessed for Ros1 phosphorylation. Levels of ROS1 phosphorylation were higher in Gpx1–/– Apoe–/– mice (P < 0.05) without differences in ROS1 protein.
Previous studies have shown that protein tyrosine phosphatases may be reversibly oxidized and that oxidation affects function of phosphatase (27), including SHP-2 (25). To differentiate the effects of s-glutathiolation from oxidation, we performed a time-course phosphatase experiment on purified SHP-2 protein. We found that both oxidation and s-glutathiolation could induce deactivation of SHP-2; however, recovery following addition of the reducing agent DTT was significantly impaired following s-glutathiolation but not oxidation (Figure 9B), consistent with the nonreducible ionic interactions that were identified in MS (Figure 8E).
Finally, to prove that SHP-2 is s-glutathiolated in vivo, we harvested vessels from experimental animals 28 days after balloon angioplasty. ROS1, SHP-1, or SHP-2 IPs from vessel homogenate under nonreducing conditions were immunoblotted with anti-GSH antibody. IP immunoblot identified the presence of glutathiolated SHP-2, but also ROS1, suggesting that ROS1 and SHP-2 physically interact in vivo (Figure 9C). Finally, in a separate group of experimental animals that underwent balloon angioplasty, we detected that Ros1 was phosphorylated in control Gpx1+/+ Apoe–/– vessels; however, levels of ROS1 phosphorylation were significantly higher in Gpx1–/– Apoe–/– vessels (Figure 9D) despite no differences in ROS1 nonphosphorylated protein.
Inactivation of ROS1 abolishes increased in-stent stenosis in Gpx1–/– Apoe–/– mice.
To test the idea that inactivation of ROS1 could modulate vascular remodeling in vivo, we administered genistein or crizotinib to a cohort of mice. We found that the increase in atherosclerotic burden in the aortic root (Figure 1B) of Gpx1–/– Apoe–/– mice was abolished with the addition of genistein, but not crizotinib (Figure 10A), implying the need for vascular injury to activate ROS1. Accordingly, the increase in in-stent neointima in stented vessels (Figure 1D) in Gpx1–/– Apoe–/– mice was abolished with the addition of genistein and crizotinib (Figure 10B) to levels observed with the mTOR inhibitor rapamycin. AdGRX1 decreased in-stent neointima to levels similar to those of crizotinib in Gpx1–/– Apoe–/–, but not in Gpx1+/+ Apoe–/– vessels (Figure 10B), suggesting deglutathiolation of SHP-2 as the mechanism. The increase in the percentage of plaque area occupied by α-actin–positive SMCs was also abolished in crizotinib-treated Gpx1–/– Apoe–/– mice, confirming the effect on VSMCs (Supplemental Table 2). Although overall neointimal cell number was higher in Gpx1–/– Apoe–/– mice, there was no difference in cell density when expressed per unit area between groups. However addition of genistein, crizotinib, or rapamycin significantly decreased cell density per unit area in both experimental groups, but AdGRX1 treatment only decreased cell density in Gpx1–/– Apoe–/– mice (Figure 10B), implicating an effect of both ROS1 inhibition and SHP-2 deglutathiolation on VSMC proliferation.
Figure 10. Increased in-stent stenosis in Gpx1–/– Apoe–/– mice is reversed by ROS1 inhibition.

(A) Relative area of atherosclerosis in the aortic root was significantly increased in Gpx1–/– Apoe–/– mice, but addition of genistein to inhibit ROS1 abolished this increase, while crizotinib did not. Scale bar: 50 μm. (B) Gpx1–/– Apoe–/– mice 4 weeks following BAS developed significantly greater neointimal hyperplasia and in-stent stenosis compared with control Gpx1+/+ Apoe–/–. Addition of genistein or crizotinib decreased neointimal hyperplasia to levels observed with the mTOR inhibitor rapamycin. AdGRX1 decreased neointimal hyperplasia to levels similar to those seen with crizotinib in Gpx1–/– Apoe–/– vessels, but not Gpx1+/+ Apoe–/–, suggesting deglutathiolation of SHP-2 as the mechanism. Scale bar: 50 μm. Black arrowheads denote neointima. Although overall neointimal cell density was higher in Gpx1–/– Apoe–/– mice, there was no difference in cell density when expressed per unit area between groups; however, addition of genistein, crizotinib, or rapamycin significantly decreased cell density per unit area in both experimental groups. Treatment with AdGrx1 reduced cell density only in Gpx1–/– Apoe–/– mice. *P < 0.05 compared with control; †P < 0.05 compared with AdGFP. n = 8–15/group. (C) Assessment of reendothelialization revealed that the ROS1 inhibitor crizotinib did not impair endothelial regeneration 4 weeks following BAS, while rapamycin did in both Gpx1–/– Apoe–/– Tie2-LacZ+ mice and control Gpx1+/+ Apoe–/– Tie2-LacZ+ mice. *P < 0.05 versus crizotinib. n = 6–10 mice/group. Gray arrowheads denote neointima. Black arrowheads denote LacZ-positive endothelial cells. Scale bar: 50 μm.
To directly investigate the therapeutic potential of crizotinib for in-stent stenosis, we assessed reendothelialization, a major determinant of stent outcome in humans (28). Twenty-eight days following BAS, treatment with crizotinib did not affect reendothelialization in either Gpx1+/+ Apoe–/– Tie2-LacZ+ or Gpx1–/– Apoe–/– Tie2-LacZ+ vessels. However, treatment with the mTOR inhibitor rapamycin, representative of the class of drug currently used clinically in DESs to prevent in-stent stenosis, significantly reduced reendothelialization (Figure 10C).
Discussion
In this study, we use network analysis to identify GPX1 as a key determinant of vascular remodeling and describe a new mechanism of effect mediated by GSH-dependent phosphorylation of ROS1 (summarized in Figure 11). Using a model of BAS in Gpx1+/+ Apoe–/– mice with or without Gpx1 deletion, we made a number of key observations. Knockout of Gpx1 increased atherosclerosis and ISS 4 weeks after BAS. In mice additionally expressing an endothelial cell–specific β-gal reporter, reendothelialization was reduced in Gpx1–/– Apoe–/– mice, yet vascular wall ROS generation was confined largely to the medium. Network analysis directed us towards the orphan protooncogene RTK ROS1 as a potential mediator. We validated the importance of this mechanism in humans by showing that SNPs in GPX1 and ROS1 are independently associated with ISS in humans and that interaction of these variants ranks highly in both interaction and allele dose models. Using in vitro and in vivo techniques, we found that ROS1 was nearly undetectable and inactive in quiescent uninjured cells, but dramatically upregulated and activated following cell injury, leading to a proproliferative antiapoptotic phenotype in VSMCs, but not endothelium. Intracellular accumulation of GSH as a result of GPX1 deficiency led to a time-dependent, dose-dependent increase in ROS1 activity. While in vivo ROS1 was upregulated following BAS, pharmacological inhibition of ROS1 abolished this increase and the greater atherosclerosis and ISS previously demonstrated in Gpx1–/– Apoe–/– mice. Together, these findings provide evidence that GPX1 is a critical regulator of the response to vascular injury and that VSMC ROS1 is a major determinant of this effect.
Figure 11. Model of ROS1 activation by reductive stress.

In VSMCs, vascular injury induces activation of ROS1 through either a yet-unidentified ligand (ligand X) or endogenous phosphorylation. (A) Concomitant vascular injury also leads to decreased transcription of GPX1 (i), which leads to a decrease in GPX1 enzymatic activity during a time of increased GSH synthesis, resulting in increased intracellular GSH (ii). Under oxidative conditions, high GSH leads to s-glutathiolation of proteins, including the ROS1 regulatory protein tyrosine phosphatase SHP-2 (iii). (B) s-Glutathiolation deactivates SHP-2, which in turn leads to sustained phosphorylation and activation of ROS1. Sustained phosphorylation of ROS1 amplifies its ability to activate kinases involved in proliferation, migration, and apoptosis through downstream target phosphorylation (iv).
Systems approaches can be powerful enablers of hypothesis generation in biology. Network models exhibiting scale-free topology identify disproportionately important roles for highly connected hubs. Hub genes revealed in this way represent attractive targets for biological and therapeutic exploration (29). Here, we took advantage of the availability of human tissue derived from coronary atherectomy to build blood vessel–specific network models identifying GPX1 as a hypothetical mediator of disease. While powerful, such large scale unbiased approaches are optimal as tools of hypothesis generation. The incorporation of a priori knowledge in our analysis helps strengthen network connections for which prior literature exists while incorporating novel associations found in the tissue of interest. Thus, network approaches represent a starting point, and further elucidation of biological mechanisms is critical to confirming the significance of the identified associations.
Consistent with such systems interactions, vascular cellular homeostasis depends on a balanced redox environment. Simplistic concepts of “oxidative stress” have failed to deliver therapeutic benefits in cardiovascular diseases (30, 31), due in large part to inadequate understanding of specific redox mechanisms that are pivotal in disease pathogenesis and may have opposing consequences. We show that oxidative stress in GPX1 deficiency also leads to a reductive redox state with pathological consequences. Starting from network analysis encompassing blind transcriptional profiling and the ability to account for a priori knowledge, we identify activation of ROS1 as a new pathological redox-dependent effector mechanism linking GSH, GPX1, and VSMC proliferation.
The finding of interaction between common genetic variants in GPX1 and ROS1 lends credence to a regulatory interplay between GPX1 and ROS1. Both variants are prevalent in the population, with minor allele frequencies of 0.25 and 0.16, respectively. They both result in coding changes to the resultant peptide. The GPX1 variant Pro198Leu has previously been noted to result in a reduced enzymatic activity of GPX1 (32), which would be consistent with increased ROS1 activation and increased risk of in-stent stenosis. The functional significance of the ROS1 variant Asp2213Asn remains unknown. Although it is tempting to postulate that both variants may indeed be functional, the possibility exists that they are instead in linkage disequilibrium with some other functional variant. There are many mechanisms by which these variants may lead to the clinical phenotype, and this is deserving of further study.
Vascular injury in Gpx1–/– Apoe–/– animals leads to a unique metabolic environment whereby GPX1 deficiency coupled with increased enzymatic synthesis of GSH leads to both oxidative and reductive stress. We found that this environment promoted s-glutathiolation of proteins. When our initial hypothesis of direct s-glutathiolation of ROS1 was refuted, peptide mass fingerprinting identified SHP-1 and SHP-2 as potential candidates. We and others have previously shown that SHP-1 (23) and SHP-2 (24) regulate ROS1 function. After confirming s-glutathiolation of SHP-2, we used MS to identify that the catalytic Cys 463 was a site of posttranslational modification. Computational molecular modeling predicted a physical obstruction of the phosphatase site of SHP-2 by s-glutathiolation, which was corroborated by our site-directed mutagenesis experiments showing a loss of SHP-2 function with perturbation of the backdoor Cys 333, but not Cys 367. These 2 backdoor cysteines protect the catalytic Cys 463 from oxidative modification (25). When Cys 367 and Cys 333 are oxidized, their physical distance favors the generation of a disulfide bridge between the two of them, protecting the catalytic domain Cys 463 from oxidation (25). In conditions in which one backdoor cysteine is irreversibly oxidized or in a mixed disulfide with another protein or molecule, it is then possible for the free backdoor cysteine to interact directly with the catalytic cysteine, rendering the phosphatase inactive (26). Our findings that s-glutathiolation of Cys 463 was reversible, while that of Cys 333 was not, support this mechanism and suggest that irreversible oxidation of Cys 333 leads the catalytic Cys 463 to be prone to s-glutathiolation.
Several features of ROS1 make it an attractive candidate for potential therapeutic intervention in vascular remodeling. ROS1 encodes a type I integral membrane protein with RTK activity (20). The ectodomain of ROS1 has high-sequence homology to the extracellular matrix protein fibronectin present in cell surface and extracellular matrix proteins, such as cell-adhesion molecules (20). The distinctive structure of the cell-binding ectodomain with intracellular kinase domain suggests a unique capability of ROS1 to transmit cell adhesion to intracellular signaling. As cell adhesion is critical to the pathogenesis of vascular remodeling, the structural properties of ROS1 make it attractive as a candidate for a role in such processes. From a clinical standpoint, ROS1 has been described predominantly in the setting of chromosomal translocations creating gene fusions in highly malignant cancers (33–38). ROS1 is an attractive therapeutic target, not only because it drives proliferation, but also because of its preferential expression. In humans, ROS1 expression is undetectable in most tissues except heart, skeletal muscle (39), and lung (20). At a cellular level, we show that ROS1 is expressed in VSMCs, but not endothelium. Current DES have reduced the problem of in-stent stenosis by targeting all proliferating cells using chemotherapeutic agents, but the beneficial reductions in neointima formation are accompanied by impaired endothelial regeneration and vascular healing that has created a host of new concerns, including stent thrombosis (40), neointimal atherosclerosis (41), and localized hypersensitivity reactions leading to peri-strut cavity and aneurysm formation (28). Our findings that crizotinib inhibited in-stent stenosis without affecting reendothelialization could thus have a major clinical impact.
In conclusion, we have shown that GPX1 critically modulates neointimal hyperplasia following vascular injury mediated by activation of ROS1. Our study identifies GSH-dependent activation of ROS1 as a critical mediator of SMC proliferation and migration, both major determinants of vessel remodeling following vascular injury. The identification of a redox mechanism with specific effects in vascular disease pathogenesis highlights the need for targeted rather than broad-spectrum therapies.
Methods
For detailed experimental procedures, see Supplemental Experimental Procedures.
Animals.
The 18- to 22-week-old male and female mice were maintained in temperature-controlled (20°C to 22°C) cages with a 12-hour light/12-hour dark cycle. Sterile water and standard chow diet were available ad libitum. Mice with ubiquitous homozygous deletion of Gpx1 (Gpx1–/–) crossed with Apoe-null (Gpx1–/– Apoe–/–) or Gpx1+/+ Apoe–/– controls were used for experiments. For endothelial survival and regeneration experiments, these mice were further crossbred with transgenic Tie2-LacZ mice expressing β-gal localized to the nucleus of endothelial cells (42), producing experimental Gpx1+/+ Apoe–/– Tie2-LacZ+ and Gpx1–/– Apoe–/– Tie2-LacZ+ mice. All experiments were performed blind. Following genotyping and prior to experiments, animals were placed in cages labeled A and B. Subsequently, all experiments were performed by a group by investigators not involved in the genotyping or blinding. Experiments were unblinded after completion of data analysis.
Gene expression analysis.
RNA preparation, hybridization to microarray, data processing, and data analysis for microarray performed on human coronary artery atherectomy specimens or mouse aorta (n = 30) were performed as described previously (14). All original microarray data were deposited in the NCBI’s Gene Expression Omnibus (GDS1597).
Network analysis.
To determine molecular targets to investigate restenosis, we performed comprehensive histological and gene network analysis of human in-stent stenosis as described previously (14) using Cytoscape (43).
Tissue GSH and cellular GPX1 activity.
Tissue concentrations of GSH (total, reduced, and oxidized) were measured in aortic homogenates after deproteinization with metaphosphoric acid in an enzymatic recycling method using GSH reductase, as described (44).
BAS procedure.
BAS was performed in 18- to 22-week-old mice as described previously (5). In order to be able to track endothelial cells by in situ hybridization for the Y chromosome, stent grafts were constructed using female donors and male recipient mice.
Gene transfer.
Freshly harvested vessels were incubated in a total of 1 × 1011 viral particles of AdGRX1 or AdGFP in 300 μl for 30 minutes at 37°C prior to implantation.
Tissue preparation, histology, and lesion quantification.
Mice were euthanized 28 days following the stent procedure. Aorta and stented vessels were excised, fixed in paraformaldehyde overnight, dehydrated in graded ethanol solution, and paraffin embedded or removed fresh and snap-frozen in either Trizol, OCT, or maintained fresh. Histomorphometric analysis of aortic atherosclerosis lesion area and stented vessel processing and analysis were performed as previously described (5). Immunohistochemistry was performed as described previously (45).
En face X-gal staining.
The procedure for en face preparation was similar to that described previously (45).
Oxidative fluorescent microtopography.
Superoxide was detected in the layers of the vessel wall using fluorescent probe DHE as described (45).
Lucigenin and luminol-enhanced chemiluminescence.
Total aortic superoxide was measured by lucigenin-enhanced chemiluminescence, as described previously (45).
Electron paramagnetic resonance spectroscopy.
Electron paramagnetic resonance (EPR) spectroscopy was used to quantify vascular NO production as previously described (45).
Cell culture.
Human coronary artery SMCs were propagated in SmGM-2 growth medium containing 5% FBS. To induce growth arrest and the expression of differentiation genes, SMCs were serum starved in basal medium (SmBM) for 72 hours, according to conventional protocols (45). In vitro ROS1 inhibition studies were completed by adding 40 μM genistein to the cell-culture media. In some experiments, cells were treated with 0.5 mM GSH monoethyl ester (Sigma-Aldrich). For overexpression of Grx1, cells were incubated with adenovirus overexpressing Grx1 (25 MOI) at a concentration of 4.5 × 1010 pfu/ml.
Proliferation and motility assays.
Proliferation and migration experiments were performed as described (46).
Survival assay.
Survival assays were performed using caspase 3/7 assays as described previously (46).
Quantitative real-time PCR.
Total RNA was extracted from cells or snap-frozen tissue and used for quantitative real-time PCR as described previously (47).
Immunoprecipitation and immunoblotting.
Human coronary artery SMCs (HCASMCs) and aorta/stented vessel lysates (n ≥ 5 per group) were used for immunoprecipitation and immunoblotting performed using a previously described method (45).
Biotinylated GSH ester and detection of s-glutathiolated proteins.
Protein s-glutathiolation was detected in tissues and in cells as described previously (48).
MS.
Samples were digested under reducing and nonreducing conditions using trypsin. Nano LC-MS was employed using in-house pulled or packed C18 reverse-phase analytical columns. The mass spectrometer was an LTQ Orbitrap Velos set to perform collision-induced dissociation, higher-energy C trap dissociation, or electron-transfer dissociation on multiple charged cations.
Modeling of s-glutathiolation by protein-ligand flexible docking.
To visualize s-glutathiolation of critical SHP-2 cysteines, structural docking was performed on the binding site of interest with GSH using Swiss Dock (49) and the docking poses were refined using GROMACS (50). A final selection of glutathiolated protein-ligand–binding modes was done by scoring refined docking poses with a knowledge-based scoring function (51), which fulfilled the criterion for s-glutathiolation.
Lipid and lipoprotein analysis.
Total plasma cholesterol and triglyceride concentrations were measured using enzymatic assay (Roche) on a Cobas Mira Plus automated analyzer (Roche).
Single nucleotide variant information gain analysis.
Attribute selection analysis was performed in the Weka (Java) environment (52), using all SNVs as attributes and using an information gain evaluator conditioned on the presence or absence of in-stent restenosis with Ranker as the attribute search method.
SNV interaction analysis.
SNV interaction analysis was performed using the Polymorphism Interaction Analysis (PIA) tool (v 2.0, available at http://www3.cancer.gov/intra/lhc/PIA2-distribution.zip) (53), which utilizes several different methods, including percentage correct, sensitivity plus specificity, PPV plus NPV, risk ratio, odds ratio, Gini index, and absolute probability difference, to evaluate for interactions between SNP combinations in case-control genotyping association studies. A 2-SNP interaction model was used for the analysis.
Linear regression model.
Fitting of a generalized linear regression model was done in R (http://www.r-project.org/), conditioning the presence or absence of in-stent stenosis on a score consisting of the number of at-risk alleles for both GPX1 and ROS1, with a minimum of 0 and a maximum of 3 (no member of the cohort had 4 at-risk alleles).
GWAS meta-analysis.
A meta-analysis was performed combining 4 separate cohorts (Cardiogene, GeneBank, deCODE, and GENDER) of cases and controls comparing patients after BAS who were found to have in-stent stenosis with those without in-stent stenosis. Cases of in-stent stenosis were clinically adjudicated, and patients were genotyped according to the protocols of each individual study. Statistical analysis was performed using inverse variance fixed effects meta-analysis, implemented in METAL (54). Genomic control correction was applied at the level of each cohort and then again at the meta-analysis level (double genomic control correction).
Statistics.
Data are presented as mean ± SEM. Data were subjected to the Kolmogorov-Smirnov test to determine distribution. Groups were compared using the Mann-Whitney U test for nonparametric data or 2-tailed Student’s t test for normally distributed data. When comparing multiple groups, data were analyzed by ANOVA with Newman-Keuls post-hoc test for parametric data or Kruskal-Wallis test with Dunn’s post-hoc test for nonparametric data. A P value of less than 0.05 was considered significant.
Study approval.
All animal studies were approved by the Stanford University or Columbia University Administrative Panel on Laboratory Animal Care (protocol 22922 and AAAE9200) and conform to NIH guidelines (Guide for the Care and Use of Laboratory Animals. NIH publication no. 85-23. Revised 1996). All human studies were performed with written informed consent and with the approval of the Stanford University Institutional Review Board.
Supplementary Material
Acknowledgments
Z.A. Ali is supported by NIH grant R00HL109256; G. Chopra is supported by a Juvenile Diabetes Research Foundation fellowship; S.L. Hazen and GeneBank are supported by NIH grant P01 HL076491; J. Wouter Jukema is supported by the Netherlands Heart Foundation (2001D032); S. Ganesh is supported by NIH grant R00HL089413; and E. Ashley is supported by the NIH Director’s New Innovator Award (DP2 OD004613) and NIH grant R01 HL105993. The CardioGene Study and deCODE analysis were supported, in part, by the Division of Intramural Research, NIH. GENDER was supported by grants from the Interuniversity Cardiology Institute of the Netherlands (ICIN) and the Durrer Center for Cardiogenetic Research, both of which are Institutes of the Netherlands Royal Academy of Arts and Sciences, the Netherlands Heart Foundation, the Center for Medical Systems Biology approved by the Netherlands Genomics Initiative/Netherlands Organization for Scientific Research, and the Netherlands Consortium for Healthy Ageing. The research leading to these results has received funding from the European Union’s Seventh Framework Program (FP7/2007-2013) under grant agreement HEALTH-F2-2009-223004. We are grateful to Reto Asmis (University of Texas San Antonio), Victoria Rimkunas (Cell Signaling), Diane Handy (Harvard University), Susumu Kobayashi (Beth Israel Deaconess Medical Center), and Jonathan Pollack (Stanford University) for their critical advice and supply of reagents. The authors would like to dedicate this article to the memory of Allan Kuchinsky, who was instumental to the development of high-throughput and network technology.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2014;124(12):5159–5174. doi:10.1172/JCI77484.
See the related Commentary beginning on page 5092.
References
- 1.Go AA, et al. Heart disease and stroke statistics — 2014 update: A report from the American Heart Association. Circulation. 2014;129(3):e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ip JH, Fuster V, Badimon L, Badimon J, Taubman MB, Chesebro JH. Syndromes of accelerated atherosclerosis: Role of vascular injury and smooth muscle cell proliferation. J Am Coll Cardiol. 1990;15(7):1667–1687. doi: 10.1016/0735-1097(90)92845-S. [DOI] [PubMed] [Google Scholar]
- 3.Weisz G, et al. Five-year follow-up after sirolimus-eluting stent implantation results of the SIRIUS (Sirolimus-Eluting Stent in De-Novo Native Coronary Lesions) Trial. J Am Coll Cardiol. 2009;53(17):1488–1497. doi: 10.1016/j.jacc.2009.01.050. [DOI] [PubMed] [Google Scholar]
- 4.Finn AV, et al. Vascular responses to drug eluting stents: importance of delayed healing. Arterioscler Thromb Vasc Biol. 2007;27(7):1500–1510. doi: 10.1161/ATVBAHA.107.144220. [DOI] [PubMed] [Google Scholar]
- 5.Ali ZA, et al. Increased in-stent stenosis in ApoE knockout mice: insights from a novel mouse model of balloon angioplasty and stenting. Arterioscler Thromb Vasc Biol. 2007;27(4):833–840. doi: 10.1161/01.ATV.0000257135.39571.5b. [DOI] [PubMed] [Google Scholar]
- 6.Blankenberg S, et al. Glutathione peroxidase 1 activity and cardiovascular events in patients with coronary artery disease. N Engl J Med. 2003;349(17):1605–1613. doi: 10.1056/NEJMoa030535. [DOI] [PubMed] [Google Scholar]
- 7.Lapenna D, et al. Glutathione-related antioxidant defenses in human atherosclerotic plaques. Circulation. 1998;97(19):1930–1934. doi: 10.1161/01.CIR.97.19.1930. [DOI] [PubMed] [Google Scholar]
- 8.Forgione MA, et al. Heterozygous cellular glutathione peroxidase deficiency in the mouse: abnormalities in vascular and cardiac function and structure. Circulation. 2002;106(9):1154–1158. doi: 10.1161/01.CIR.0000026820.87824.6A. [DOI] [PubMed] [Google Scholar]
- 9.Galasso G, et al. Impaired angiogenesis in glutathione peroxidase-1–deficient mice is associated with endothelial progenitor cell dysfunction. Circ Res. 2006;98(2):254–261. doi: 10.1161/01.RES.0000200740.57764.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Torzewski M, et al. Deficiency of glutathione peroxidase-1 accelerates the progression of atherosclerosis in apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol. 2007;27(4):850–857. doi: 10.1161/01.ATV.0000258809.47285.07. [DOI] [PubMed] [Google Scholar]
- 11.Lewis P, et al. Lack of the antioxidant enzyme glutathione peroxidase-1 accelerates atherosclerosis in diabetic apolipoprotein e–deficient mice. Circulation. 2007;115(16):2178–2187. doi: 10.1161/CIRCULATIONAHA.106.664250. [DOI] [PubMed] [Google Scholar]
- 12.Yusuf S, Dagenais G, Pogue J, Bosch J, Sleight P. Vitamin E supplementation and cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342(3):154–160. doi: 10.1056/NEJM200001203420302. [DOI] [PubMed] [Google Scholar]
- 13.Lange H, et al. Folate therapy and in-stent restenosis after coronary stenting. N Engl J Med. 2004;350(26):2673–2681. doi: 10.1056/NEJMoa032845. [DOI] [PubMed] [Google Scholar]
- 14.Ashley EA, et al. Network analysis of human in-stent restenosis. Circulation. 2006;114(24):2644–2654. doi: 10.1161/CIRCULATIONAHA.106.637025. [DOI] [PubMed] [Google Scholar]
- 15.Schwartz RS, et al. Restenosis and the proportional neointimal response to coronary artery injury: results in a porcine model. J Am Coll Cardiol. 1992;19(2):267–274. doi: 10.1016/0735-1097(92)90476-4. [DOI] [PubMed] [Google Scholar]
- 16.Inoue T, Croce K, Morooka T, Sakuma M, Node K, Simon DI. Vascular inflammation and repair: implications for re-endothelialization, restenosis, and stent thrombosis. JACC Cardiovasc Interv. 2011;4(10):1057–1066. doi: 10.1016/j.jcin.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forgione MA, et al. Cellular glutathione peroxidase deficiency and endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2002;282(4):H1255–H1261. doi: 10.1152/ajpheart.00598.2001. [DOI] [PubMed] [Google Scholar]
- 18.Oelze M, et al. Glutathione peroxidase-1 deficiency potentiates dysregulatory modifications of endothelial nitric oxide synthase and vascular dysfunction in aging. Hypertension. 2014;63(2):390–396. doi: 10.1161/HYPERTENSIONAHA.113.01602. [DOI] [PubMed] [Google Scholar]
- 19.Oguri M, et al. Genetic risk for restenosis after coronary stenting. Atherosclerosis. 2007;194(2):e172–e178. doi: 10.1016/j.atherosclerosis.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 20.Acquaviva J, Wong R, Charest A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim Biophys Acta. 2009;1795(1):37–52. doi: 10.1016/j.bbcan.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 21.Rikova K, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131(6):1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 22.Lu SC. Regulation of hepatic glutathione synthesis: current concepts and controversies. FASEB J. 1999;13(10):1169–1183. [PubMed] [Google Scholar]
- 23.Keilhack H, et al. Negative regulation of Ros receptor tyrosine kinase signaling. An epithelial function of the SH2 domain protein tyrosine phosphatase SHP-1. J Cell Biol. 2001;152(2):325–334. doi: 10.1083/jcb.152.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Charest A, et al. ROS fusion tyrosine kinase activates a SH2 domain-containing phosphatase-2/phosphatidylinositol 3-kinase/mammalian target of rapamycin signaling axis to form glioblastoma in mice. Cancer Res. 2006;66(15):7473–7481. doi: 10.1158/0008-5472.CAN-06-1193. [DOI] [PubMed] [Google Scholar]
- 25.Chen C-Y, Willard D, Rudolph J. Redox regulation of SH2-domain-containing protein tyrosine phosphatases by two backdoor cysteines. Biochemistry. 2009;48(6):1399–1409. doi: 10.1021/bi801973z. [DOI] [PubMed] [Google Scholar]
- 26.Tanner JJ, Parsons ZD, Cummings AH, Zhou H, Gates KS. Redox regulation of protein tyrosine phosphatases: structural and chemical aspects. Antioxid Redox Signal. 2011;15(1):77–97. doi: 10.1089/ars.2010.3611. [DOI] [PubMed] [Google Scholar]
- 27.Östman A, Frijhoff J, Sandin Å, Böhmer F-D. Regulation of protein tyrosine phosphatases by reversible oxidation. J Biochem. 2011;150(4):345–356. doi: 10.1093/jb/mvr104. [DOI] [PubMed] [Google Scholar]
- 28.Joner M, et al. Pathology of drug-eluting stents in humans: delayed healing and late thrombotic risk. J Am Coll Cardiol. 2006;48(1):193–202. doi: 10.1016/j.jacc.2006.03.042. [DOI] [PubMed] [Google Scholar]
- 29.Ashley EA, et al. The endogenous peptide apelin potently improves cardiac contractility and reduces cardiac loading in vivo. Cardiovasc Res. 2005;65(1):73–82. doi: 10.1016/j.cardiores.2004.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vivekananthan DP, Penn MS, Sapp SK, Hsu A, Topol EJ. Use of antioxidant vitamins for the prevention of cardiovascular disease: meta-analysis of randomised trials. Lancet. 2003;361(9374):2017–2023. doi: 10.1016/S0140-6736(03)13637-9. [DOI] [PubMed] [Google Scholar]
- 31.Stephens NG, Parsons A, Schofield PM, Kelly F, Cheeseman K, Mitchinson MJ. Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS). Lancet. 1996;347(9004):781–786. doi: 10.1016/S0140-6736(96)90866-1. [DOI] [PubMed] [Google Scholar]
- 32.Hu YJ, Diamond AM. Role of glutathione peroxidase 1 in breast cancer: loss of heterozygosity and allelic differences in the resonse to selenium. Cancer Research. 2003;63(12):3347–3351. [PubMed] [Google Scholar]
- 33.Suehara Y, et al. Identification of KIF5B-RET and GOPC-ROS1 fusions in lung adenocarcinomas through a comprehensive mRNA-based screen for tyrosine kinase fusions. Clinical Cancer Research. 2012;18(24):6599–6608. doi: 10.1158/1078-0432.CCR-12-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee H, et al. ROS1 receptor tyrosine kinase, a druggable target, is frequently overexpressed in non-small cell lung carcinomas via genetic and epigenetic mechanisms. Ann Surg Oncol. 2013;20(1):200–208. doi: 10.1245/s10434-012-2553-6. [DOI] [PubMed] [Google Scholar]
- 35.Zhao J-F, Sharma S. Expression of the ROS1 oncogene for tyrosine receptor kinase in adult human meningiomas. Cancer Genet Cytogenet. 1995;83(2):148–154. doi: 10.1016/0165-4608(95)00043-O. [DOI] [PubMed] [Google Scholar]
- 36.Charest A, et al. Fusion of FIG to the receptor tyrosine kinase ROS in a glioblastoma with an interstitial del(6)(q21q21). Genes Chromosomes Cancer. 2003;37(1):58–71. doi: 10.1002/gcc.10207. [DOI] [PubMed] [Google Scholar]
- 37.Gu TL, et al. Survey of tyrosine kinase signaling reveals ROS kinase fusions in human cholangiocarcinoma. PLoS One. 2011;6(1):e15640. doi: 10.1371/journal.pone.0015640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee J, et al. Identification of ROS1 rearrangement in gastric adenocarcinoma. Cancer. 2013;119(9):1627–1635. doi: 10.1002/cncr.27967. [DOI] [PubMed] [Google Scholar]
- 39.Su AI, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A. 2004;101(16):6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Finn AV, et al. Pathological correlates of late drug-eluting stent thrombosis. Circulation. 2007;115(18):2435–2441. doi: 10.1161/CIRCULATIONAHA.107.693739. [DOI] [PubMed] [Google Scholar]
- 41.Nakazawa G, et al. The pathology of neoatherosclerosis in human coronary implants bare-metal and drug-eluting stents. J Am Coll Cardiol. 2011;57(11):1314–1322. doi: 10.1016/j.jacc.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schlaeger TM, et al. Uniform vascular-endothelial-cell-specific gene expression in both embryonic and adult transgenic mice. Proc Natl Acad Sci U S A. 1997;94(7):3058–3063. doi: 10.1073/pnas.94.7.3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baker MA, Cerniglia GJ, Zaman A. Microtiter plate assay for the measurement of glutathione and glutathione disulfide in large numbers of biological samples. Anal Biochem. 1990;190(2):360–365. doi: 10.1016/0003-2697(90)90208-Q. [DOI] [PubMed] [Google Scholar]
- 45.Chun HJ, et al. Apelin signaling antagonizes Ang II effects in mouse models of atherosclerosis. J Clin Invest. 2008;118(10):3343–3354. doi: 10.1172/JCI34871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perez VA, et al. BMP promotes motility and represses growth of smooth muscle cells by activation of tandem Wnt pathways. J Cell Biol. 2011;192(1):171–188. doi: 10.1083/jcb.201008060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ali ZA, et al. CCR2-mediated antiinflammatory effects of endothelial tetrahydrobiopterin inhibit vascular injury-induced accelerated atherosclerosis. Circulation. 2008;118(14 suppl 1):S71–S77. doi: 10.1161/CIRCULATIONAHA.107.753558. [DOI] [PubMed] [Google Scholar]
- 48.Adachi T, et al. S-glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J Biol Chem. 2004;279(28):29857–29862. doi: 10.1074/jbc.M313320200. [DOI] [PubMed] [Google Scholar]
- 49.Grosdidier A, Zoete V, Michielin O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011;39(suppl 2):W270–W277. doi: 10.1093/nar/gkr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4(3):435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 51.Bernard B, Samudrala R. A generalized knowledge-based discriminatory function for biomolecular interactions. Proteins. 2009;76(1):115–128. doi: 10.1002/prot.22323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hall M, Frank E, Holmes G, Pfahringer B, Reutemann P, Witten IH. The WEKA data mining software: an update. SIGKDD Explor Newsl. 2009;11(1):10–18. doi: 10.1145/1656274.1656278. [DOI] [Google Scholar]
- 53.Mechanic LE, Luke BT, Goodman JE, Chanock SJ, Harris CC. Polymorphism Interaction Analysis (PIA): a method for investigating complex gene-gene interactions. BMC Bioinformatics. 2008;9:146. doi: 10.1186/1471-2105-9-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26(17):2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.