

 With the aim of discovering novel G9a inhibitory chemotypes, we have identified a new quinoline inhibitor scaffold and better defined the pharmacophoric features of the central heterocycle.
With the aim of discovering novel G9a inhibitory chemotypes, we have identified a new quinoline inhibitor scaffold and better defined the pharmacophoric features of the central heterocycle.
Abstract
G9a is a histone lysine methyltransferase (HKMT) involved in epigenetic regulation via the installation of histone methylation marks. 6,7-Dimethoxyquinazoline analogues, such as BIX-01294, are established as potent, substrate competitive inhibitors of G9a. With an objective to identify novel chemotypes for substrate competitive inhibitors of G9a, we have designed and synthesised a range of heterocyclic scaffolds, and investigated their ability to inhibit G9a. These studies have led to improved understanding of the key pharmacophoric features of BIX-01294 and the identification of a new core quinoline inhibitory scaffold, which retains excellent potency and high selectivity. Molecular docking was carried out to explain the observed in vitro data.
Introduction
G9a (also known as EHMT2) is a histone-lysine N-methyltransferase (HMKT), which catalyses the addition of one or two methyl groups to lysine 9 of histone H3 (H3K9me1 and H3K9me2) within a chromatin environment.1 Due to its central role in epigenetic control, the methylation of H3K9 is associated with many biological pathways and is aberrantly regulated in several diseases including cancer2 and AIDS.3 According to a genome-wide analysis of histone modifications,4 mono-methylation of histone H3 (H3K9me1) is associated with permissive chromatin, while di- and tri-methylation (H3K9me2/3) label a repressed chromatin state. Overexpression of G9a has been found in many types of cancer and is associated with a poor prognosis.5,6 Furthermore, G9a has been reported the methylate a variety of non-histone targets including the chromatin-modifying factor Tip49a7 and CDYL1.8
Like the majority of HKMTs, the SET (Su(var), E(z) and Trithorax) domain of G9a mediates catalysis and is comprised of two binding pockets; one for the cofactor S-adenosylmethionine (SAM), the other for the protein substrate. It is increasingly being realised that both pockets are ‘druggable’ and occupancy of either with a small molecule inhibitor is an effective strategy to block HKMT mediated methylation.9 The first substrate-competitive inhibitor of G9a, BIX-01294, was discovered using high throughput screening (Fig. 1).10 Focused medicinal chemistry efforts from a number of groups has led to a series of optimised diaminoquinazoline analogues including UNC0224,11 UNC0321,12 UNC0631,13 UNC0638,14 UNC0646,13 UNC0965,15 E72,16 and UNC0642,17 which is suitable for in vivo work. Recently, 2-amino-indole derivatives have also been reported as substrate competitive G9a inhibitors.18 There is little doubt that the provision of such high quality inhibitors has dramatically facilitated the study of G9a biology,14,15,19,20 and related targets, especially in a disease context.21–24
Fig. 1. Representative examples of substrate-competitive G9a inhibitors.

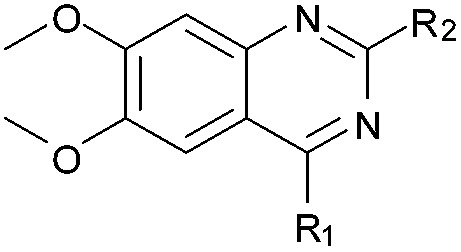
BIX-01294 and its optimised analogues are composed of a quinazoline heterocyclic core, substituted at positions 2, 4 and 7 (Fig. 1). The co-crystallized structure of UNC0224 with G9a (PDB code ; 3K5K)11 reveals important interactions between the inhibitor and the substrate pocket of G9a. Key binding interactions include (Fig. 2): (1) a salt bridge between N-1 of the quinazoline core, which is expected to be protonated at physiological pH,25,26 and Asp1088; (2) a hydrogen bond between the C-4 NH functionality and Asp1083; (3) a hydrogen bond between the protonated ‘lysine mimic’ amine at C-7 with the backbone of Leu1086, as well as a cation–π interaction between the same protonated amine and Tyr1154.
Fig. 2. A summary of the interactions between UNC0224 and G9a (PDB code 3K5K). Salt bridges, cation–π interactions and hydrogen bonds are depicted as pink, golden and green dashed lines, respectively.

Whilst the prior studies, particularly those of Jin and co-workers,11–17 have established important G9a structure–activity relationships (SAR) with respect to the side chains of the quinazoline core, it was apparent to us that the pharmacophoric features of the central heterocycle were yet to be determined. Thus we set out to better define the important features of the central inhibitor scaffold, while attempting to maintain the aforementioned interactions within the substrate pocket of the enzyme.
Chemistry
BIX-01294 (1) and other quinazoline derivatives (2–4, Table 1), were synthesised following the established two step synthesis.11,27
Table 1. SAR, biological and computational results of the BIX-01294 derivatives a .
| Compound ID | Scaffold | R2 | Activity% compound
b
(μM) |
IC50 c (μM) | c log P d | N-1 pK a e | Docking score (kcal mol–1) |
|||
| 50 | 10 | 1 | SP | XP | ||||||
| 1 (BIX-01294) |
|
|
6 | 6 | 11 | 0.067 ± 0.003 | 3.9 | 8.13 (±2.22) | –7.859 | na |
| 2 (HKMTI-1-005) |
|
5 | 6 | 19 | 0.101 ± 0.010 | 3.5 | 5.60 (±2.22) | –6.995 | –6.380 | |
| 3 (HKMTI-1-022) |
|
7 | 9 | 35 | 0.472 ± 0.017 | 4.7 | 7.47 (±1.47) | –5.789 | –4.244 | |
| 4 (HKMTI-1-011) |
|
15 | 31 | 79 | 3.190 ± 0.080 | 4.4 | 8.10 (±1.47) | na | na | |
| 12 |
|
|
99 | 102 | 99 | — | 2.9 | 4.57 (±0.70) | na | na |
| 13 |
|
98 | 100 | 107 | — | 2.5 | 4.66 (±0.70) | na | na | |
| 14 |
|
97 | 96 | 101 | — | 3.7 | 5.31 (±0.70) | na | na | |
| 15 |
|
102 | 98 | 107 | — | 3.4 | 5.77 (±0.70) | na | na | |
| 16 |
|
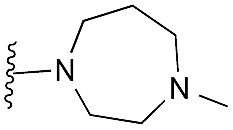
|
47 | 82 | 91 | — | 3.9 | 5.68 (±0.70) | na | na |
| 17 |
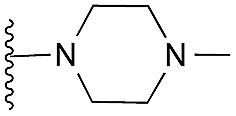
|
62 | 83 | 105 | — | 3.5 | 5.77 (±0.70) | na | na | |
| 18 |
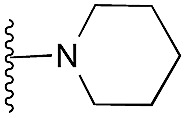
|
91 | 102 | 114 | — | 4.8 | 6.42 (±0.70) | na | na | |
| 19 |
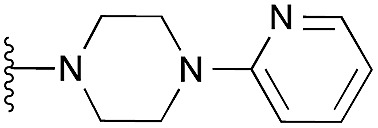
|
104 | 105 | 98 | — | 4.5 | 6.88 (±0.70) | na | na | |
| 20 |
|
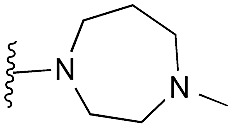
|
81 | 96 | 91 | — | 3.9 | 3.28 (±2.22) | na | na |
| 21 |
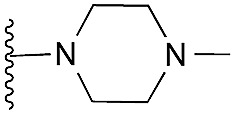
|
88 | 92 | 105 | — | 3.5 | 3.35 (±2.22) | na | na | |
| 22 |
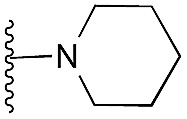
|
93 | 105 | 105 | — | 4.8 | 3.86 (±2.22) | na | na | |
| 23 |
|
|
67 | 90 | 101 | — | 2.6 | 3.15 (±2.22) | na | na |
| 24 |
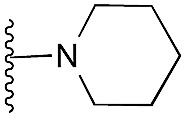
|
101 | 90 | 97 | — | 3.4 | 3.73 (±2.22) | na | na | |
| 25 |
|
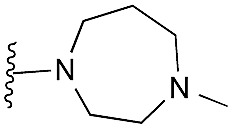
|
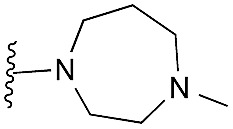
55 | 84 | 96 | — | 3.2 | 6.62 (±0.70) | na | na |
| 31 |
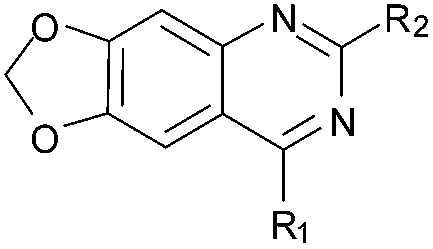
|
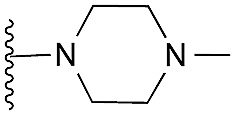
|
63 | 91 | 98 | — | 3.2 | 5.58 (±2.22) | na | na |
| 32 |
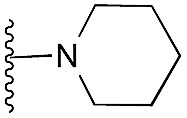
|
78 | 95 | 96 | — | 4.4 | 7.45 (±1.47) | na | na | |
| 33 |
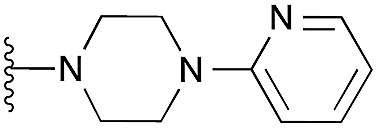
|
109 | 98 | 96 | — | 4.2 | 8.08 (±1.47) | na | na | |
| 34 |
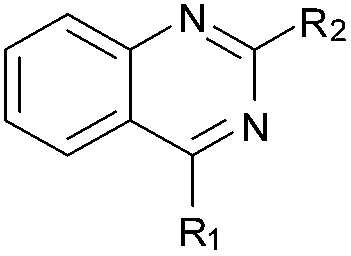
|
|
18 | 46 | 93 | — | 3.8 | 8.06 (±2.22) | na | na |
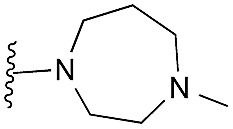
| 35 |
|
27 | 59 | 94 | — | 3.5 | 5.53 (±2.22) | na | na | |
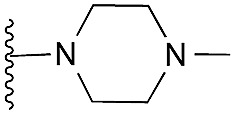
| 36 |
|
72 | 82 | 94 | — | 4.7 | 7.40 (±2.22) | na | na | |
| 37 |
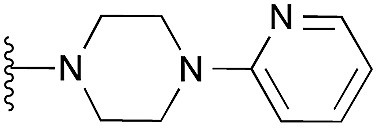
|
97 | 108 | 104 | — | 4.4 | 8.03 (±2.22) | na | na | |
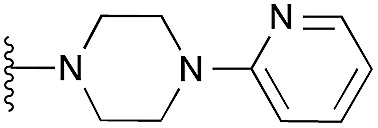
| 41 (HKMTI-1-248) |
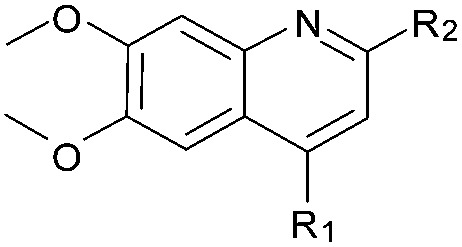
|
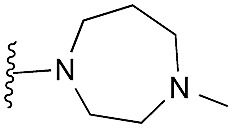
|
5 | 4 | 6 | 0.013 ± 0.001 | 4.5 | 10.57 (±2.22) | –7.522 | –6.904 |
| 42 (HKMTI-1-247) |
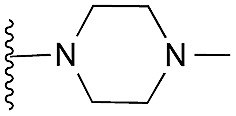
|
5 | 6 | 7 | 0.031 ± 0.003 | 4.1 | 9.84 (±2.22) | na | –6.476 | |
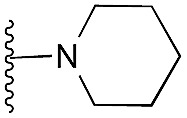
aR1 is the same for all compounds: R1 = 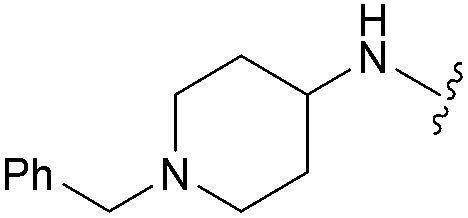 , SP = standard precision mode, XP = extra precision mode, na = no desired pose found.
, SP = standard precision mode, XP = extra precision mode, na = no desired pose found.
bThe experiment was conducted in duplicate.
cThe assay was conducted in triplicate at K m of both substrates (0.8 μM peptide [H3 1–25] and 8 μM SAM) for G9a (5 nM).17
d c log P values were calculated using the freely available program RDkit.34
e‘Sequential’ pK a values were calculated at pH 7.0, with water as the solvent model, using the Epik 2.7 program implemented in Schrodinger (see ESI).
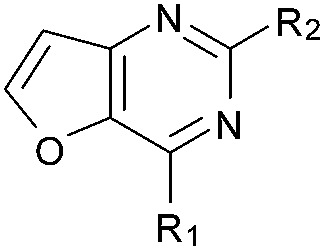
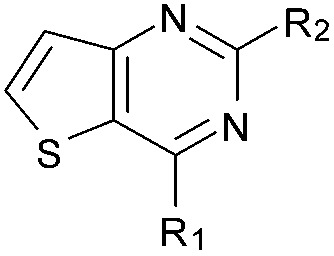
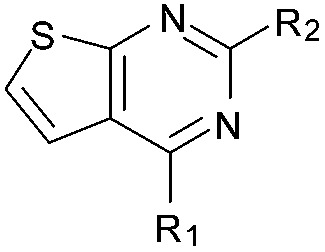
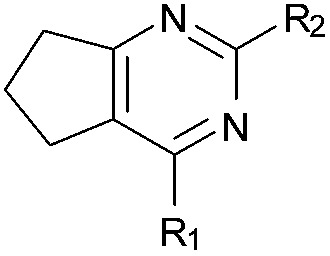
Initially the importance of the dimethoxy-benzenoid ring of BIX-01294 was explored, while retaining the substituted pyrimidine ring. Hence, compounds 12 to 25 were synthesised (Table 1) containing a pyrimidine ring fused to a furan, thiophene, imidazole, or cyclopentane ring.
Amino furan 8a was synthesised using a slight modification of a previously reported method (Scheme 1).28 The carboxyl group of furan 5 was converted to a Boc protected amine 6 in a single step via a Curtius rearrangement. Treatment of amino furan 6 with n-butyllithium, followed by a methyl chloroformate quench, resulted in the isolation of product 7. Boc deprotection of 7 yielded the desired intermediate 8a with a free amino group at position 3. Amino furan 8a and its commercially available analogues 8b and 8c were then converted to the corresponding fused-pyrimidinediones 9a–9c following reported reaction conditions.28,29 The desired dichloropyrimidines 10a–10c were obtained by heating 9a–9c to reflux in the presence of phosphoryl chloride, while the commercially available fused pyrimidinediones 9d and 9e were converted to 10d and 10e using pyrophosphoryl chloride30 and phosphoryl chloride, respectively. Key intermediates 10a–10e were then substituted with 4-amino-1-benzylpiperidine at C-4 to yield 11a–11e which were further heated with a variety of secondary amines to obtain the fully substituted heterocyclic compounds 12–25 (Table 1).
Scheme 1. The synthesis of fused-pyrimidine derivatives. Reagents and conditions: (a) DPPA, TEA, t-BuOH, reflux; (b) CH3OCOCl, TMEDA, n-BuLi, THF; (c) TFA, DCM, rt, (d) ClSO2NCO, DCM, aq. NaOH, aq. HCl or urea, 190 °C (neat), microwave; (e) POCl3, DMF, reflux or O(POCl2)2, 145 °C, sealed tube, 2 day; (f) 4-amino-1-benzylpiperidine, DIEA, THF, rt, 24 h or 4-amino-1-benzylpiperidine, TEA, n-butanol, sealed tube, overnight; (g) cyclic amine, 185 °C (neat), microwave, 30 min.

As an alternative strategy to completely replacing the benzenoid ring of the original quinazoline analogues, a number of derivatives were prepared where the dimethoxy functionality had been altered or removed. Thus, the commercially available benzonitrile 26 was nitrated, followed by reduction of the nitro-group with sodium dithionite to give amine 27 (Scheme 2). Acylation of 27 using methyl chloroformate, followed by cyclisation under basic conditions, resulted in quinazoline-dione 28a. The synthesised quinazoline-dione 28a and commercially available desmethoxyquinazoline-dione 28b were both chlorinated and substituted with amines as described previously11,27 to provide 31–37 (Table 1).
Scheme 2. The synthesis of desmethoxy and dioxoloquinazoline derivatives. Reagents and conditions: (a) HNO3, Ac2O, rt; (b) Na2S2O4, TBAB, DCM/H2O, rt; (c) ClCOOCH3, DIEA, DMA/DCM; (d) H2O, NaOH, EtOH, reflux; (e) POCl3, PhNEt2, MeCN, reflux; (f) 4-amino-1-benzylpiperidine, DIEA, THF, rt; (g) cyclic amine, 185 °C (neat), microwave, 30 min or amine, toluene, reflux, overnight.

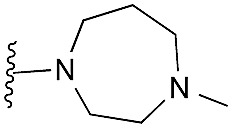
Finally, the pyrimidine ring portion of the quinazoline scaffold was varied. In light of the ligand–G9a interaction analysis above (Fig. 2), we decided to remove the N-3 nitrogen, since obvious interactions with G9a were not apparent. Thus, dimethoxyquinoline derivatives 41 and 42 were synthesised (Scheme 3). Unlike the synthesis of the related quinazoline derivatives, the regioselective synthesis of our target 2,4-diaminoquinolines was challenging. We found very few examples of such compounds in the literature, one example being from Campbell and co-workers, who have previously reported the synthesis of quinoline analogues of the antihypertensive diaminoquinazoline drug, prazosin.26,31 With a slight modification of their methodology, we were able to prepare free-amino quinoline derivatives 40a and 40b. Thus, the commercially available 38 was converted to an intermediate aminobenzonitrile and it was then treated with triethylorthoacetate under slightly reduced pressure to yield an imidate, which was heated with either methylpiperazine or methylhomopiperazine to yield corresponding derivatives 39a and 39b, respectively (Scheme 3). The ring closure of 39a and 39b was carried out by treatment with zinc chloride, which occurs presumably via the enamine tautomer.32,33 Finally, the free-amino quinoline analogues 40a and 40b were converted to target compounds 41 and 42 via reductive amination with 1-benzyl-4-piperidone.
Scheme 3. The synthesis of quinoline derivatives. Reagents and conditions: (a) HNO3, Ac2O, rt; (b) Na2S2O4, TBAB, DCM/H2O, rt; (c) CH3C(OCH2CH3)3, 150 °C, (d) 1-methylhomopiperazine or 1-methylpiperazine, pTsOH, 100 °C; (e) ZnCl2, DMA, reflux; (f) 1-benzyl-4-piperidone, AcOH, toluene, reflux, Dean–Stark, then a reducing agent (NaBH4 or NaBH(OAc)3), AcOH, THF, reflux.

Results and discussion
All the compounds synthesised were evaluated for G9a inhibitory activity using a previously reported SPA assay.17 The preliminary screen was conducted at three concentrations (50, 10, 1 μM), with full dose response data derived for compounds with >20% inhibition at the 1 μM dose. UNC0638 (Fig. 1) was used as a positive control and gave a comparable activity to that observed previously (IC50 ∼ 2.5 nM). Expectedly, BIX-01294 and the other quinazolines (2–4) exhibited IC50 values in the nanomolar to low micromolar range. For the derivatives where the benzenoid ring had been replaced with a furan, thiophene, imidazole or a cyclopentane ring (12–25), no significant G9a inhibitory activity was observed. While obvious interactions are not apparent for this region of the quinazoline G9a inhibitors (vide supra), except when a ‘lysine mimic’ is present at C-7, it would seem the benzenoid ring does play an important role in binding. Interestingly, the recently reported G9a inhibitor A-366 shares a similar structural feature with BIX-01294 in this regard.18
Since the benzenoid ring is important for inhibitory activity, the role of the dimethoxy substituents on this ring should be questioned. Desmethoxyquinazoline derivatives (34–37) were found to be far less active against G9a, which is in agreement with a recent report on related analogues,35–42 and demonstrates that these substituents are also of importance. A number of theoretical and experimental studies have shown that ortho-dimethoxybenzenes prefer to adopt a co-planar conformation, with the sp2 oxygen lone pairs projecting towards each other and the corresponding methyl groups angled away from each other. Indeed, such a conformation is apparent in the inhibitor-G9a/GLP crystal structures (such as PDB ; 3FPD,43; 3K5K (Fig. 2),11 and ; 3RJW14). Thus, analogues 31–33 (Table 1) were prepared where the methoxy groups were replaced by a dioxalone ring; forcing the oxygen lone pairs to point in the opposite direction. Interestingly, these analogues were also found to be largely devoid of G9a inhibitory activity, suggesting the conformational preference of the dimethoxy functionality to be important for G9a activity.
In inhibitor designs where the dimethoxybenzenoid moiety was retained, but N-3 (Fig. 1) replaced by carbon atom (41 and 42), significant activity was observed. Indeed, to our delight, 41 and 42 were found to be ∼5-fold more potent than the parental inhibitor BIX-01294. This result is clearly in agreement with our initial inhibitor-G9a crystal structure analysis (vide supra), which suggested that the N-3 nitrogen of the quinazoline scaffold might not be essential for G9a binding (Fig. 2). Comparison of the calculated pK as revealed that N-1 of the quinolines 41 and 42 is more basic than the corresponding nitrogen of quinazolines 1 and 2 (Table 1). This computational prediction is in agreement with experimental data which demonstrates diaminoquinolines to be more basic than the analogous quinazolines.25,26 N-1 of the quinoline derivatives were predicted to have the highest pK as (10.57 and 9.84 for 41 and 42, respectively) of all the analogues prepared (Table 1). Interestingly, the N-1 atom in all inactive molecules (12–25, Table 1) were predicted to have lower pK a values. A similar analysis between quinazoline and quinoline activity based on pK a dependent interactions has been described earlier for the design of α1-adrenoreceptor antagonists.25,26
In light of this trend, we measured the experimental pK a values for compounds 1 (BIX-01294) and 41 in the pH range 2–12 using UV-vis spectroscopy. As expected, both compounds were found to be tribasic due to the presence of two aliphatic basic centres (the benzylpiperidine nitrogen and the homo piperazine nitrogen) in addition to N-1. The experimental pK a values of BIX-01294 (1) were 6.94 ± 0.01, 8.24 ± 0.05 and 9.22 ± 0.04. The experimental pK a values of quinoline 41 were 7.14 ± 0.01, 8.05 ± 0.02 and 9.50 ± 0.01. Since the largest spectral change for 1 and 41 would be expected upon ring protonation, the UV-vis spectra observed upon each protonation event can be used to assign the pK a of N-1 (see Fig. S5 and S6, ESI†). We therefore assign the pK a of N-1 of quinazoline 1 to be 6.94 ± 0.01 and N-1 of quinoline 41 to be 9.50 ± 0.01.
This data is highly suggestive that a protonated N-1 position is required for good G9a binding, through a strong electrostatic interaction with Asp1088. Indeed, using the Henderson–Hasselbalch equation, under the given assay conditions (pH 8), 96.9% of 41 will be in the N-1 protonated form, whereas for BIX-01294 (1), 8.0% of the molecule will be N-1 protonated. We note however that there is an alternative explanation of the excellent potency of our quinoline hit compounds: the enthalpy loss due to breakage of a hydrogen bond between N-3 and water before ligand binding is not been compensated for upon ligand binding to G9a.44 Despite this trend in N-1 pK a, inactive quinazolines 31–37 are predicted to have N-1 pK a values that are similar to active derivatives 1–4. Therefore clearly both the dimethoxybenzenoid ring system and basic N-1 functionality are important features for G9a binding and hence inhibitory activity.
Our initial design of the new fused heterocyclic scaffolds was based on the hypothesis that they could bind to the substrate binding pocket of G9a with a pose comparable to that of the quinazoline co-crystallized ligand (Fig. 2). In light of the in vitro data obtained, all molecules prepared were docked without constraints into the substrate pocket of G9a to see if active and inactive molecules could be differentiated computationally. The G9a X-ray structure co-crystallised with UNC0224 (PDB code ; 3K5K)11 was used, employing both standard precision (SP) and extra precision (XP) modes of the Glide program (Schrodinger, see ESI†).
Interestingly, analysis of the top scoring poses revealed that none of the inactive molecules could reproduce the expected pose in either SP or XP mode (Table 1). On the other hand all active derivatives were predicted to bind to G9a in a comparable manner to UNC0224, in at least one of the precision modes. For example, the poses of quinolines 41 (Fig. 3) and 42 (see ESI†) overlaid perfectly with UNC0224, with the protonated N-1 functionality interacting with Asp1088. Similarly, other active compounds 1–3 exhibited a similar binding mode (see ESI, Fig. S1–S4†). Overall, the docking scores gave a qualitative correlation with the IC50 data; compounds 1 and 41 giving higher scores than 2 and 3.
Fig. 3. Docking pose of quinoline analogue 41 (grey sticks) overlaid with the co-crystallized quinazoline derivative, UNC0224 (pink sticks; PDB ; 3K5K), in the G9a substrate binding pocket. Purple dashed lines display H-bonds. As expected, protonated the N-1 and 4-amino moiety of 41 were shown to interact with Asp1088 and Asp1083, respectively.

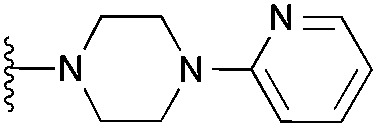
Interestingly, the docking study reinforced the importance of dimethoxy structural feature in acquiring the correct pose. For example, quinazoline derivatives either lacking dimethoxy groups (34–37) or with the bridged methoxy groups (31–33) did not display the desired pose which is in agreement to their lack of in vitro activity. Also, moderately active derivative 4 could not reproduce the expected pose in spite of possessing dimethoxy groups, plausibly due to the large pyridylpiperazine substituent at position 2.
In light of the excellent potency of our quinoline inhibitors 41 and 42 against G9a, the selectivity of these compounds was examined in a methyltransferase enzyme panel, and compared to BIX-01294. This panel consisted of twenty three additional methyltransferases including sixteen HKMTs, six protein arginine methyltransferases (PRMTs), and one DNA methyltransferase (DNMT) (see ESI, Table S1†).17 Compound 41 and 42 were found to be equipotent against G9a and GLP (G9a-like protein, EHMT1), which is not surprising given the high degree of homology between the G9a and GLP SET domains. Such dual G9a/GLP activity is a common feature of the diaminoquinazoline inhibitors17 and was shared by BIX-01294. However to our delight, quinoline inhibitors 41 and 42 were found to be inactive against all other methyltransferases in the panel, with the exception of SETD2 and EZH2 for which a moderate inhibitory activity was at the highest concentrations surveyed (50 μM).
Conclusions
In summary, we have designed and synthesised a variety of heterocyclic derivatives in order to identify novel G9a inhibitors and better define the pharmacophoric features associated with the core heterocycle. These efforts resulted in the identification of potent and selective G9a/GLP inhibitors 41 and 42 based on a quinoline scaffold. Activity and computational data highlight the importance of the dimethoxy groups on the benzenoid ring of this scaffold and a basic nitrogen at position 1 for potent G9a activity. The ability of molecular docking to predict inhibitor activity based on the UNC0224 binding pose, once further validated, may be helpful in the future for predicting and prioritising novel G9a/GLP inhibitors.
Acknowledgments
N.S. was supported by a Royal Thai Government Scholarship and the EPSRC-funded Institute of Chemical Biology Doctoral Training Centre. S.S. acknowledges European Commission for awarding Marie Curie International Incoming Fellowship (Agreement no. 299857). The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Boehringer Ingelheim, the Canada Foundation for Innovation, the Canadian Institutes for Health Research, Genome Canada through the Ontario Genomics Institute [OGI-055], GlaxoSmithKline, Janssen, Lilly Canada, the Novartis Research Foundation, the Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and the Wellcome Trust [092809/Z/10/Z].
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c4md00274a
References
- Tachibana M., Sugimoto K., Fukushima T., Shinkai Y. J. Biol. Chem. 2001;276:25309–25317. doi: 10.1074/jbc.M101914200. [DOI] [PubMed] [Google Scholar]
- Tao H., Li H., Su Y., Feng D., Wang X., Zhang C., Ma H., Hu Q. Mol. Cell. Biochem. 2014;394:23–30. doi: 10.1007/s11010-014-2077-4. [DOI] [PubMed] [Google Scholar]
- Bouchat S., Gatot J. S., Kabeya K., Cardona C., Colin L., Herbein G., De Wit S., Clumeck N., Lambotte O., Rouzioux C., Rohr O., Van Lint C. AIDS. 2012;26:1473–1482. doi: 10.1097/QAD.0b013e32835535f5. [DOI] [PubMed] [Google Scholar]
- Barski A., Cuddapah S., Cui K., Roh T. Y., Schones D. E., Wang Z., Wei G., Chepelev I., Zhao K. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Ding J., Li T., Wang X., Zhao E., Choi J. H., Yang L., Zha Y., Dong Z., Huang S., Asara J. M., Cui H., Ding H. F. Cell Metabolism. 2013;18:896–907. doi: 10.1016/j.cmet.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X., Chen X., Guan X., Zhang H., Ma Y., Zhang S., Wang E., Zhang L., Han Y. Histopathology. 2014 doi: 10.1111/his.12456. [DOI] [PubMed] [Google Scholar]
- Lee J. S., Kim Y., Kim I. S., Kim B., Choi H. J., Lee J. M., Shin H. J., Kim J. H., Kim J. Y., Seo S. B., Lee H., Binda O., Gozani O., Semenza G. L., Kim M., Kim K. I., Hwang D., Baek S. H. Mol. Cell. 2010;39:71–85. doi: 10.1016/j.molcel.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathert P., Dhayalan A., Murakami M., Zhang X., Tamas R., Jurkowska R., Komatsu Y., Shinkai Y., Cheng X., Jeltsch A. Nat. Chem. Biol. 2008;4:344–346. doi: 10.1038/nchembio.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen K. T., Li F., Poda G., Smil D., Vedadi M., Schapira M. J. Chem. Inf. Model. 2013;53:681–691. doi: 10.1021/ci300596x. [DOI] [PubMed] [Google Scholar]
- Kubicek S., O'Sullivan R. J., August E. M., Hickey E. R., Zhang Q., Teodoro M. L., Rea S., Mechtler K., Kowalski J. A., Homon C. A., Kelly T. A., Jenuwein T. Mol. Cell. 2007;25:473–481. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Liu F., Chen X., Allali-Hassani A., Quinn A. M., Wasney G. A., Dong A., Barsyte D., Kozieradzki I., Senisterra G., Chau I., Siarheyeva A., Kireev D. B., Jadhav A., Herold J. M., Frye S. V., Arrowsmith C. H., Brown P. J., Simeonov A., Vedadi M., Jin J. J. Med. Chem. 2009;52:7950–7953. doi: 10.1021/jm901543m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F., Chen X., Allali-Hassani A., Quinn A. M., Wigle T. J., Wasney G. A., Dong A., Senisterra G., Chau I., Siarheyeva A., Norris J. L., Kireev D. B., Jadhav A., Herold J. M., Janzen W. P., Arrowsmith C. H., Frye S. V., Brown P. J., Simeonov A., Vedadi M., Jin J. J. Med. Chem. 2010;53:5844–5857. doi: 10.1021/jm100478y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F., Barsyte-Lovejoy D., Allali-Hassani A., He Y., Herold J. M., Chen X., Yates C. M., Frye S. V., Brown P. J., Huang J., Vedadi M., Arrowsmith C. H., Jin J. J. Med. Chem. 2011;54:6139–6150. doi: 10.1021/jm200903z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedadi M., Barsyte-Lovejoy D., Liu F., Rival-Gervier S., Allali-Hassani A., Labrie V., Wigle T. J., Dimaggio P. A., Wasney G. A., Siarheyeva A., Dong A., Tempel W., Wang S. C., Chen X., Chau I., Mangano T. J., Huang X. P., Simpson C. D., Pattenden S. G., Norris J. L., Kireev D. B., Tripathy A., Edwards A., Roth B. L., Janzen W. P., Garcia B. A., Petronis A., Ellis J., Brown P. J., Frye S. V., Arrowsmith C. H., Jin J. Nat. Chem. Biol. 2011;7:566–574. doi: 10.1038/nchembio.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konze K. D., Pattenden S. G., Liu F., Barsyte-Lovejoy D., Li F., Simon J. M., Davis I. J., Vedadi M., Jin J. ChemMedChem. 2014;9:549–553. doi: 10.1002/cmdc.201300450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y., Ganesh T., Horton J. R., Spannhoff A., Liu J., Sun A., Zhang X., Bedford M. T., Shinkai Y., Snyder J. P., Cheng X. J. Mol. Biol. 2010;400:1–7. doi: 10.1016/j.jmb.2010.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F., Barsyte-Lovejoy D., Li F., Xiong Y., Korboukh V., Huang X. P., Allali-Hassani A., Janzen W. P., Roth B. L., Frye S. V., Arrowsmith C. H., Brown P. J., Vedadi M., Jin J. J. Med. Chem. 2013;56:8931–8942. doi: 10.1021/jm401480r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweis R. F., Pliushchev M., Brown P. J., Guo J., Li F. L., Maag D., Petros A. M., Soni N. B., Tse C., Vedadi M., Michaelides M. R., Chiang G. G., Pappano W. N. ACS Med. Chem. Lett. 2014;5:205–209. doi: 10.1021/ml400496h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Desponts C., Do J. T., Hahm H. S., Scholer H. R., Ding S. Cell Stem Cell. 2008;3:568–574. doi: 10.1016/j.stem.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Imai K., Togami H., Okamoto T. J. Biol. Chem. 2010;285:16538–16545. doi: 10.1074/jbc.M110.103531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dembele L., Franetich J. F., Lorthiois A., Gego A., Zeeman A. M., Kocken C. H., Le Grand R., Dereuddre-Bosquet N., van Gemert G. J., Sauerwein R., Vaillant J. C., Hannoun L., Fuchter M. J., Diagana T. T., Malmquist N. A., Scherf A., Snounou G., Mazier D. Nat. Med. 2014;20:307–312. doi: 10.1038/nm.3461. [DOI] [PubMed] [Google Scholar]
- Upadhyay A. K., Rotili D., Han J. W., Hu R., Chang Y., Labella D., Zhang X., Yoon Y. S., Mai A., Cheng X. J. Mol. Biol. 2012;416:319–327. doi: 10.1016/j.jmb.2011.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmquist N. A., Moss T. A., Mecheri S., Scherf A., Fuchter M. J. Proc. Natl. Acad. Sci. U. S. A. 2012;109:16708–16713. doi: 10.1073/pnas.1205414109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundriyal S., Malmquist N. A., Caron J., Blundell S., Liu F., Chen X., Srimongkolpithak N., Jin J., Charman S., Scherf A., Fuchter M. ChemMedChem. 2014 doi: 10.1002/cmdc.201402098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordner J., Campbell S. F., Palmer M. J., Tute M. S. J. Med. Chem. 1988;31:1036–1039. doi: 10.1021/jm00400a026. [DOI] [PubMed] [Google Scholar]
- Campbell S. F., Hardstone J. D., Palmer M. J. J. Med. Chem. 1988;31:1031–1035. doi: 10.1021/jm00400a025. [DOI] [PubMed] [Google Scholar]
- Brown R., Fuchter M., Chapman-Rothe N., Srimongkolpithak N., Caron J., Synder J., Ganesh T., Liu J. and Sun A., EU Pat., WO/2013/140148, 2013.
- Heffron T. P., Wei B., Olivero A., Staben S. T., Tsui V., Do S., Dotson J., Folkes A. J., Goldsmith P., Goldsmith R., Gunzner J., Lesnick J., Lewis C., Mathieu S., Nonomiya J., Shuttleworth S., Sutherlin D. P., Wan N. C., Wang S., Wiesmann C., Zhu B. Y. J. Med. Chem. 2011;54:7815–7833. doi: 10.1021/jm2007084. [DOI] [PubMed] [Google Scholar]
- Zhu W., Liu Y., Zhai X., Wang X., Zhu Y., Wu D., Zhou H., Gong P., Zhao Y. Eur. J. Med. Chem. 2012;57:162–175. doi: 10.1016/j.ejmech.2012.09.002. [DOI] [PubMed] [Google Scholar]
- Chiriac M., Axente D., Palibroda N., Craescu C. T. J. Labelled Compd. Radiopharm. 1999;42:377–385. [Google Scholar]
- Campbell S. F., Hardstone J. D., Palmer M. J. Tetrahedron Lett. 1984;25:4813–4816. [Google Scholar]
- Schafer H., Sattler K., Gewald K. J. Prakt. Chem. 1979;321:695–698. [Google Scholar]
- Moore J. A., Kornreich L. D. Tetrahedron Lett. 1963;4:1277–1281. [Google Scholar]
- http://www.rdkit.org/, accessed December, 2013.
- Rotili D., Tarantino D., Marrocco B., Gros C., Masson V., Poughon V., Ausseil F., Chang Y., Labella D., Cosconati S., Di Maro S., Novellino E., Schnekenburger M., Grandjenette C., Bouvy C., Diederich M., Cheng X., Arimondo P. B., Mai A. PLoS One. 2014;9:e96941. doi: 10.1371/journal.pone.0096941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson G. M., Kollman P. A., Domelsmith L. N., Houk K. N. J. Am. Chem. Soc. 1979;101:2344–2352. [Google Scholar]
- Caillet J., Caillet J. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1982;38:1786–1791. [Google Scholar]
- Schaefer T., Sebastian R., Lemire A., Penner G. H. Can. J. Chem. 1990;68:1393–1398. [Google Scholar]
- Gerzain M., Buchanan G. W., Driega A. B., Facey G. A., Enright G., Kirby R. A. J. Chem. Soc., Perkin Trans. 2. 1996:2687–2693. [Google Scholar]
- Emsley J. W., Foord E. K., Lindon J. C. J. Chem. Soc., Perkin Trans. 2. 1998:1211–1218. [Google Scholar]
- Tsuzuki S., Houjou H., Nagawa Y., Hiratani K. J. Chem. Soc., Perkin Trans. 2. 2002:1271–1273. [Google Scholar]
- Velde C. V., Bultinck E., Tersago K., Alsenoy C. V., Blockhuys F. Int. J. Quantum Chem. 2007;107:670–679. [Google Scholar]
- Chang Y., Zhang X., Horton J. R., Upadhyay A. K., Spannhoff A., Liu J., Snyder J. P., Bedford M. T., Cheng X. Nat. Struct. Mol. Biol. 2009;16:312–317. doi: 10.1038/nsmb.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H., Huang D. PLoS One. 2011;6:e19923. doi: 10.1371/journal.pone.0019923. [DOI] [PMC free article] [PubMed] [Google Scholar]