

Abstract
In this study, a rat model of inflammatory pain was produced by injecting complete Freund's adjuvant into the hind paw, and the expression of acetylated histone 3 in the spinal cord dorsal horn was examined using immunohistochemical staining. One day following injection, there was a dramatic decrease in acetylated histone 3 expression in spinal cord dorsal horn neurons. However, on day 7, expression recovered in adjuvant-injected rats. While acetylated histone 3 labeling was present in dorsal horn neurons, it was more abundant in astrocytes and microglial cells. The recovery of acetylated histone 3 expression was associated with a shift in expression of the protein from neurons to glial cells. Morphine injection significantly upregulated the expression of acetylated histone 3 in spinal cord dorsal horn neurons and glial cells 1 day after injection, especially in astrocytes, preventing the transient downregulation. Our results indicate that inflammatory pain induces a transient downregulation of acetylated histone 3 in the spinal cord dorsal horn at an early stage following adjuvant injection, and that this effect can be reversed by morphine. Thus, the downregulation of acetylated histone 3 may be involved in the development of inflammatory pain.
Keywords: inflammation, hyperalgesia, acetylated histone 3, spinal cord, morphine, neurobiology
Abbreviations:
CFA, complete Freund's adjuvant; HDAC, histone deacetylase; ACH3, acetylated histone 3
INTRODUCTION
Recent studies have shown that gene expression changes in the nociceptive pathway are crucial for the induction and maintenance of inflammatory pain[1,2]. Most research on the mechanisms of pathological pain have focused on dynamic changes in steady-state levels of mRNAs and proteins in the peripheral and central nervous system. Animals with knockout or knockdown of specific genes exhibit changes in nociceptive response and altered susceptibility to the development of pathological pain[1]. There are a number of different transcription factors involved in the development of pathological pain, such as downstream regulatory element antagonistic modulator[3], cAMP response element-binding protein[4] and nuclear factor kappa B[5]. These effectors activate signaling pathways and are associated with pain hypersensitivity. Environmental and developmental factors can influence gene transcription during the course of a painful illness or during treatment[6]. This modulation is independent of genomic DNA sequences per se, and is known as epigenetic regulation. Epigenetics can be regarded as a heritable phenotype resulting from changes in chromosomes, but without alteration in the DNA sequence. Epigenetic mechanisms mainly include modifications of the DNA (e.g., methylation) and histones (e.g., acetylation, methylation or phosphorylation)[7]. Histones are DNA-packaging proteins that can undergo post-translational modification involving changes in their N-terminal tail[8]. One such modification is the acetylation of conserved lysine residues which can regulate transcription and facilitate neuronal plasticity in various neurobiological processes[9]. Five major classes of histones exist: H1, H2A, H2B, H3 and H4. Among these, H3 is considered to play a major role in epigenetic regulation. Acetylation of histone H3 occurs at several different lysine positions, and is catalyzed by histone acetyltransferases, while removal is catalyzed by histone deacetylases (HDACs)[10]. Studies suggest that inhibitors of HDACs, which act on the epigenome by indirectly remodeling the spatial conformation of the chromatin, may provide a new therapeutic approach for neuropathic and other types of pain. For example, two selective HDAC inhibitors, 2-aminobenzamides (MS-275) and suberoyl anilide bishydroxamide, reduce nociceptive responses in the second phase of the formalin assay[11]. Trichostatin A, another HDAC inhibitor, induces increased thermal response latency[12]. The expression of class IIa HDAC proteins in the spinal dorsal horn was upregulated following complete Freund’s adjuvant (CFA) injection, and the inhibition of class II HDACs was important for attenuating pain hypersensitivity[13].
It has been suggested that epigenetic regulation in the spinal cord participates in the development of inflammatory pain. Epigenetic mechanisms, especially histone acetylation and deacetylation, are involved in the modulation of nociception and pathological pain. However, the expression profiles of acetylated histones in the development of pathological pain are still unknown. Since the mechanisms underlying chronic pain remain largely unclear (e.g., whether the nociceptive pathway is involved), there has been difficulty in developing the proper treatment[14]. Our study focuses on changes in acetylated histone 3 (ACH3) in the spinal dorsal horn during inflammatory pain induced by CFA and explores a novel target for treatment of pathological pain.
RESULTS
Treatment and group management of experimental animals
CFA intervention experiment: A total of 24 rats were randomly divided into three groups: naive (normal feeding), CFA 1 day and CFA 7 day, with eight rats in each group.
Morphine intervention experiment: A total of 15 rats were randomly divided into three groups: normal control (normal feeding), CFA 1 day and morphine treatment (CFA 1 day + morphine), with five rats in each group. In the final analysis, 39 rats were included, with no dropouts.
CFA injection induces ACH3 downregulation in the spinal dorsal horn
CFA injection into the left hind paw induced localized swelling and redness. As shown in Figure 1A, CFA injection resulted in a 50% reduction in the paw withdrawal threshold within 1 hour (P < 0.01, P < 0.05, vs. baseline), demonstrating the presence of allodynia. The threshold reached a minimal value between 6 hours and 1 day (P < 0.01), slightly increased at 3 days, and the value at 14 days was still lower than the baseline (P < 0.05).
Figure 1.
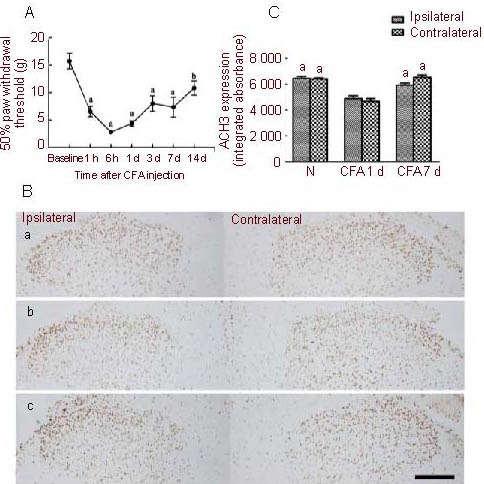
Downregulation of acetylated histone 3 in the spinal cord after complete Freund's adjuvant (CFA) injection into the hind paw (light microscopy).
(A) Change in the 50% paw withdrawal threshold of the left hind paw at different time points after injection. Data are expressed as mean ± SD, n = 8; one-way analysis of variance and Dunnett's test. aP < 0.01, bP < 0.05, vs. baseline.
(B) Immunohistochemical analysis of acetylated histone 3 expression in the spinal dorsal horn after injection. (a) Naive rat; (b) CFA 1 d; (c) CFA 7 d. An acetylated histone 3 positive cell was determined by examining brown granules in the nucleus (diaminobenzidine staining; scale bar: 200 μm).
(C) Quantitative analysis of acetylated histone 3 expression, using integrated absorbance, in the dorsal horn. Data are expressed as mean ± SEM, n = 5; two-way analysis of variance. aP < 0.05, vs. CFA 1 d group. CFA 1 d: Day 1 after CFA hind paw injection; CFA 7 d: day 7 after CFA hind paw injection; N: naive group.
In the naive rat, ACH3 was highly expressed in the nucleus in the spinal dorsal horn (Figure 1B-a). After CFA hind paw injection, ACH3 immunoreactivity dramatically decreased at day 1 post-injection (Figures 1B-b, c). No difference was observed in ACH3 expression between the ipsilateral and contralateral spinal dorsal horns (Figures 1B, C). However, at 7 days after CFA injection, ACH3 expression recovered and was comparable to that in naive rats (Figures 1B-c, C). These results suggest that CFA-induced inflammatory pain results in the transient downregulation of ACH3 expression in the spinal dorsal horn.
Phenotypic shift in ACH3 expression in the spinal dorsal horn in distinct phases of CFA-induced inflammatory pain
To better understand the changes in ACH3 expression, double immunofluorescence labeling was performed for ACH3 and markers associated with nociceptive processing (neurons, marked by NeuN; microglia, marked by CD11b; and astrocytes, marked by GFAP). As shown in Figure 2A, intense ACH3 labeling was observed in NeuN-labeled cells. In addition, some ACH3 immunoreactivity was also localized to microglia (CD11b positive staining) and astrocytes (GFAP positive staining) in naive rats (Figures 2E, I).
Figure 2.
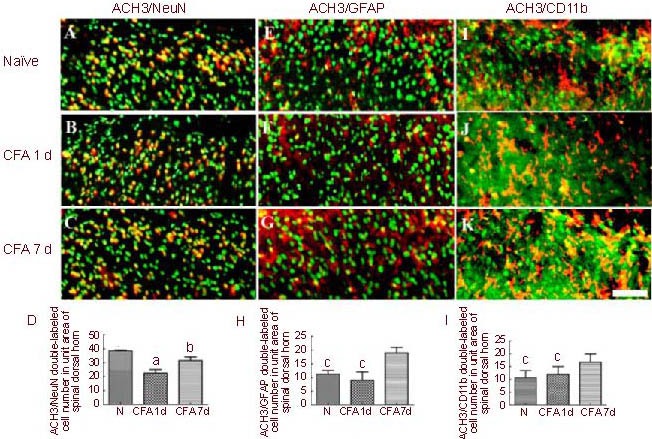
Double immunofluorescence co-localization of acetylated histone 3 (ACH3) with neuronal nuclei (NeuN) (A–C), glial firbrillary acidic protein (GFAP; E-G) and CD11b (I–K) in the ipsilateral spinal dorsal horn after complete Freund's adjuvant (CFA) injection (immunofluorescence microscope; fluorescent dyes: Alexa Fluor 488, Cy3, Hoechst; scale bar: 100 μm).
(D, H, L) Quantitative analysis of double-labeled cell numbers in a unit area of the spinal dorsal horn.
Data are expressed as mean ± SEM, n = 8; one-way analysis of variance, Dunnett's test. aP < 0.01, vs. N; bP < 0.05, vs. CFA 1 d group; cP < 0.05, vs. CFA 7 d group. N: Naive group.
ACH3 expression was significantly reduced 1 day after CFA hind paw injection (Figure 1B-b). The number of ACH3/NeuN double-labeled cells was also lower (Figure 2B). However, the number of ACH3/GFAP (Figure 2F) and ACH3/CD11b (Figure 2J) double-labeled cells did not change significantly 1 day after CFA injection. These results suggest that the downregulation of ACH3 expression observed in the early phase of CFA-induced inflammatory pain is largely due to decreased expression of ACH3 by neurons, and is not due to reduced expression in microglia or astrocytes.
At 7 days after CFA injection, the number of ACH3/NeuN double labeled cells recovered to an extent, but was still less compared with the naive rats (Figure 2C). However, the number of ACH3/GFAP (Figure 2G) and ACH3/CD11b (Figure 2K) double labeled cells was greatly increased compared with naive rats on day 7, and in comparison to 1 day after CFA injection. These data strongly suggest that there is a phenotypic change in ACH3 expression in the later phase of inflammatory pain, i.e., the expression of ACH3 shifts from neurons to microglia and astrocytes.
Effects of morphine on ACH3 expression in the spinal dorsal horn after CFA-induced inflammatory pain (Figure 3)
Figure 3.
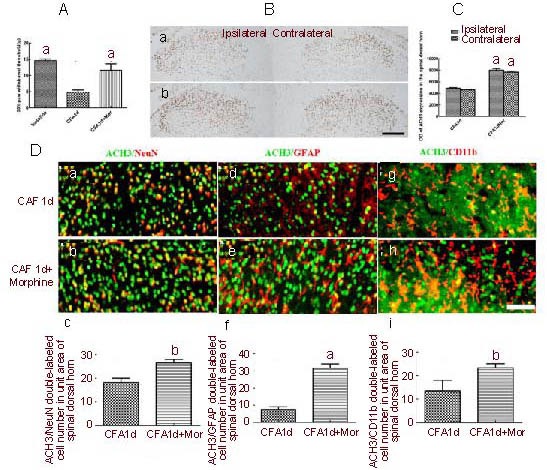
Effects of morphine on acetylated histone 3 (ACH3) expression in spinal dorsal horns in complete Freund's adjuvant (CFA)-induced inflammatory pain (immunofluorescence microscope; fluorescent dyes: Alexa Fluor 488, Cy3, Hoechst).
(A) Effects of morphine on 50% paw withdrawal threshold in CFA-induced pain (aP < 0.01, vs. CFA 1 d).
(B) ACH3 expression in spinal dorsal horn in CFA 1 d (Ba) or CFA 1 d + Mor (B-b) (diaminobenzidine staining; scale bar: 200 μm).
(C) Quantitative analysis of ACH3 expression in spinal dorsal horn (two-way analysis of variance. aP < 0.01, vs. CFA 1 d group, n = 5).
(D) Double labeling of ACH3 with neuronal nuclei (NeuN), glial fibrillary acidic protein (GFAP) or CD11b in ipsilateral spinal dorsal horn, and quantitative analysis of double labeled cell number. Scale bar: 100 μm. (a, d, g) CFA 1 d; (b, e, h) CFA 1 d + Mor; (c, f, i) quantitative analysis of double-labeled cell numbers in a unit area of spinal dorsal horn. Data are expressed as mean ± SEM, one-way analysis of variance, Dunnett's test. aP < 0.01, bP < 0.05, vs. CFA 1 d group.
CFA 1 d: Day 1 after CFA injection; CFA 1 d + Mor: day 1 after CFA injection treated by morphine (Mor).
The downregulation of ACH3 in the spinal cord dorsal horns may be inhibited by analgesia. To test this hypothesis, we examined whether morphine, the most common painkiller, could modify ACH3 expression in CFA-induced inflammatory pain.
We found that intraperitoneal injection of morphine significantly suppressed the 50% reduction in paw withdrawal threshold observed at day 1 post-CFA injection (Figure 3A), and the decrease in ACH3 expression in the spinal dorsal horn was significantly attenuated in the morphine treatment group at day 1 post-CFA injection (Figures 3B, C). Interestingly, double immunofluorescence labeling showed that morphine could increase the expression of ACH3 in neurons and glial cells, having the most robust effect on astrocytes at day 1 post-CFA injection (Figure 3D). These findings suggest that the transient downregulation of ACH3 expression in the spinal dorsal horn can be reversed by morphine, and that its effect mainly targets glial cells.
DISCUSSION
There are several interesting findings in the present study. First, there is a transient downregulation of ACH3 expression in the spinal dorsal horns in CFA-induced inflammatory pain. Second, there is a phenotypic change in ACH3 expression during the development of inflammatory pain; i.e., ACH3 expression in the early phase shifts from neurons to microglia and astrocytes in the later phase. Third, morphine, the most extensively used analgesic, could prevent the downregulation of ACH3 by increasing expression in neurons and glial cells, especially in astrocytes, in the early phase of inflammatory pain.
Chromatin remodeling through histone modification has emerged as an important epigenetic mechanism regulating gene expression. Histones can undergo, at specific sites in their N-terminus, acetylation, methylation, phosphorylation and ubiquitination[8]. The result can be either transcriptional activation or repression, depending on the type and site of the modification (e.g., acetylation mostly induces activation, deacetylation mostly induces repression). Accumulating evidence indicates that histone acetylation is involved in nociceptive processes. Several HDAC inhibitors, which result in a net increase in histone acetylation, have been reported to attenuate the nociceptive response in inflammatory pain[11] 13]. However, it is still unknown whether the anti-nociceptive effect of HDAC inhibitors can be attributed to the activation of histone acetylation or by inhibiting the reduction of histone acetylation.
In the present study, ACH3 expression was transiently downregulated in inflammatory pain induced by CFA, further supporting a role for histone acetylation in the induction of inflammatory pain by the adjuvant. Furthermore, the anti-nociceptive effect of HDAC inhibitors is likely due to their ability to suppress the reduction in histone acetylation in response to nociceptive stimuli.
Peripheral inflammation leads to central sensitization in the spinal cord, which contributes to the hyperalgesia and allodynia typically associated with inflammation. The spinal dorsal horn is a major target of small-diameter afferent nerve fibers that predominantly transmit nociceptive signals. Therefore, gene expression changes in the spinal dorsal horn play a critical role in the induction and maintenance of inflammatory pain. The reduced histone acetylation (as indicated by the downregulation of ACH3) strongly suggests that in the early phase of inflammatory pain, gene transcription is reduced in the spinal dorsal horn. This may repress anti-nociceptive genes, such as opiate receptors, GABA receptors, glycine receptors and endorphin receptors[15,16,17]. Supporting this hypothesis, several studies have shown that inhibitory GABA neurons are lost during nociceptive processing[18]. However, this does not exclude the possibility that other epigenetic mechanisms are involved in gene regulation during pain, such as DNA methylation. In fact, some pro-nociceptive cytokines are upregulated during the development of pain, such as substance P, calcitonin gene related peptide, nerve growth factor and brain derived neurotrophic factor[19,20]. It is likely that the transcription of these genes is affected by DNA methylation or other factors, but not by histone H3 acetylation regulation. Overall, during the early phase of pain, a reduction in gene transcription appears to be primarily responsible for the reduced ACH3 levels. However, in the late phase of pain, ACH3 expression recovered, indicating that the level of gene expression gradually returned to normal, which may include synergistic effects of analgesic genes and pain-causing genes.
In the present study, the downregulation of ACH3 mainly occurred in neurons in the early phase, whereas the upregulation primarily occurred in glial cells in the late phase of inflammatory pain. These findings suggest that in the early phase of inflammatory pain, changes in neuronal gene transcription play a major role in the nociceptive process. Previous studies have shown that glial cells, including microglia and astrocytes, are activated in the spinal cord in sub-acute and chronic phases of CFA-induced inflammatory pain[21]. In the present study, increased ACH3 expression in glial cells at day 7 post-CFA injection suggests that dynamic changes in glial cells play a crucial role in the maintenance of inflammatory pain. Increased histone acetylation in glial cells may trigger the expression of pain-related genes. It is well known that cytokines released from glial cells contribute to the maintenance of pain[21,22]. Future studies are required to elucidate the changes in gene expression profile due to decreased histone acetylation in neurons in the early phase and that due to increased histone acetylation in glial cells in the late phase of inflammatory pain.
Morphine is the most commonly used analgesic for pain management, and it works by binding to opioid receptors. A recent study demonstrated that acute morphine pretreatment can induced histone hyperacetylation in the nucleus accumbens, which can be inhibited by HDAC inhibitors[23]. However, another recent study showed that HDAC inhibitors can reverse the disruptive effect of morphine-related memory reconsolidation induced by nuclear factor kappa B inhibition[24]. These studies suggest that different morphine treatment regimens have distinct effects on histone acetylation in different brain regions. In the present study, morphine was able to reverse the downregulation of ACH3, suggesting that morphine promotes histone acetylation in the spinal cord in inflammatory pain. The increased histone acetylation may subsequently induce the activation of analgesic genes and the attenuation of pain hypersensitivity through opioid receptors. Thus, epigenetic regulation may play an important role in the analgesic effect of morphine.
In summary, CFA hind paw injection results in a transient downregulation of ACH3 in the spinal cord, which can be reversed by morphine. There is a phenotypic change in ACH3 expression during the development of inflammatory pain. Epigenetic mechanisms may play important roles in the induction and maintenance of inflammatory pain, and morphine’s analgesic effect may be mediated, in part, by its ability to modulate spinal histone acetylation.
MATERIALS AND METHODS
Design
Randomized, controlled animal experiments.
Time and setting
This study was performed at the Department of Human Anatomy and Neurobiology, Xiangya Medical School, Central South University, China, from May to October 2010.
Materials
Adult male Sprague-Dawley rats, weighing 180–250 g, were obtained from Animal Services of Xiangya School of Medicine, Central South University, Changsha, China (license No. SCXK (Xiang) 2006-0002). All efforts were made to minimize the number of rats used and their suffering according to the Guidance Suggestions for the Care and Use of Laboratory Animals, issued by the Ministry of Science and Technology of China[25]. All behavioral experiments were conducted blindly with respect to drug administration.
Methods
Preparation of inflammatory pain model
Peripheral inflammation[4] was produced by intraplantar injection of 100 μL CFA (Sigma, St. Louis, MO, USA) suspended in an oil/saline (1:1) emulsion under 1.5% isoflurane anesthesia (Shandong Keyuan Pharmaceutical Co., Ltd., Jinan, Shandong Province, China). This procedure produced persistent peripheral inflammation in the injected hindpaw that was characterized by mechanical hyperalgesia.
Determination of 50% paw withdrawal threshold
Mechanical hyperalgesia was assessed in naive and CFA-injected rats by using the up-and-down method as described previously[16]. The experiment was performed by two authors who were blinded to the treatment. Rats were placed on wire mesh platforms in clear cylindrical plastic enclosures and allowed to acclimate for 20 minutes. A series of calibrated von Frey filaments were applied to the center of the swollen hind paw. A positive paw withdrawal response was recorded if the animal briskly lifted the hindpaw. In the absence of a paw withdrawal response to the initially selected filament (2.0 g), the next stronger filament was presented; however, in the event of a positive withdrawal response, the next weaker stimulus in the series was used during the next trial. The resulting pattern of positive and negative responses was tabulated as X = withdrawal and O = no withdrawal. The 50% response threshold was interpolated using the following formula: 50% g threshold = (10[xf+kδ]/10 000); where xf = value (in log units) of the final von Frey hair used, k = tabular value for the pattern of positive/negative responses, and δ = mean difference (in log units) between stimuli. Mechanical hyperalgesia was measured at 30 minutes before and 6 hours, 1 day, 3 days, 7 days and 14 days after CFA injection. For morphine-treated rats, the 50% paw withdrawal threshold was measured again at 1 hour after morphine administration.
Spinal cord tissue processing
Rats were deeply anesthetized with 10% chloral hydrate (0.4 mL/100 g) and perfused intracardially with 4% paraformaldehyde in phosphate-buffered saline (PBS), pH 7.4. The L4-5 segment of the spinal cord was removed and immersed in the same fixative for 2 hours at 4°C, then stored in PBS solution containing 30% sucrose overnight. When tissues sank to the bottom of the container, the spinal cord was cut into 30-μm transverse sections on a cryostat and collected in 0.01 M PBS.
ACH3 expression as detected by immunohistochemistry and immunofluorescence staining
The floating sections were immersed in 3% hydrogen peroxide for 15 minutes, and then washed in 0.01 M PBS. After incubation in 0.01 M PBS containing 5% bovine serum and 0.1% Triton X-100 for 2 hours, the sections were incubated with primary antibodies: ACH3 (rabbit anti-ACH3 polyclonal antibody, 1:1 000; Cell Signaling Technology, CA, USA) overnight at 4°C. The next day, the sections were washed in 0.01 M PBS, and then incubated with secondary antibody (goat anti-rabbit IgG; Vector Laboratories, Burlingame, CA, USA) for 2 hours. The sections were again washed three times with 0.01 M PBS and then incubated for 2 hours with streptavidin-horseradish peroxidase (Vector Laboratories). Subsequently, sections were washed and incubated with DAB (Vector Laboratories) for 1–3 minutes, and then the reaction was stopped by washing in 0.01 M PBS. Finally, the sections were mounted on slides, dehydrated, cleared, and coverslipped with neutral gum. Negative control sections from each animal received identical staining, except that the primary antibody was omitted. The evaluation of positive cells was performed by examining the number of brown granules in the nucleus. Pictures of the dorsal horn region were taken with a light microscope (Olympus, Tokyo, USA). Six spinal cord sections were randomly taken for each sample.
Quantification of integrated optical density of immunoreactive area was performed with Image-Pro Plus 6.0 (Media Cybernetics, Silver Spring, MD, USA). For double immunofluorescence labeling, the sections were mounted on gelatin coated slides, incubated with rabbit anti-ACH3 polyclonal antibody (1:500; Cell Signaling Technology, USA) and mouse anti-NeuN (1:1 000, Millipore Corporation, Billerica, MA, USA) or GFAP (1:1 000, Millipore) or CD11b (1:1 000, Millipore) polyclonal antibody at 4°C overnight. Sections were then washed in 0.01 M PBS, followed by incubation with secondary antibodies conjugated with fluorescent dyes, Alexa Fluor 488 goat anti-rabbit antibody and Cy3-conjugated goat anti-mouse antibody (1:200; Invitrogen Life Technologies, Carlsbad, CA, USA), for 2 hours. Finally the sections were incubated in Hoechst for 15 minutes, cleared, coverslipped with 50% glycerite, and imaged by fluorescence microscopy (Olympus). The number of double-labeled cells was counted under a unit area (about 150 000 square micrometers, including lamina I and II, and part of lamina III, of the spinal dorsal horns). Six spinal cord sections were randomly taken for each sample.
Statistical analysis
Data were presented as mean ± SEM and analyzed using SPSS 13.0 software (SPSS, Chicago, IL, USA). Analysis was accomplished using one-way analysis of variance followed by Dunnett’s post hoc test for nociceptive testing and double immunofluorescence staining, or two-way analysis of variance followed by Bonferroni testing for ACH3 expression (diaminobenzidine staining). A value of P < 0.05 was considered to indicate a significant difference.
Acknowledgments:
We would like to thank Dr. Xinfu Zhou from Flinders University for his editorial guidance.
Footnotes
Funding: This study was supported by the National Natural Science Foundation of China, No. 81070897, 81102726.
Ethical approval: The experimental protocol was approved by the Animal Care and Use Committee of Central South University in China.
(Edited by Fang MR, Zhu WJ/Yang Y/Song LP)
REFERENCES
- [1].Lacroix-Fralish ML, Ledoux JB, Mogil JS. The pain genes database: an interactive web browser of pain-related transgenic knockout studies. Pain. 2007;131(1-2):1–4. doi: 10.1016/j.pain.2007.04.041. [DOI] [PubMed] [Google Scholar]
- [2].Geranton SM, Morenilla-Palao C, Hunt SP. A role for transcriptional repressor methyl-CpG-binding protein 2 and plasticity-related gene serum and glucocorticoid-inducible kinase 1 in the induction of inflammatory pain states. J Neurosci. 2007;27(23):6163–6173. doi: 10.1523/JNEUROSCI.1306-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Penninger JM, Cheng HY, Pitcher GM, et al. DREAM is a critical transcriptional repressor for pain modulation. Cell. 2002;108(1):31–43. doi: 10.1016/s0092-8674(01)00629-8. [DOI] [PubMed] [Google Scholar]
- [4].Duric V, McCarson KE. Neurokinin-1 (NK-1) receptor and brain-derived neurotrophic factor (BDNF) gene expression is differentially modulated in the rat spinal dorsal horn and hippocampus during inflammatory pain. Mol Pain. 2007;3(1):32. doi: 10.1186/1744-8069-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tegeder I, Niederberger E, Schmidt R, et al. Specific Inhibition of IkappaB kinase reduces hyperalgesia in inflammatory and neuropathic pain models in rats. J Neurosci. 2004;24(7):1637–1645. doi: 10.1523/JNEUROSCI.3118-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Doehring A, Geisslinger G, Lotsch J. Epigenetics in pain and analgesia: an imminent research field. Eur J Pain. 2011;15(1):11–16. doi: 10.1016/j.ejpain.2010.06.004. [DOI] [PubMed] [Google Scholar]
- [7].Berger SL, Kouzarides T, Shiekhattar R, et al. An operational definition of epigenetics. Genes Dev. 2009;23(7):781–783. doi: 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- [9].Thomas EA. Focal nature of neurological disorders necessitates isotype selective histone deacetylase (HDAC) inhibitors. Mol Neurobiol. 2009;40(1):33–45. doi: 10.1007/s12035-009-8067-y. [DOI] [PubMed] [Google Scholar]
- [10].Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10(1):32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chiechio S, Zammataro M, Morales ME, et al. Epigenetic modulation of mGlu2 receptors by histone deacetylase inhibitors in the treatment of inflammatory pain. Mol Pharmacol. 2009;75(5):1014–1020. doi: 10.1124/mol.108.054346. [DOI] [PubMed] [Google Scholar]
- [12].Lu Y, Nie J, Liu X, et al. Trichostatin A, a histone deacetylase inhibitor, reduces lesion growth and hyperalgesia in experimentally induced endometriosis in mice. Hum Reprod. 2010;25(4):1014–1025. doi: 10.1093/humrep/dep472. [DOI] [PubMed] [Google Scholar]
- [13].Bai G, Wei D, Zou S, et al. Inhibition of class II histone deacetylases in the spinal cord attenuates inflammatory hyperalgesia. Mol Pain. 2010;6(1):51. doi: 10.1186/1744-8069-6-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Forde G, Stanos S. Practical management strategies for the chronic pain patient. J Fam Pract. 2007;(Suppl 56):21–30. [PubMed] [Google Scholar]
- [15].Li CQ, Xu JM, Liu D, et al. Brain derived neurotrophic factor (BDNF) contributes to the pain hypersensitivity following surgical incision in the rats. Mol Pain. 2008;4(1):27. doi: 10.1186/1744-8069-4-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Obara I, Parkitna JR, Korostynski M, et al. Local peripheral opioid effects and expression of opioid genes in the spinal cord and dorsal root ganglia in neuropathic and inflammatory pain. Pain. 2009;141(3):283–291. doi: 10.1016/j.pain.2008.12.006. [DOI] [PubMed] [Google Scholar]
- [17].Keller AF, Coull JA, Chery N, et al. Region-specific developmental specialization of GABA-glycine cosynapses in laminas I-II of the rat spinal dorsal horn. J Neurosci. 2001;21(20):7871–7880. doi: 10.1523/JNEUROSCI.21-20-07871.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Polgár E, Hughes DI, Riddell JS, et al. Selective loss of spinal GABAergic or glycinergic neurons is not necessary for development of thermal hyperalgesia in the chronic constriction injury model of neuropathic pain. Pain. 2003;104(1-2):229–239. doi: 10.1016/s0304-3959(03)00011-3. [DOI] [PubMed] [Google Scholar]
- [19].Merighi A, Carmignoto G, Gobbo S, et al. Neurotrophins in spinal cord nociceptive pathways. Prog Brain Res. 2004;146(1):291–321. doi: 10.1016/s0079-6123(03)46019-6. [DOI] [PubMed] [Google Scholar]
- [20].Bird GC, Han JS, Fu Y, et al. Pain-related synaptic plasticity in spinal dorsal horn neurons: role of CGRP. Mol Pain. 2006;2(1):31. doi: 10.1186/1744-8069-2-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci. 2004;20(2):467–473. doi: 10.1111/j.1460-9568.2004.03514.x. [DOI] [PubMed] [Google Scholar]
- [22].Lee S, Zhao YQ, Ribeiro-da-Silva A, et al. Distinctive response of CNS glial cells in oro-facial pain associated with injury, infection and inflammation. Mol Pain. 2010;6(1):79. doi: 10.1186/1744-8069-6-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jing L, Luo J, Zhang M, et al. Effect of the histone deacetylase inhibitors on behavioural sensitization to a single morphine exposure in mice. Neurosci Lett. 2011;494(2):169–173. doi: 10.1016/j.neulet.2011.03.005. [DOI] [PubMed] [Google Scholar]
- [24].Yang J, Yu J, Jia X, et al. Inhibition of nuclear factor-κB impairs reconsolidation of morphine reward memory in rats. Behav Brain Res. 2011;216(2):592–596. doi: 10.1016/j.bbr.2010.08.047. [DOI] [PubMed] [Google Scholar]
- [25].The Ministry of Science and Technology of the People's Republic of China. Guidance Suggestions for the Care and Use of Laboratory Animals. 2006-09-30 [Google Scholar]