

Abstract
Human cytomegalovirus (HCMV) infection is the most common cause of congenital viral infections and a major source of morbidity and mortality after organ transplantation. NK cells are pivotal effector cells in the innate defense against CMV. Recently, hallmarks of adaptive responses, such as memory-like features, have been recognized in NK cells. HCMV infection elicits the expansion of an NK cell subset carrying an activating receptor heterodimer, comprising CD94 and NKG2C (CD94/NKG2C), a response that resembles the clonal expansion of adaptive immune cells. Here, we determined that expansion of this NKG2C+ subset and general NK cell recovery rely on signals derived from CD14+ monocytes. In a coculture system, a subset of CD14+ cells with inflammatory monocyte features produced IL-12 in response to HCMV-infected fibroblasts, and neutralization of IL-12 in this model substantially reduced CD25 upregulation and NKG2C+ subset expansion. Finally, blockade of CD94/NKG2C on NK cells or silencing of the cognate ligand HLA-E in infected fibroblasts greatly impaired expansion of NKG2C+ NK cells. Together, our results reveal that IL-12, CD14+ cells, and the CD94/NKG2C/HLA-E axis are critical for the expansion of NKG2C+ NK cells in response to HCMV infection. Moreover, strategies targeting the NKG2C+ NK cell subset have the potential to be exploited in NK cell–based intervention strategies against viral infections and cancer.
Introduction
NK cells are a critical part of the multilayered innate defense line against infectious agents and malignancies. Their control relies on the integration of multiple signals received via inhibitory receptors mainly binding to MHC class I molecules and activating receptors recognizing ligands primarily expressed on infected or transformed cells. Studies on NK cell deficiencies in humans highlight their pivotal role in the control of herpesvirus infections including human cytomegalovirus (HCMV), herpes simplex virus (HSV), vesicular stomatitis virus (VSV), and EBV (1–3). A recent case report revealed that NK cells were able to control HCMV infection even in the absence of T cells (4). Whereas infections usually remain asymptomatic in healthy persons, immunocompromised individuals, e.g., HIV-infected patients and organ transplant recipients, are at high risk of developing disease. Congenital HCMV infection occurs with an incidence of 0.2% and 2.5% depending on the country and socioeconomic status (5, 6), often causes permanent disabilities, and represents a serious disease with high costs to society.
HCMV dedicates a considerable number of genes to immune evasion from NK cell–mediated immune responses, e.g., by interfering with ligands for the activating NK cell receptors NKG2D, DNAM-1, and NKp30 (7). In addition, certain HCMV-encoded genes provide inhibitory signals that compensate for the downregulation of MHC class I, which would otherwise render infected cells susceptible to NK cell responses (7). While the molecular determinants for the direct recognition of HCMV-infected cells by NK cells are well studied, comparably little is known about the long-term consequences of interactions between NK cell (sub)populations and infected cells.
An initial report by Gumá et al. (8) described a skewing of the NK cell repertoire toward NK cells expressing the activating heterodimeric receptor CD94/NKG2C in HCMV seropositive individuals. Usually only around 10% of NK cells in peripheral blood carry this receptor, which binds to HLA-E, a nonclassical MHC class I molecule, whereas the remaining 90% express the inhibitory heterodimer CD94/NKG2A. In a follow-up study, the same group demonstrated that up to 50% of all NK cells expressed NKG2C after 10 days of in vitro exposure of peripheral blood leukocytes (PBLs) to HCMV-infected fibroblasts (9). This effect was not observed when UV-inactivated virus or an HCMV deletion mutant deficient for the gene region US2-11, which generates a high density of surface MHC class I molecules, was used (9).
Several longitudinal clinical studies described an increase of NKG2C+ NK cells after HCMV infection or reactivation. The NKG2C+ NK cell subset expressing the terminal differentiation marker CD57 was expanded during acute HCMV infection following solid organ transplantation (10), and similar results were obtained during episodes of HCMV reactivation after hematopoietic cell transplantation (11, 12) or after umbilical cord blood transplantation (13).
Functionally, NKG2C+ NK cells produce higher amounts of IFN-γ in response to K562 cells than NKG2C– cells from the same donor (11). In a follow-up study, NKG2C+ NK cells from CMV-seropositive donors expanded more during HCMV reactivation in the recipient than NKG2C+ NK cells from CMV-seronegative donors and also displayed stronger IFN-γ responses in vitro (12), suggesting the possible existence of a memory-like response of the NKG2C+ NK cells after secondary HCMV exposure.
Moreover, a recent report showed that NKG2C+ NK cells are highly potent effectors against HCMV-infected autologous macrophages in the presence of HCMV-specific antibodies that trigger cytotoxicity via CD16 (14).
While HCMV was the first pathogen that was shown to promote the expansion of the NKG2C+ NK cell subset, similar observations were made in the context of hantavirus (15) and chikungunyavirus infection (16) as well as in EBV, HBV, HCV (17–19), and HIV (20–22). Notably, in EBV, hepatitis, and HIV infection, expansion of NKG2C+ NK cells was only detected when the patients were also seropositive for HCMV, and Björkström and coworkers likewise speculate that previous CMV infection might have “primed” the NKG2C+ NK cells for a subsequent expansion during hantavirus infection in the majority of their patients (15).
The requirements underlying the expansion of human NKG2C+ NK cells in viral infection remain elusive (23). The aim of our study was to define the molecular mechanisms that drive the expansion of NKG2C+ NK cells in response to HCMV-infected cells.
Results
NKG2C+ NK cells expand in response to HCMV-infected fibroblasts.
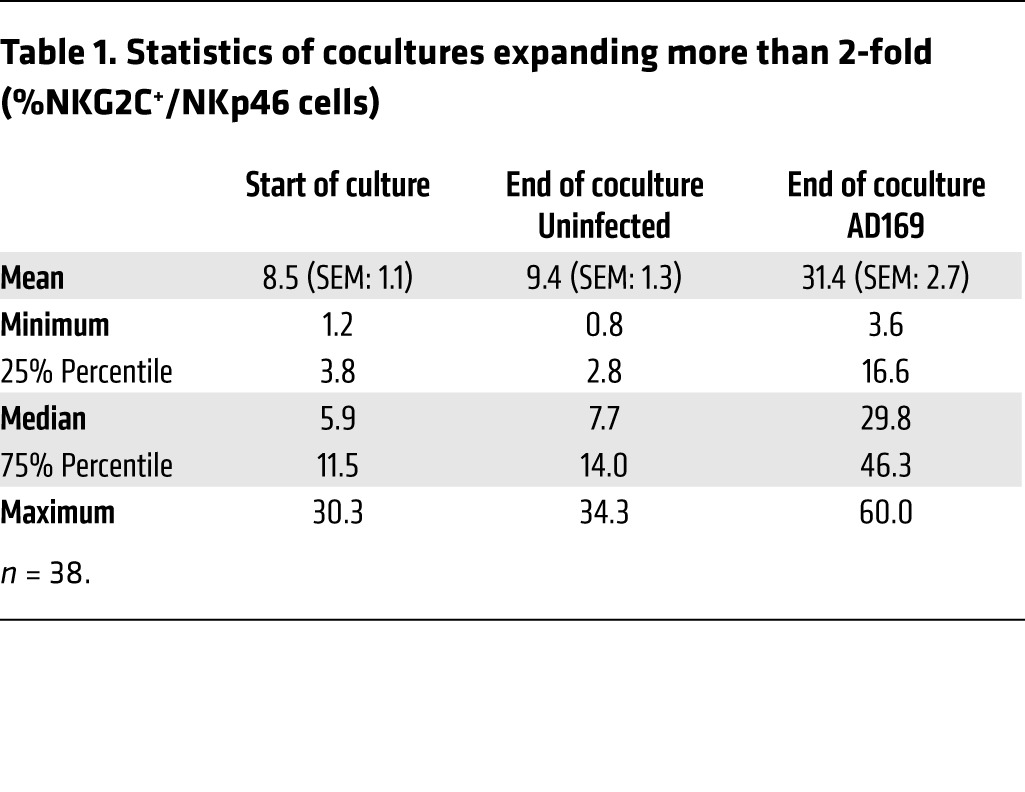
To dissect the molecular mechanisms underlying the expansion of NKG2C+ NK cells, we applied an in vitro coculture system as previously described (9). MRC-5 fibroblasts were infected with the HCMV laboratory strain AD169 or left uninfected (Figure 1A). After 6 to 8 hours, peripheral blood mononuclear cells (PBMCs) from healthy donors were added and cocultures were supplemented with low doses of IL-2 (20 U/ml). Flow cytometric analysis was performed between day 8 and 12 post infection (p.i.). In 71 cocultures, the percentage of NKG2C+ NK cells among all NK cells (NKp46+CD3–) increased significantly, from 14.1% (± 1.7%) at day 0 to 24.8% (± 1.9%), in infected cultures after 8 to 12 days (Supplemental Table 1; supplemental material available online with this article; doi:10.1172/JCI77440DS1). The individual cocultures, however, displayed a remarkably high donor-to-donor variability in terms of percentage and absolute number of NKG2C+ NK cells (Figure 1, B and C). We defined a 2-fold increase in the percentage of NKG2C+ NK cells in infected compared with uninfected cocultures as the criterion for inclusion in our analysis, and this criterion was fulfilled by 38 cocultures (Figure 1, B and C, and Table 1). In these cultures, NKG2C+ NK cells increased significantly, from 8.5% (± 1.1%) to 31.4% (± 2.7%), during infection (Figure 1, B and C). We did not detect a significant correlation between the expansion of NKG2C+ NK cells and age, sex, or CMV-serostatus of the donor (Supplemental Figure 1, A–C). When PBMCs were plated in the absence of MRC-5 fibroblasts or in uninfected cocultures, only minor changes in the percentage of NKG2C+ NK cells were observed over time (Figure 1D and Table 1). CFSE labeling of PBMCs revealed that, in infected cocultures, almost the entire NKG2C+ NK cell subset proliferated, whereas approximately 55% of the NKG2C– cells had diluted the dye (Figure 1D). This preferential proliferation of NKG2C+ cells was not observed in uninfected cocultures. In concordance with the literature (10, 11), most of the NKG2C+ NK cells in infected cocultures expressed CD57 (Supplemental Figure 2), a terminal differentiation marker (24, 25). In PBMC cultures without fibroblasts, no proliferation of NKG2C+ NK cells was detectable and marginal proliferation occurred among the NKG2C– NK cells.
Figure 1. NKG2C+ NK cells expand in response to HCMV-infected fibroblasts.

(A) MRC-5 fibroblasts were infected with HCMV strain AD169 at an MOI of 10 or left untreated. PBMCs were added 6 to 8 hours p.i. to uninfected or infected fibroblasts and cocultured for 8 to 12 days in the presence of 20 U IL-2/ml. (B) Graph depicts the percentage of NKG2C+ cells among all NK cells (NKp46+CD3–) at the indicated time points. Wilcoxon matched pairs signed-rank test, ****P ≤ 0.0001; n = 38; error bars indicate ± SEM. (C) Graph depicts the normalized absolute cell numbers of NKG2C+NKp46+ cells at the indicated time points. Wilcoxon matched pairs signed-rank test, **P = 0.0017, ****P ≤ 0.0001; n = 20; error bars indicate ± SEM. (D) Representative example of NK cell phenotype at day 10 p.i. CFSE-labeled PBMCs cultured alone, with uninfected, or with infected MRC-5 fibroblasts were stained for NKp46, NKG2C, and CD3 at day 10 and analyzed by flow cytometry. Dot plots were gated on live CD3– cells. Numbers indicate the percentage of NKG2C+ cells among all NKp46+ cells (upper panels) or the percentage of NKG2C+CFSE– cells among all NKp46+ cells (lower panels).
Table 1.
Statistics of cocultures expanding more than 2-fold (%NKG2C+/NKp46 cells)
Soluble factors from PBMCs are involved in the expansion of NKG2C+ NK cells in response to HCMV-infected fibroblasts.
To reduce the complexity of the cocultures, we set up experiments using purified NK cells instead of PBMCs. However, consistent with previously reported results (9), at the low IL-2 doses added in our experimental system, viable NK cells were recovered after the 10-day culture period only in cocultures with uninfected fibroblasts but not when cultivated with HCMV-infected fibroblasts, which disintegrated after 48 and 72 hours of coculture (Figure 2, B and C, and data not shown). This observation prompted us to investigate whether soluble factors secreted by PBMC mediated NK cell recovery and NKG2C+ subset expansion in HCMV-infected cocultures or whether cell-to-cell contact between NK cells and accessory cells was required. Using Transwell plates, which permit free passage of viral particles and cytokines, we demonstrated that PBMCs in the upper chamber were necessary and sufficient to ensure NK cell recovery and thus permit the expansion of the NKG2C+ subset in cultures containing purified NK cells and infected MRC-5 fibroblasts in the lower chamber (Figure 2, A–C).
Figure 2. Soluble factors from PBMCs are involved in the expansion of NKG2C+ NK cells in response to HCMV-infected fibroblasts.

(A) Transwell plates were used with purified NK cells on infected fibroblasts in the lower and autologous PBMCs in the upper chamber. (B) PBMCs or purified NK cells were cultured alone or with uninfected or AD169-infected MRC-5 fibroblasts. Transwell plates were used when indicated. At the end of the coculture, cells were stained for NKp46, NKG2C, and CD3 and analyzed by flow cytometry. Dot plots were gated on live CD3– cells. Numbers indicate the percentage of NKG2C+ cells among all NKp46+ cells (1 representative donor out of 3 is shown). (C) Summary of 3 independent experiments. Depicted are the absolute numbers of NKG2C+NKp46+ cells per well at the end of the coculture. Paired t test, P = 0.048, n = 3. Error bars indicate ± SEM.
IL-12 neutralization reduces NKG2C+ subset expansion in HCMV-infected cocultures.
In order to identify the PBMC-derived soluble factors that drive expansion of the NKG2C+ subset, we monitored the expansion of the NKG2C+ NK cells in infected cocultures in the presence or absence of neutralizing antibodies directed against cytokines implicated in NK cell proliferation/activation. When neutralizing IL-12, the fold increase of the percentage of NKG2C+ cells among NK cells in infected versus uninfected cocultures dropped significantly, from 11.7% (± 2.3%) to 6.0% (± 1.3%) (P = 0.0463) (Figure 3, A and B). In contrast, neutralization of IL-15 (Figure 3C) or IL-18 (Figure 3D) or blockade of the IL-15 receptor (data not shown) did not have a significant impact on the expansion of NKG2C+ NK cells in response to HCMV-infected fibroblasts. To corroborate our finding, we determined the concentration of biologically active IL-12p70 in coculture supernatant at 24, 48, and 72 hours p.i. (Figure 3E). Increasing levels of IL-12p70 were detected over time, with the highest levels occurring after 72 hours. HCMV infection of MRC-5 fibroblasts induced production of type I IFN starting after 4 to 5 hours p.i. (data not shown). However, blockade of the type I IFN receptor using a blocking anti–IFN-α/βR mAb did not affect the expansion of the NKG2C+ NK cell subset (data not shown). Taken together, our data identify IL-12 as an important cytokine contributing to NKG2C+ subset expansion in response to HCMV.
Figure 3. IL-12 neutralization reduces NKG2C+ subset expansion in HCMV-infected cocultures.

(A) PBMCs were cultured with uninfected or AD169-infected MRC-5 fibroblasts in the presence of neutralizing mAb against IL-12 or isotype control. At the end of the coculture, cells were stained for NKp46, NKG2C, and CD3 and analyzed by flow cytometry. Dot plots were gated on live CD3– cells. Numbers indicate the percentage of NKG2C+ cells among all NKp46+ cells (1 representative donor out of 10 is shown). (B–D) Summary of cocultures with neutralizing mAbs against IL-12, IL-15, and IL-18. Depicted is the fold increase of percentage of NKG2C+ NKp46+CD3– cells in infected versus uninfected cocultures. Wilcoxon matched pairs signed-rank test (B) IL-12 neutralization. **P = 0.0039; n = 10. (C) IL-15 neutralization (n = 5) or (D) IL-18 neutralization (n = 5). (E) ELISA for IL-12p70 with coculture supernatant at the indicated time points. n = 5–7. Error bars indicate ± SEM (B–E).
Upregulation of CD25 on NK cells in HCMV-infected cocultures is partially IL-12 dependent.
IL-12 has previously been described as upregulating CD25 on NK cells (26–28). CD25 represents the α chain of the high-affinity receptor for IL-2 and could therefore contribute to NK cell expansion in our coculture system containing low amounts of IL-2. When we assessed the expression of CD25 on CD56+CD3– NK cells, we observed a marked increase of CD25+ NK cells, from 12.76% (± 1.49%) in uninfected to 50.63% (± 8.84%) in infected cocultures. Neutralization of IL-12 had no significant impact on the percentage of CD25+ NK cells in uninfected cocultures, but resulted in a highly significant reduction to 37.78% (± 7.89%) in infected cocultures (Figure 4, A and B). This effect was also reflected by the mean fluorescence intensity of CD25 expression on NK cells (Figure 4C).
Figure 4. Upregulation of CD25 on NK cells in infected cocultures is partially IL-12 dependent.

PBMCs were cultured with uninfected or AD169-infected MRC-5 fibroblasts in the presence of neutralizing mAb against IL-12 or isotype control. Cells were stained for CD56, CD25, CD19, CD14, and CD3 and analyzed by flow cytometry at day 7. Histograms were gated on live CD56+CD3–CD19–CD14– cells. (A) Numbers indicate the percentage of CD25+ cells among all CD56+ cells (1 representative donor out of 7 at day 7 p.i.). (B) Summary of cocultures with neutralizing mAbs against IL-12 at day 7 p.i. Depicted are the percentages of CD56+CD25+ cells. **P = 0.0044; ***P = 0.0004. (C) Geometric MFI of CD25+ on CD56+ cells in uninfected or AD169-infected cocultures at day 7 p.i. **P = 0.0016; ***P = 0.0001. Two-tailed paired t test; n = 7; error bars indicate ± SEM (B and C).
Impact of depletion of CD14+ cells on NKG2C+ subset expansion in HCMV-infected cocultures.
In a next step, we attempted to identify the cellular source of IL-12 in our cocultures by intracellular staining for IL-12. Remarkably, within all PBMCs, a subset of CD14+ cells (around 4%) represented the only population producing IL-12 p.i. (Figure 5A). The IL-12–producing CD14+ cells expressed CD120b, high levels of HLA-DR, and low levels of CD38, which is consistent with the phenotypic definition of nonclassical, “inflammatory” monocytes (Figure 5B and refs. 29, 30). The depletion of CD14+ cells from PBMCs before the initiation of cocultures moderately, but not significantly, reduced the percentage of NKG2C+ cells among all NK cells in infected versus uninfected cultures (Figure 5, C and D). However, there was a significant difference between cocultures of infected fibroblasts with PBMCs and cocultures with PBMCs depleted of CD14+ cells in terms of absolute numbers of NKG2C+ NK cells. Depletion of CD14+ cells substantially reduced the fold increase of absolute cell numbers of NKG2C+ NK cells in infected cocultures from 26.1 (± 14.7) to 4.7 (± 2.2) (Figure 5, C and E). In contrast, depletion of T cells did not have a substantial impact on the expansion of NKG2C+ NK cells (Supplemental Figure 3, A and B), and the absence of exogenously added IL-2 to our cocultures did not affect the expansion of the NKG2C+ NK cell subset significantly (Supplemental Figure 3C), while a neutralizing antibody, blocking both exogenous and endogenous IL-2 strongly impaired overall NK cell survival, thereby precluding subset expansion (data not shown).
Figure 5. Impact of depletion of CD14+ cells on NKG2C+ subset expansion in HCMV-infected cocultures.

(A) PBMCs were cultured with uninfected or AD169-infected MRC-5 fibroblasts. Cells were stained for cell-surface–expressed CD14 and intracellular IL-12 at 36 hours p.i. Displayed are cells within the monocyte gate. Numbers indicate the percentages of IL-12–producing monocytes (1 representative donor out of 6 is shown). (B) Monocytes were stained for HLA-DR, CD38, CD120b (black line), or the respective isotype controls (shaded). Histograms are gated on IL-12+CD14+ cells (1 representative donor out of 3 is shown). (C) PBMCs or PBMCs depleted of CD14+ cells (dCD14) were cocultured with uninfected or infected fibroblasts. At the end of the coculture, cells were stained for NKp46, NKG2C, and CD3 and analyzed by flow cytometry. Dot plots were gated on live CD3– cells. Numbers indicate the percentages of NKG2C+ cells among all NKp46+CD3–cells (1 representative donor out of 10 is shown). (D and E) Summary of cocultures of PBMC or PBMC with CD14 depletion (dCD14) is shown. Depicted are the fold increases of percentage (D) and absolute numbers (E) of NKG2C+ among NKp46+ cells in infected versus uninfected cocultures. Wilcoxon matched pairs signed-rank test: **P = 0.002; n = 10; error bars indicate ± SEM.
Addition of CD14+ cells increases NK cell recovery and permits NKG2C+ subset expansion in purified NK cultures.
We then asked whether CD14+ cells were sufficient to restore NKG2C+ subset expansion in cocultures of purified NK cells with infected MRC-5 fibroblasts. To this end, we set up cocultures with NK cells and purified CD14+ cells from the same donors at different ratios. We observed a restoration of NKG2C+ subset expansion in response to HCMV-infected fibroblasts in purified NK cell cultures when CD14+ cells were added (Figure 6A). This restoration was reflected in the absolute cell numbers of both types, NK cells (Figure 6B) as well as the NKG2C+ NK cell subset (Figure 6C). Thus, our data indicate that CD14+ cells are indispensable for overall NK cell recovery and contribute to NKG2C+ subset expansion.
Figure 6. Addition of CD14+ cells increases NK cell recovery and permits NKG2C+ subset expansion in purified NK cultures.

(A) PBMCs or purified NK cells were cocultured with AD169-infected fibroblasts. CD14+ cells from the same donor were added to NK cells in infected cocultures at a ratio of 1:1. At the end of the coculture, cells were stained for NKp46, NKG2C, and CD3 and analyzed by flow cytometry. Dot plots were gated on live CD3– cells (1 representative out of 5 donors is shown). (B and C) Graphs depict the absolute number of all NKp46+CD3– cells (B) or NKG2C+NKp46+CD3– (C) cells per 24 wells (Mann-Whitney test: *P = 0.0317; **P = 0.0079; n = 5; error bars indicate ± SEM).
Blockade of CD94 or NKG2C reduces expansion of NKG2C+ NK cells in response to HCMV-infected fibroblasts.
As described previously (2), HCMV infection of fibroblasts leads to the upregulation of HLA-E, the only known cellular ligand of the CD94/NKG2C heterodimer. In our experimental system, we also detected an upregulation of HLA-E on MRC-5 fibroblasts in response to infection starting after 6 to 8 hours, with a maximum after 24 hours (Figure 7A). Expression levels were maintained until the fibroblasts disintegrated in infected cocultures (data not shown).
Figure 7. Blockade of CD94 or NKG2C reduces expansion of NKG2C+ NK cells in response to HCMV-infected fibroblasts.

(A) Fibroblasts were stained with anti–HLA-E mAb or isotype control at the indicated time points before and after infection. (B) PBMCs were cultured with uninfected or AD169-infected fibroblasts in the presence of anti-CD94 F(ab)2 fragments or anti-NKG2C mAb or respective isotype controls. At the end of the coculture, cells were stained for NKp46, NKG2C, and CD3 and analyzed by flow cytometry. Dot plots were gated on live CD3– cells (1 representative donor out of 4 is depicted). Numbers indicate the percentages of NKG2C+ cells among all NKp46+ cells. (C) Summary of cocultures with anti-CD94 F(ab)2 fragments or (D) anti-NKG2C mAbs (paired t test: *P = 0.0314, n = 3 in C; *P = 0.0311, n = 4 in D; error bars indicate ± SEM) is shown. Plotted is the fold increase of percentage of NKG2C+NKp46+CD3– cells in infected versus uninfected cocultures.
We then tested the effect of blocking CD94 or NKG2C at the beginning of the cocultures on expansion of the NKG2C+ subset (Figure 7B). Blocking either with an anti-CD94 F(ab)2 fragment or with a mAb directed against NKG2C greatly reduced subset expansion from 3.82-fold (± 0.69) to 1.88-fold (± 0.36) and from 4.21-fold (± 1.04) to 0.20-fold (± 0.08), respectively (Figure 7, C and D). To assess whether the blocking mAb directed against NKG2C would interfere with anti-NKG2C mAb used for staining, we added secondary mAb by itself and failed to detect any residual bound blocking anti-NKG2C mAb after 10 days of coculture (data not shown).
shRNA-mediated knockdown of HLA-E on infected cells critically impairs NKG2C+ subset expansion.
We then wanted to assess whether expression levels of HLA-E, the ligand for CD94/NKG2C, would affect subset expansion. The HCMV open-reading frame UL40 has been described as upregulating HLA-E expression in a variety of experimental systems (31, 32). We hypothesized that infection using a mutant strain of HCMV, deficient for UL40, would fail to upregulate HLA-E. However, we did not detect any differences in HLA-E levels on MRC-5 fibroblasts, depending on whether they were infected with wild-type AD169 virus or its UL40-deletion mutant at different multiplicities of infection (Supplemental Figure 4), indicating that in our experimental system, UL40 does not modulate HLA-E levels on infected MRC-5 cells. To investigate whether shRNA-mediated knockdown of HLA-E expression would impair expansion of NKG2C+ NK cells, we generated MRC-5 fibroblasts transduced with vectors containing 2 different shRNAs directed against HLA-E or with an empty vector control. Upon infection, upregulation of HLA-E was strongly impaired in MRC-5 fibroblasts expressing shRNAs directed against HLA-E, but not in fibroblasts carrying the vector control (Figure 8A). Most importantly, the fibroblasts transduced with shRNAs directed against HLA-E failed to drive expansion of NKG2C+ cells after infection (Figure 8, B and C). shRNA-mediated knockdown of HLA-E in uninfected fibroblasts expressing very low levels of HLA-E did not affect the percentage of NKG2C+ NK cells in uninfected cocultures (Figure 8, B and C). In conclusion, our data show that HLA-E upregulation upon infection is indispensable for NKG2C+ subset expansion.
Figure 8. shRNA-mediated knockdown of HLA-E on infected cells critically impairs NKG2C+ subset expansion.

(A) MRC-5 fibroblasts were transduced with 2 different shRNAs (no. 1, no. 2) directed against HLA-E or with an empty vector (VC). Transduced fibroblasts were infected with AD169 at MOI 10 and stained with anti–HLA-E mAb (black line) or isotype control (shaded) at 24 hours p.i. (B) PBMCs were cultured with uninfected or AD169-infected MRC-5 fibroblasts that were either transduced with an empty vector or with a vector encoding shRNAs directed against HLA-E (no. 1, no. 2). At the end of the coculture, cells were stained for NKp46, NKG2C, and CD3 and analyzed by flow cytometry. Graphs are gated on CD3– cells. Numbers indicate the percentage of NKG2C+ cells among all NKp46+ cells (1 representative donor out of 6 donors is shown). (C) Summary of cocultures with fibroblasts transduced with vector control or with shRNA against HLA-E is shown. Wilcoxon matched pairs signed-rank test: *P = 0.031 (no. 1) and *P = 0.031 (no. 2); n = 6; error bars indicate ± SEM.
Discussion
During viral infections, such as HCMV, a remarkable expansion of an NKG2C+ subset of NK cells has been described in numerous studies (8–11, 15–22). The molecular mechanisms driving this expansion, however, have remained largely elusive. Here, we demonstrate that the expansion of NKG2C+ NK cells in HCMV infection relies on crosstalk between NK cells and CD14+ monocytes, involves IL-12, and is dependent on the interaction of the activating CD94/NKG2C receptor heterodimer on NK cells with upregulated HLA-E on infected cells.
We observed that shRNA-mediated knockdown of HLA-E in infected fibroblasts or blockade of NKG2C largely abrogated NKG2C+ subset expansion in our experiments. The impact of IL-12 neutralization, however, was less pronounced, possibly indicating a redundancy with other proinflammatory cytokines. A preferential expansion of NKG2C+ cells with exogenously added IL-15 has been demonstrated in cocultures of purified NK cells and HCMV-infected fibroblasts (9) or HLA-E–transfected K562 cells (15). However, in our cocultures of PBMCs and infected MRC-5 fibroblasts, we did not observe a substantial effect by neutralizing IL-15 or IL-18 or by blocking the type I IFN receptor or the IL-15 receptor (data not shown), indicating that these cytokines play a subordinate role in NKG2C+ subset expansion in our system.
DCs and monocytes are the major source of IL-12 in a broad range of infectious conditions, including HCMV infection (33). It is well established that, among other innate receptors, TLR2, TLR4, and CD14 recognize HCMV virions (34, 35), leading to downstream activation of NF-κB. Recognition of HCMV by monocytes has also been shown to occur via an alternative pathway involving scavenger receptor A1, endosomal TLR-3, and the adaptor protein TRIF, resulting in IL-12 and TNF-α transcription (36). In our cocultures, a small subset of CD14+ cells was the only cell population within PBMCs displaying detectable IL-12 production in an early time window of 24 to 36 hours after infection of cocultured MRC-5 cells. It is tempting to speculate that this monocytic population belongs to the so-called “nonclassical” or “inflammatory” monocytes characterized by low levels of CD14, as observed in our experiments. However, we did not detect CD16 expression — considered to represent an “inflammatory” monocyte marker — on IL-12+CD14+ cells. A possibility is that CD16 was lost due to cellular activation via TLRs, as previously shown (29). We therefore defined the IL-12–producing subpopulation as HLA-DR+, CD38lo, and CD120b+, which is consistent with the phenotype of inflammatory monocytes (29, 30). Notably, the production of IL-12 by CD16+CD14+ monocytes has also been described in other conditions (37), e.g., multiple sclerosis (38) and Trypanosoma cruzi infection (39), but so far — to our knowledge — was not observed during HCMV infection or linked to the expansion of NKG2C+ NK cells. Polymorphisms in innate cytokine signaling, e.g., in TLR2, in the IL-12 promoter, or in the IL-12 receptor, have been shown to affect immune responses in different infections including HCMV (40–43). It is possible that these polymorphisms partially account for the remarkable donor variability in our study.
Depletion of CD14+ cells in infected cocultures not only reduced the numbers of NKG2C+ NK cells, but also greatly diminished overall NK cell recovery, at least at the low IL-2 amounts added in our system. Of note, IL-12 secretion in response to MCMV infection has been linked to the induction of CD25 and the consecutive formation of the high-affinity IL-2 receptor on NK cells (44), resulting in increased sensitivity to IL-2. Consistent with this study, we also observed a marked increase of CD25 expression on NK cells in HCMV-infected cocultures, and this increase was partially IL-12 dependent. In our experimental set-up, we envisaged a scenario in which IL-12 — secreted by CD14+ monocytes in response to infection — contributes to an upregulation of CD25 on NK cells. The presence of the high-affinity receptor for IL-2 then permits and facilitates the proliferation of all CD25+ NK cells. Accordingly, neutralization of IL-2 severely impaired overall NK cell survival, precluding NKG2C subset expansion. The specific expansion of the NKG2C+ NK cell subset is triggered if sufficient additional signals, such as the critical engagement of CD94/NKG2C by HLA-E that is upregulated on infected cells, are provided. The discrepancy between the impact of IL-12 neutralization that specifically impairs expansion of NKG2C+ NK cells and of CD14 depletion that reduces numbers of both NKG2C+ and NKG2C– NK cells implies that CD14+ cells engage in a more complex crosstalk with NK cells in HCMV-infected cocultures and provide more signals than only IL-12 (45, 46). Our Transwell experiments indicated that cell-to-cell contact was not primarily required for the NK cell/monocyte crosstalk. Thus, we hypothesize that additional soluble factors might contribute to the observed NKG2C+ subset expansion.
The variability of NK cell responses from different donors in our study was substantial. Only NK cells from 38 out of 71 donors responded with a robust expansion (≥ 2-fold expansion of the NKG2C+ NK cell subset in infected vs. uninfected cocultures) of the NKG2C+ subset to HCMV-infected fibroblasts. Although most of our donors were CMV-seropositive, we have neither information about the frequency of previous exposures to HCMV (or other viruses) nor about the strains that the individual NK cell compartments encountered. Clinical isolates of HCMV display considerable polymorphism in the UL40 gene, yielding different HLA-E–binding peptides that can affect the interaction between HLA-E and CD94/NKG2 receptors (47), potentially affecting subset expansion. Thus, the observed variability might at least partially result from different previous infection records of the donors.
Another explanation for the high degree of donor variability is the distribution of differentially expressed inhibitory receptors of the Killer-cell immunoglobulin-like receptor (KIR) family on human NK cells. KIRs are not only highly polymorphic, but they are also stochastically expressed in every individual. In cocultures with MRC-5 fibroblasts (expressing HLA-Cw, recognized, e.g., by KIR2DL3/2), different KIR combinations could result in different levels of NK cell inhibition affecting subset expansion. Clinical studies have described a predominant expression of KIR2DL receptors or, more specifically, of KIR2DL3/2 among expanded NKG2C+ NK cells (11), and a similar skewing was also observed in chikungunyavirus, hantavirus, and HBV infection (15, 16, 19). Another study highlighted the lack of KIR3DL1 expression on NKG2C+ NK cells in individuals who carry the cognate HLA-Bw4 ligand (10). More recently, a role for activating KIRs in the expansion of NKG2C+ NK cells emerged as well (48, 49). However, in preliminary experiments, we could not detect a bias toward KIR2DL3/2, KIR2DL1, or KIR3DL1 on the NKG2C+ NK cell subset on day 10 after coculture.
The HCMV protein UL18 has been described as a low-affinity ligand for CD94/NKG2C (50). However, using a viral mutant deficient for UL18, Gumá et al. demonstrated that expansion of NKG2C+ NK cells did not depend on this viral gene product (9). Instead, the broad range of structurally very diverse viruses that were reported to trigger the expansion of NKG2C+ NK cells seems to favor induced- or altered-self structures rather than pathogen-derived antigens as driving factors for subset expansion. This is consistent with our data that demonstrate that HLA-E expression on infected cells and IL-12 induction are key elements in promoting the expansion of NKG2C+ NK cells.
In general, surface expression of HLA-E depends on the availability of peptides derived from classical MHC class I leader sequences. HCMV upregulates HLA-E by means of the viral gene product UL40, which encodes a stretch that is identical to the leader sequences of most HLA-C allotypes, thereby ensuring HLA-E expression or even upregulation in spite of MHC class I downmodulation during infection (31, 32, 51). When comparing a wild-type AD169 strain of HCMV with a strain deficient for UL40 (BAC2-ΔUL40), we did, to our surprise, not detect any differences in terms of HLA-E expression on the infected fibroblasts (Supplemental Figure 4) and — consistent with that finding — detected no impact of the UL40 deletion on NKG2C+ subset expansion (data not shown). Another possibility is that peptides other than those derived from MHC class I or UL40 are presented on HLA-E during infection and that they in turn represent the decisive triggering structure for NKG2C+ subset expansion rather than HLA-E expression levels per se. In fact, it has been shown that NK cells can be activated by subtle peptide differences (52–54).
In recent years, adaptive features of NK cells, such as clonal expansion and long-lasting functional alterations, became increasingly appreciated (23). In mice, these similarities extend to NK cell–mediated memory that was demonstrated initially for haptens and later for influenza, VSV, HIV, and MCMV (55–57). These findings sparked renewed interest in the expansion of the human NKG2C+ NK cell subset in HCMV infection. Our results strongly support a functional role for NKG2C in the process leading to the expansion of NKG2C+ NK cells, since blockade of the receptor subunits on NK cells as well as knockdown of the cellular ligand HLA-E on infected cells reduced NKG2C subset expansion. Of note, in our experiments, blockade of NKG2C had a more pronounced effect than blockade of CD94. While this observation may simply reflect different efficiencies of the blocking antibodies, it could also be related to structural aspects of the CD94/NKG2C–HLA-E interaction. A critical involvement of NKG2C in the antiviral response is also supported by a report that investigated a cohort of HIV patients carrying a deletion in the KLRC2 gene, which encodes the NKG2C protein. The patients lacking NKG2C suffered from an increased risk of contracting HIV, a more rapid disease progression, and higher viral titers prior to initiation of treatment (58). A more recent study describes an influence of NKG2C zygosity on surface receptor levels and NKG2C+ NK cell numbers in HCMV-seropositive individuals, implying that the receptor plays an active role in shaping the NK cell compartment by HCMV infection (59).
In the well-characterized infection model of MCMV, the expansion of an NK cell subset carrying the activating receptor Ly49H was described and direct interaction of Ly49H with the viral ligand m157 was mandatory for the expansion, leading to the formation of m157-specific NK cell memory (55). Moreover, intact proinflammatory signaling via the IL-12 receptor and STAT4 was indispensable for the expansion of Ly49H+ NK cells (60). The requirements for the NK cell response to MCMV bears remarkable conceptual resemblance to the mechanisms underlying the expansion of human NKG2C+ NK cells in HCMV infection uncovered by our study: in both model systems, the binding of a DAP12-coupled activating receptor — Ly49H in MCMV, CD94/NKG2C in HCMV — to a cell-surface molecule with MHC class I–like fold — m157 in MCMV, HLA-E in HCMV — was necessary for NK cell subset expansion in response to viral infection; in both experimental systems, IL-12 acted as a key cytokine.
In our study, we defined molecular mechanisms that govern the expansion of NKG2C+ NK cells during viral infection: we show that (a) the expansion of NKG2C+ NK cells in response to HCMV-infected fibroblasts is partially driven by IL-12, (b) IL-12 is produced by CD14+ monocytes in response to infection, (c) IL-12 contributes to a marked increase of CD25 expression on NK cells, (d) the presence of CD14+ monocytes is indispensable for NK cell survival, thus enabling subset expansion in purified NK cell cultures at low IL-2 levels, and (e) the CD94/NKG2C-HLA-E axis is functionally involved and indispensable for the expansion of the NKG2C+ NK cell subset.
It remains to be investigated whether NKG2C+ NK cells are also endowed with generally improved functions or whether they even possess a degree of specificity for yet-unknown pathogenic structures. A more comprehensive understanding of NK cell subset biology and the mechanisms leading to the formation of NK cell subsets might ultimately inspire a new generation of adoptive NK cell therapies.
Methods
Cell preparation.
PBMCs were isolated from buffy coats, using LSM 1077 Lymphocyte Separation Medium (PAA). NK cells were isolated from PBMCs by negative selection using the human NK Cell Isolation Kit (Miltenyi Biotec). CD14+ cells and PBMCs depleted of CD14+ cells (dCD14) were obtained by magnetic activated cell sorting using CD14 MicroBeads (Miltenyi Biotec).
Cell culture.
MRC-5 human fetal lung fibroblasts were purchased from ATCC and cultured in DMEM (Sigma-Aldrich) completed with 10% FCS (Life Technologies) and 1% penicillin/streptomycin (Sigma-Aldrich).
Viruses.
The HCMV wild-type strain AD169 was propagated using MRC-5 cells (ATCC CCL171). The UL40 deletion mutant (BAC2-ΔUL40) was reconstituted from its respective BAC clone, BAC2. For preparation of purified HCMV stocks, cells were infected with a low MOI (ca. 0.05), and supernatants were harvested when the cells showed a strong cytopathic effect. After disposal of cell debris by centrifugation, supernatants were centrifuged at 20,000 g for 3 hours at 10°C. Pellets were resuspended in PBS, dounced, and centrifuged through a 20% sorbitol cushion at 65,000 g for 1 hour at 10°C. The virus pellet was homogenized in PBS and stored at –80°C. Virus titers were determined by standard plaque assay.
Cocultures.
MRC-5 fibroblasts were seeded at a density of 1 × 105 cells per well in a 24-well plate. Twenty-four hours later, fibroblasts were either infected with HCMV strain AD169 at an MOI of 10 or left uninfected. After 6 to 8 hours p.i., virus-containing medium was removed and 2 × 106 freshly isolated PBMCs, purified NK cells, or purified NK cells in the presence of autologous CD14+ cells in complete DMEM containing 20 U/ml recombinant human IL-2 (NIH) was added to infected or uninfected fibroblasts or cultured alone. Medium was exchanged every 2 to 3 days.
For Transwell experiments, 6.5-mm Transwell plates with 0.4-μm Pore Polyester Membrane Inserts (Corning Life Sciences) were used. After 8 to 12 days, cells were harvested and analyzed.
Antibodies and flow cytometry.
PBMCs were harvested and fixed using the Permeabilization/Fixation Kit (eBioscience) according to the manufacturer’s instructions. Fixed cells were stained with α–NKG2C-PE (clone 134591, R&D Systems), α–NKp46–Alexa Fluor 647 (clone 9E2), α–CD25-APC (clone BC96), α–CD56-PE-Cy7 (clone HCD56), α–CD14-APC-Cy7 (clone HCD14), α–CD19-APC-Cy7 (clone HIB19), α–CD3-APC-Cy7 (clone HIT3a), α–CD57-Pacific Blue (clone HCD57, all BioLegend), or the respective isotype controls. In addition, cells were stained with a fixable live/dead cell stain (Life Technologies) according to the manufacturer’s protocol. For CFSE labeling, 1 × 107 PBMCs were incubated for 10 minutes with 1 μM CFSE (Sigma-Aldrich) in PBS. For intracellular staining, cells were first stained with α–CD14-FITC (clone HCD14, BioLegend), then permeabilized with BD Cytofix/Cytoperm Fixation/Permeabilization Solution Kit (BD Biosciences) and subsequently stained with α–human IL12-APC (clone C11.5, recognizing IL-12p40, BD Biosciences — Pharmingen). HLA-E was detected using an α–HLA-E–PE antibody (clone 3D12, BioLegend).
For blocking experiments, 5 μg/ml α–IL-12 antibody or 1.5 μg/ml (Human IL-12 Affinity Purified Polyclonal Goat Ab, recognizing IL-12p35, p40 and p70, R&D Systems), 5 μg/ml α–IL-15 antibody (clone 34559, R&D Systems), 2.5 μg/ml α–IL-18 antibody (clone 125-2H, MBL International), 5 μg/ml α–IL-15R antibody (Purified Polyclonal Goat Ab, #AF247, R&D Systems), 10–20 μg/ml α–IL-2 antibody (clone MQ1-17H12, BioLegend), 4 μg/ml F(ab)2 fragments of α-CD94 antibody (clone HP-3B1, Beckman Coulter Inc.), 5 μg/ml α-NKG2C antibody (clone 134522, R&D Systems), 5 μg/ml α–IFN- α/βR (PBL Interferon Source), or the respective isotype controls were added in the beginning of the cocultures.
Stained cells were analyzed using FACSCanto II or LSRFortessa (both BD Biosciences). Data were analyzed with FlowJo version 9.6 (Tree Star Inc.). All NK cell phenotype analyses were gated on live, CD3– singlet lymphocytes unless stated otherwise.
ELISA.
Coculture supernatants were assessed for biologically active IL-12p70 at the indicated time points using a BioLegend ELISA Max Deluxe Kit and following the manufacturer’s instructions.
shRNA.
HLA-E knockdown in MRC-5 cells was achieved using 2 different oligonucleotides encoding for shRNAs synthesized by Sigma-Aldrich, as follows: HLA-E (no. 1) 5′-GATCCCCATTTGCTAGAGATGTGCTGCCCCCTTCAAGAGAGGGGGCAGCACATCTCTAGCAAATTTTTTGGAAC-3′; HLA-E (no. 2) 5′-GATCCCCATTAACCCATGAATGAAGGCCCCCTTCAAGAGAGGGGGCCTTCATTCATGGGTTAATTTTTTGGAAC-3′.
These oligonucleotides were annealed and ligated into a pSUPER.retro.neo+gfp expression vector (OligoEngine), and retroviral transduction was conducted according to standard protocols. In short, retrovirus was produced in Phoenix-AMPHO cells after CaPO4 transfection. After 2 days, retrovirus was harvested and filtered; MRC-5 cells were infected and stable transfectants selected using 250 μg/ml G418 (Sigma-Aldrich).
Statistics.
Statistical analysis was performed with Graphpad Prism 5.0/6.0 (Graphpad Prism Software) using the Wilcoxon signed-rank test for paired samples or the Mann-Whitney test for unpaired samples of nonnormal distribution, if not stated otherwise.
Study approval.
Buffy coats or fresh blood, collected according to the principles of the Declaration of Helsinki, were provided by Deutsches Rotes Kreuz DRK-Blutspendedienst Baden-Württemberg-Hessen gGmbH (Mannheim, Germany). Written informed consent was obtained from all human subjects prior to blood donation and ethical approval 87/04 was granted by the Ethik Kommission II of the Medical Faculty Mannheim (Mannheim, Germany).
Supplementary Material
Acknowledgments
We would like to thank Åsa Hidmark for performing bioassays for type I IFNs, Isabel Poschke for helpful discussions on advanced flow cytometry, Annette Kopp-Schneider for excellent advice on statistical analysis, and Nicole Sitzmann and Karin Janetzko for providing statistical data on our blood donors. The work was supported by a KI-DKFZ joint project grant by StratCan (Strategic Research Programme in Cancer) at Karolinska Institute and the German Cancer Center (to A. Cerwenka) and the Helmholtz Association through VISTRIE VH-VI-242 (to H. Hengel). A. Rölle was the recipient of a scholarship of the Fritz-Thyssen Foundation.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2014;124(12):5305–5316. doi:10.1172/JCI77440.
References
- 1.Biron CA, Byron KS, Sullivan JL. Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med. 1989;320(26):1731–1735. doi: 10.1056/NEJM198906293202605. [DOI] [PubMed] [Google Scholar]
- 2.Etzioni A, Eidenschenk C, Katz R, Beck R, Casanova JL, Pollack S. Fatal varicella associated with selective Natural Killer cell deficiency. J Pediatr. 2005;146(3):423–425. doi: 10.1016/j.jpeds.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 3.Eidenschenk C, et al. A novel primary immunodeficiency with specific natural-killer cell deficiency maps to the centromeric region of chromosome 8. Am J Hum Genet. 2006;78(4):721–727. doi: 10.1086/503269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuijpers TW, et al. Human NK cells can control CMV infection in the absence of T cells. Blood. 2008;112(3):914–915. doi: 10.1182/blood-2008-05-157354. [DOI] [PubMed] [Google Scholar]
- 5.Ludwig A, Hengel H. Epidemiological impact and disease burden of congenital cytomegalovirus infection in Europe. Euro Surveill. 2009;14(9):26–32. [PubMed] [Google Scholar]
- 6.Kenneson A, Cannon MJ. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev Med Virol. 2007;17(4):253–276. doi: 10.1002/rmv.535. [DOI] [PubMed] [Google Scholar]
- 7.Wilkinson GWG, et al. Modulation of natural killer cells by human cytomegalovirus. J Clin Virol. 2008;41(3):206–212. doi: 10.1016/j.jcv.2007.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gumá M, et al. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood. 2004;104(12):3664–3671. doi: 10.1182/blood-2004-05-2058. [DOI] [PubMed] [Google Scholar]
- 9.Gumá M, et al. Expansion of CD94/NKG2C+ NK cells in response to human cytomegalovirus-infected fibroblasts. Blood. 2006;107(9):3624–3631. doi: 10.1182/blood-2005-09-3682. [DOI] [PubMed] [Google Scholar]
- 10.Lopez-Verges S, et al. Expansion of a unique CD57+NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. Proc Natl Acad Sci U S A. 2011;108(36):14725–14732. doi: 10.1073/pnas.1110900108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foley B, et al. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood. 2011;119(11):2665–2674. doi: 10.1182/blood-2011-10-386995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foley B, et al. Human cytomegalovirus (CMV)-induced memory-like NKG2C(+) NK cells are transplantable and expand in vivo in response to recipient CMV antigen. J Immunol. 2012;189(10):5082–5088. doi: 10.4049/jimmunol.1201964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Della Chiesa M, et al. Phenotypic and functional heterogeneity of human NK cells developing after umbilical cord blood transplantation: a role for human cytomegalovirus? Blood. 2011;119(2):399–410. doi: 10.1182/blood-2011-08-372003. [DOI] [PubMed] [Google Scholar]
- 14.Wu Z, et al. Human cytomegalovirus-induced NKG2C(hi) CD57(hi) natural killer cells are effectors dependent on humoral antiviral immunity. J Virol. 2013;87(13):7717–7725. doi: 10.1128/JVI.01096-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Björkström NK, et al. Rapid expansion and long-term persistence of elevated NK cell numbers in humans infected with hantavirus. J Exp Med. 2011;208(1):13–21. doi: 10.1084/jem.20100762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petitdemange C, et al. Unconventional repertoire profile is imprinted during acute chikungunya infection for natural killer cells polarization toward cytotoxicity. PLoS Pathog. 2011;7(9):e1002268. doi: 10.1371/journal.ppat.1002268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saghafian-Hedengren S, et al. Epstein-Barr virus coinfection in children boosts cytomegalovirus-induced differentiation of natural killer cells. J Virol. 2013;87(24):13446–13455. doi: 10.1128/JVI.02382-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oliviero B, et al. Natural killer cell functional dichotomy in chronic hepatitis B and chronic hepatitis C virus infections. Gastroenterology. 2009;137(3):1151–1160. doi: 10.1053/j.gastro.2009.05.047. [DOI] [PubMed] [Google Scholar]
- 19.Béziat V, et al. CMV drives clonal expansion of NKG2C(+) NK cells expressing self-specific KIRs in chronic hepatitis patients. Eur J Immunol. 2011;42(2):447–457. doi: 10.1002/eji.201141826. [DOI] [PubMed] [Google Scholar]
- 20.Gumá M, et al. Human cytomegalovirus infection is associated with increased proportions of NK cells that express the CD94/NKG2C receptor in aviremic HIV-1-positive patients. J Infect Dis. 2006;194(1):38–41. doi: 10.1086/504719. [DOI] [PubMed] [Google Scholar]
- 21.Goodier MR, et al. NKG2C+ NK cells are enriched in AIDS patients with advanced-stage Kaposi’s sarcoma. J Virol. 2007;81(1):430–433. doi: 10.1128/JVI.01567-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brunetta E, et al. Chronic HIV-1 viremia reverses NKG2A/NKG2C ratio on natural killer cells in patients with human cytomegalovirus co-infection. AIDS. 2010;24(1):27–34. doi: 10.1097/QAD.0b013e3283328d1f. [DOI] [PubMed] [Google Scholar]
- 23.Rölle A, Pollmann J, Cerwenka A. Memory of infections: an emerging role for natural killer cells. PLoS Pathog. 2013;9(9):e1003548. doi: 10.1371/journal.ppat.1003548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Björkström NK, et al. Expression patterns of NKG2A, KIR, and CD57 define a process of CD56 dim NK-cell differentiation uncoupled from NK-cell education. Blood. 2010;116(19):3853–3864. doi: 10.1182/blood-2010-04-281675. [DOI] [PubMed] [Google Scholar]
- 25.Lopez-Verge S, et al. CD57 defines a functionally distinct population of mature NK cells in the human CD56dimCD16+ NK-cell subset. Blood. 2010;116(19):3865–3874. doi: 10.1182/blood-2010-04-282301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uharek L, et al. High lytic activity against human leukemia cells after activation of allogeneic NK cells by IL-12 and IL-2. Leukemia. 1996;10(11):1758–1764. [PubMed] [Google Scholar]
- 27.Leong JW, et al. Preactivation with IL-12, IL-15, and IL-18 induces CD25 and a functional high-affinity IL-2 receptor on human cytokine-induced memory-like natural killer cells. Biol Blood Marrow Transplant. 2014;20(4):463–473. doi: 10.1016/j.bbmt.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ni J, Miller M, Stojanovic A, Garbi N, Cerwenka A. Sustained effector function of IL-12/15/18-preactivated NK cells against established tumors. J Exp Med. 2012;209(13):2351–2365. doi: 10.1084/jem.20120944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Picozza M, Battistini L, Borsellino G. Mononuclear phagocytes and marker modulation: when CD16 disappears, CD38 takes the stage. Blood. 2013;122(3):456–7. doi: 10.1182/blood-2013-05-500058. [DOI] [PubMed] [Google Scholar]
- 30.Hijdra D, Vorselaars AD, Grutters JC, Claessen AM, Rijkers GT. Differential expression of TNFR1 (CD120a) and TNFR2 (CD120b) on subpopulations of human monocytes. J Inflamm (Lond). 2012;9(1):38. doi: 10.1186/1476-9255-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tomasec P, et al. Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science. 2000;287(5455):1031–1033. doi: 10.1126/science.287.5455.1031. [DOI] [PubMed] [Google Scholar]
- 32.Ulbrecht M, et al. Cutting edge: the human cytomegalovirus UL40 gene product contains a ligand for HLA-E and prevents NK cell-mediated lysis. J Immunol. 2000;164(10):5019–5022. doi: 10.4049/jimmunol.164.10.5019. [DOI] [PubMed] [Google Scholar]
- 33.Renneson J, et al. IL-12 and type I IFN response of neonatal myeloid DC to human CMV infection. Eur J Immunol. 2009;39(10):2789–2799. doi: 10.1002/eji.200939414. [DOI] [PubMed] [Google Scholar]
- 34.Compton T, et al. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J Virol. 2003;77(8):4588–4596. doi: 10.1128/JVI.77.8.4588-4596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yew K-H, Carpenter C, Duncan RS, Harrison CJ. Human cytomegalovirus induces TLR4 signaling components in monocytes altering TIRAP, TRAM and downstream interferon-β and TNF-α expression. PLoS One. 2012;7(9):e44500. doi: 10.1371/journal.pone.0044500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yew KH, Harrison CJ. Blockade of Lyn kinase upregulates both canonical and non-canonical TLR-3 pathways in THP-1 monocytes exposed to human cytomegalovirus. Acta Virol. 2011;55(3):243–253. doi: 10.4149/av_2011_03_243. [DOI] [PubMed] [Google Scholar]
- 37.Sciaraffia E, et al. Human monocytes respond to extracellular cAMP through A2A and A2B adenosine receptors. J Leukoc Biol. 2014;96(1):113–122. doi: 10.1189/jlb.3A0513-302RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chuluundorj D, Harding S a, Abernethy D, La Flamme AC. Expansion and preferential activation of the CD14(+)CD16(+) monocyte subset during multiple sclerosis. Immunol Cell Biol. 2014;92(6):509–517. doi: 10.1038/icb.2014.15. [DOI] [PubMed] [Google Scholar]
- 39.Guilmot A, Bosse J, Carlier Y, Truyens C. Monocytes play an IL-12-dependent crucial role in driving cord blood NK cells to produce IFN-g in response to Trypanosoma cruzi. PLoS Negl Trop Dis. 2013;7(6):e2291. doi: 10.1371/journal.pntd.0002291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown RA, Gralewski JH, Razonable RR. The R753Q polymorphism abrogates toll-like receptor 2 signaling in response to human cytomegalovirus. Clin Infect Dis. 2009;49(9):e96–e99. doi: 10.1086/644501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoffmann TW, et al. Association between a polymorphism in the IL-12p40 gene and cytomegalovirus reactivation after kidney transplantation. Transplantation. 2008;85(10):1406–1411. doi: 10.1097/TP.0b013e31816c7dc7. [DOI] [PubMed] [Google Scholar]
- 42.Morahan G, et al. A promoter polymorphism in the gene encoding interleukin-12 p40 (IL12B) is associated with mortality from cerebral malaria and with reduced nitric oxide production. Genes Immun. 2002;3(7):414–418. doi: 10.1038/sj.gene.6363909. [DOI] [PubMed] [Google Scholar]
- 43.De Paus RA, Geilenkirchen MA, van Riet S, van Dissel JT, van de Vosse E. Differential expression and function of human IL-12Rβ2 polymorphic variants. Mol Immunol. 2013;56(4):380–389. doi: 10.1016/j.molimm.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 44.Lee SH, Fragoso MF, Biron CA. Cutting edge: a novel mechanism bridging innate and adaptive immunity: IL-12 induction of CD25 to form high-affinity IL-2 receptors on NK cells. J Immunol. 2012;189(6):2712–2716. doi: 10.4049/jimmunol.1201528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Romo N, et al. Natural killer cell-mediated response to human cytomegalovirus-infected macrophages is modulated by their functional polarization. J Leukoc Biol. 2011;90(4):717–726. doi: 10.1189/jlb.0311171. [DOI] [PubMed] [Google Scholar]
- 46.Renneson J, et al. IL-12 and type I IFN response of neonatal myeloid DC to human CMV infection. Eur J Immunol. 2009;39(10):2789–2799. doi: 10.1002/eji.200939414. [DOI] [PubMed] [Google Scholar]
- 47.Heatley SL, et al. Polymorphism in human cytomegalovirus UL40 impacts on recognition of HLA-E by natural killer cells. J Biol Chem. 2013;288(12):8679–8690. doi: 10.1074/jbc.M112.409672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Charoudeh HN, et al. Modulation of the natural killer cell KIR repertoire by cytomegalovirus infection. Eur J Immunol. 2013;43(2):480–487. doi: 10.1002/eji.201242389. [DOI] [PubMed] [Google Scholar]
- 49.Béziat V, et al. NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood. 2013;121(14):2678–2688. doi: 10.1182/blood-2012-10-459545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaiser BK, Pizarro JC, Kerns J, Strong RK. Structural basis for NKG2A/CD94 recognition of HLA-E. Proc Natl Acad Sci U S A. 2008;105(18):6696–6701. doi: 10.1073/pnas.0802736105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pietra G, et al. HLA-E-restricted recognition of cytomegalovirus-derived peptides by human CD8+ cytolytic T lymphocytes. Proc Natl Acad Sci U S A. 2003;100(19):10896–10901. doi: 10.1073/pnas.1834449100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Michaelsson J, et al. A signal peptide derived from hsp60 binds HLA-E and interferes with CD94/NKG2A recognition. J Exp Med. 2002;196(11):1403–1414. doi: 10.1084/jem.20020797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoare HL, et al. Subtle changes in peptide conformation profoundly affect recognition of the non-classical MHC class I molecule HLA-E by the CD94-NKG2 natural killer cell receptors. J Mol Biol. 2008;377(5):1297–1303. doi: 10.1016/j.jmb.2008.01.098. [DOI] [PubMed] [Google Scholar]
- 54.Fadda L, et al. Peptide antagonism as a mechanism for NK cell activation. Proc Natl Acad Sci U S A. 2010;107(22):10160–10165. doi: 10.1073/pnas.0913745107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature. 2009;457(7229):557–561. doi: 10.1038/nature07665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.O’Leary JG, Goodarzi M, Drayton DL, von Andrian UH. T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat Immunol. 2006;7(5):507–516. doi: 10.1038/ni1332. [DOI] [PubMed] [Google Scholar]
- 57.Paust S, et al. Critical role for the chemokine receptor CXCR6 in NK cell-mediated antigen-specific memory of haptens and viruses. Nat Immunol. 2010;11(12):1127–1135. doi: 10.1038/ni.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thomas R, Low HZ, Kniesch K, Jacobs R, Schmidt RE, Witte T. NKG2C deletion is a risk factor of HIV infection. AIDS Res Hum. 2012;28(8):844–851. doi: 10.1089/aid.2011.0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Muntasell A, et al. NKG2C zygosity influences CD94/NKG2C receptor function and the NK cell compartment redistribution in response to human cytomegalovirus. Eur J Immunol. 2013;43(5):1133–1141. doi: 10.1002/eji.201243117. [DOI] [PubMed] [Google Scholar]
- 60.Sun JC, Beilke JN, Bezman NA, Lanier LL. Homeostatic proliferation generates long-lived natural killer cells that respond against viral infection. J Exp Med. 2011;208(2):357–368. doi: 10.1084/jem.20100479. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.